

Abstract
Recent developments in instrumentation and bioinformatics have led to new quantitative mass spectrometry platforms including LC-MS/MS with data-independent acquisition (DIA) and targeted analysis using parallel reaction monitoring mass spectrometry (LC-PRM), which provide alternatives to well-established methods, such as LC-MS/MS with data dependent acquisition (DDA) and targeted analysis using multiple reaction monitoring mass spectrometry (LC-MRM). These tools have been used to identify signaling perturbations in lung cancers and other malignancies, supporting the development of effective kinase inhibitors and, more recently, providing insights on therapeutic resistance mechanisms and drug repurposing opportunities. However, detection of kinases in biological matrices can be challenging; therefore, activity-based protein profiling enrichment of ATP-utilizing proteins was selected as a test case for exploring the limits of detection of low abundance analytes in complex biological samples. To examine the impact of different MS acquisition platforms, quantification of kinase ATP-uptake following kinase inhibitor treatment was analyzed by four different methods: LC-MS/MS with DDA and DIA, LC-MRM, and LC-PRM. For discovery datasets, DIA increased the number of identified kinases by 21% and reduced missingness when compared to DDA. In this context, MRM and PRM were most effective at identifying global kinome responses to inhibitor treatment, highlighting the value of a priori target identification and manual evaluation of quantitative proteomics datasets. We compare results for a selected set of desthiobiotinylated peptides from PRM, MRM, and DIA and identify considerations for selecting a quantification method and post-processing steps that should be used for each data acquisition strategy.
Keywords: Lung Cancer, Targeted Therapy, Activity-Based Protein Profiling, Quantitative Mass Spectrometry
INTRODUCTION
Many cancers, e.g. lung adenocarcinomas, are driven by genetic and epigenetic abnormalities and a dysfunctional microenvironment, resulting in altered signal transduction pathways; these complex networks require phosphorylation profiling and other proteomics approaches to enable comprehensive examination.1–2 Protein phosphorylation, through regulation of protein-protein interactions and enzyme activity levels, leads to changes to cellular phenotype in response to external cues.3–4 Because kinases modulate signaling events vital to cancer progression, including proliferation, motility, and cell survival, their activity is frequently altered in cancer cells.1, 4 Improvements in detection of these alterations have led to a renaissance in targeted therapy development, primarily kinase inhibitors (KIs), over the last decade. While dozens of newly approved inhibitors hold great promise in improving patient outcomes, the effectiveness of treatments relies largely on predictive biomarkers.5–6 Clinically significant responses have been observed for targeted therapy in non-small cell lung cancer (NSCLC) patients harboring KI sensitizing mutations, such as Erlotinib treatment of EGFR mutant tumors.7 However, drug resistance is eventually acquired through numerous mechanisms.8–9 Therefore, quantification of kinase activity and changes in phosphorylation are necessary to identify effective biomarkers, elucidate drug resistance mechanisms, repurpose existing FDA approved drugs, and define new avenues for patient treatment.
However, kinases present a challenge for global profiling methods, like proteomics. Therefore, enrichment methods including activity-based protein profiling (ABPP) have emerged as useful approaches to identify active signaling pathways in model systems and patient tumors.10–14 Activity-based protein profiling uses an ATP mimetic probe to desthiobiotinylate lysines near the active site in order to use avidin-biotin capture chemistry for enrichment.12 The chemical probe enriches all ATP-binding proteins, pulling down a large number of other enzyme classes, including those involved with metabolism and stress-response. While 100-200 kinases can be typically be observed in ABPP experiments of NSCLC cell line samples using data dependent acquisition LC-MS/MS, kinase peptides represent less than 10% of the total observed peptides and datasets may have large numbers of missing values for desthiobiotinylated kinase peptides. Therefore, this ABPP kinase model system serves as a microcosm of the general proteomics problems related to quantification of low abundance components of a complex mixture. Here, we quantify desthiobiotinylated kinase peptides amongst the complex background of higher abundance labeled peptides from all ATP-binding proteins using multiple approaches to compare sensitivity, reproducibility of detection, and quantification.
Multiplexed liquid chromatography-multiple reaction monitoring mass spectrometry (LC-MRM or selected reaction monitoring, SRM) is a useful tool in translational cancer research, providing novel biological insights including drug resistance mechanisms, which can be used to examine tumor biology in situ or measure correlates in clinical trials.15–18 Using discovery datasets from LC-MS/MS with data-dependent acquisition (DDA), relevant targets are selected for LC-MRM analysis on a triple-quadrupole mass spectrometer. LC-MRM panels, consisting of specific precursor and fragment ion pairs, can be designed to quantify peptides from proteins involved in the same biological process or signaling network, resulting in pathway analysis.19 In order to interrogate perturbations resulting from promiscuous kinase inhibitors that cause diverse changes in signaling, scheduled, multiplexed LC-MRM panels have been developed to interrogate about half the kinome.11, 13 LC-MRM is the established method for quantitative proteomics, but the advent of novel hybrid mass spectrometers with high resolution and accurate mass measurement capabilities has led to the development of new quantitative approaches. Parallel reaction monitoring mass spectrometry (PRM) uses precursor selection to generate full-scan high resolution MS/MS spectra with mass measurement error for the fragment ions expected to be less than 10 ppm. LC-MRM using a triple-quadrupole instrument, on the other hand, uses comparatively low resolution mass filtering for both precursors and fragments (often widths of 0.7 Th), which can result in interference. LC-PRM has emerged as a powerful tool to profile cellular phenotypes using protein assay panels with similar capabilities to LC-MRM that relies on high resolution accurate mass measurements to reduce or remove interference.20–23
Building on improvements in instrumentation, a new acquisition strategy has been developed for LC-MS/MS: data-independent acquisition (DIA).24–26 Instead of attempting to isolate and fragment individual precursors (i.e. DDA), DIA samples all peptides across specified m/z windows sequentially selected to cover the desired mass range.25 The fundamental nature of DDA, which samples the most abundant ions in each scan, results in undersampling as well as a decrease in repeated identification of lower abundance peptides; ultimately, the challenges of missing data will result in the reduction of sample number, negatively impacting precision. With DIA, undersampling can be reduced, yielding more consistent and comprehensive peptide datasets leading to more effective quantification. The improvement in peptide quantification using DIA compared to DDA has been demonstrated.27–29 However, the resulting chimeric spectra are more challenging to interpret; therefore, specific software and a high-quality spectral library are necessary to determine peptide identifications.30 The goal is to achieve the precision of quantification expected for LC-MRM/PRM (shortened to MRM or PRM throughout the paper) approaches, while retaining the ability to detect and quantify other peptide by library matching or make novel discoveries using additional database search strategies. To compare the four methods described above, quantification of low abundance desthiobiotinylated peptides from kinases in a complex mixture is evaluated in this study using cell line models of lung cancer treatment. The strengths and weaknesses of each platform are explored using a lung cancer cell line, H1993, treated with different multi-target kinase inhibitors to examine the ability to detect known drug targets (as positive controls) and changes in downstream signaling.
METHODS
Cell Culture and Lysis
NCI-H1993 cells were obtained from the Moffitt Cancer Center Lung Cancer Center of Excellence Cell Line Core, where they have been authenticated with short tandem repeat analysis and routinely tested to certify that they are free of mycoplasma contamination. Cells were grown in RPMI-1640 media containing L-glutamine and HEPES (Gibco), supplemented with 10% fetal bovine serum (HyClone), 10 U/mL Penicillin/Streptomycin (Gibco), and 2.5 g/L glucose (Sigma). Cells were grown to 70% confluence for about 24 hours, and then treated with DMSO, 200 nM BEZ-235, or 500 nM Crizotinib (Selleck Chemicals) for 24 hours before harvesting. Plates were washed twice with ice cold PBS containing 1 mM Sodium orthovanadate (Sigma) prior to scraping the cells off the culture dish in 600 μL of Pierce IP Lysis Buffer containing protease inhibitors (Thermo). Cell extracts were sonicated on ice and cleared by centrifugation at 18,000 × g for 20 minutes at 4° C prior to desalting using Zeba Spin Columns (Thermo). The protein concentration in each lysate was determined using Bradford Assays (Coomassie Plus Protein Assay, Thermo).
Activity-based Protein Profiling and MS Sample Prep
Cell lysates were labeled for activity-based protein profiling (ABPP) following the manufacturer’s recommendations for the Pierce Kinase Enrichment Kit with ActivX Probes (Thermo).12 Briefly, 1 mg of total protein from each desalted lysate was used in quadruplicate for each treatment condition. Following incubation with 20 mM MnCl2 for 5 minutes, lysates from cells (from the vehicle controls described above) were treated with DMSO, Dasatinib, or Erlotinib (Selleck Chemicals) at a final concentration of 10 μM for 10 minutes. Lysates were then incubated with 10 μM desthiobiotin-ATP probe for an additional 10 minutes. All incubation steps were performed at room temperature. Labeled lysates were denatured in 5 M urea and 5 mM DTT for 30 minutes at 65° C, and then alkylated with 40 mM iodoacetamide (Sigma). Samples were desalted again into a HEPES digestion buffer and digested overnight with 20 μg Trypsin (Worthington) at 37° C. Digested lysates were incubated for 2 hours at room temperature with high-capacity streptavidin beads to enrich for desthiobiotinylated peptides. Beads were sequentially washed multiple times with lysis buffer, 1x PBS, and water; labeled peptides were eluted using aqueous 50% acetonitrile (ACN) with 0.1% TFA. Eluted peptides were vacuum concentrated (SpeedVac, Thermo) and reconstituted in 20 μL of A solvent (2% ACN with 0.1% formic acid) containing 1 fmol/μL Pierce Retention Time Calibrator (PRTC) standard peptides (Thermo). Samples were pooled to form two biological replicates to allow for multiple injections using different LC-MS acquisition methods (see Supplemental Figure 1 for a simplified workflow diagram).
To examine the reliability of ion signal observed in each method when using lower amounts of input as a functional estimate of LOQ, an untreated H1993 cell line bulk ABPP sample was serially diluted with solvent A to create the following series: Sample A is undiluted, Sample B = 1:3, Sample C = 1:12, Sample D = 1:36, Sample E = 1:108. PRTC standard peptides were spiked into each sample for quality control.
Mass Spectrometry Analysis
Kinase peptide selection for targeted analysis and LC-MRM was performed as previously described.13 All samples were separated by reversed phase nano-liquid chromatography (Dionex RSLC, Thermo) and analyzed by DDA, DIA with quantification using extracted ion chromatograms of peptide precursors (previously described as pSMART),27 PRM, and MRM. Each condition was analyzed with the four LC-MS methods a total of three times from two biological replicates. For DDA and PRM analysis, peptides were separated using a two step gradient, ramping from 5 – 15% B solvent (90% ACN containing 0.1% formic acid) in 5 minutes, followed by a 90 minute linear gradient from 15 – 35% B Solvent in A solvent prepared as described above, on a C18 column 75 μM ID × 50 cm (Acclaim PepMap, Thermo). To be consistent with the gradient used to create the spectral library, peptides were separated using a gradient from 5 – 15% B over 5 minutes, then15 – 50% B solvent for the DIA experiment. Prior to MRM acquisition, peptides were separated on a 90 minute gradient from 5 – 35% B solvent on a C18 column, 75 μM ID × 25 cm (Acclaim PepMap, Thermo).
A different gradient was used for DDA and PRM compared to DIA, because the gradient was optimized specifically for desthiobiotinylated peptides after original datasets were acquired for the spectral library. A longer column (50 cm) is used for discovery data acquisition on the Q Exactive, compared to the 25 cm column used for LC-MRM. Because of this difference in column length, the starting amount of B solvent was decreased to improve hydrophilic peptide separation on the shorter column.
MRM analysis was performed on a TSQ Quantiva (Thermo) with a capillary temperature of 275° C and a spray voltage of 2100 V. The Q1 and Q3 resolution values were set to 0.4 and 0.7, respectively, with a dwell time of 10 ms per transition. A method containing 1,627 transitions (Table S1, Supporting Information) corresponding to 409 desthiobiotin-labeled peptides was scheduled using iRT in Skyline.31–32 Variable retention time windows were used, with the center of the gradient utilizing 10 minute acquisition windows. Collision energy (CE) values were calculated using CE optimization equations empirically derived with previous datasets in Skyline.33
DDA, pSMART/DIA, and PRM samples were analyzed using a Q-Exactive Plus MS with a capillary temperature of 250° C and spray voltage of 2400 V. A DDA “top 16” method with an isolation window of 2 Th around the precursor (0.5 Th offset) and 30 normalized collision energy value (NCE) were used. MS1 resolution was 70,000 and MS/MS resolution was 17,500, with ion accumulation times of 100 and 80 ms, respectively.
DIA was performed using the isolation list in Table S2 (Supporting Information), using variable steps of 5, 8, and 10 m/z with a 1 m/z offset, covering the mass range from m/z 450 to m/z 1332 in 7 cycles. The instrument performed MS1 scans with a resolution of 70,000 and ion accumulation time of 200 ms, followed by eighteen MS/MS scans at a resolution of 17,500 with an ion accumulation time of 80 ms, in each ~2.5 s cycle. PRM analysis used the same kinase desthiobiotin-labeled peptide panel previously described for MRM; the scheduled PRM precursor list is shown in Table S3 (Supporting Information). To reduce crowding, PRM retention time windows were set to 8 minutes around the expected precursor detection time. PRM analysis did not include MS1 data acquisition, and MS/MS resolution was 17,500 with a maximum accumulation time of 100 ms. DDA and DIA data are available through PRIDE34 and ProteomeXchange35; MRM and PRM data are available through Panorama36 (see Notes).
Data Processing and Statistical Analysis
The spectral library used for DIA analysis was generated using DDA files acquired on the same LC-MS/MS platform described, from many lung cancer cell lines. The spectral library was built using Proteome Discoverer 2.1 (Thermo) with the Percolator algorithm (q-value cut off = 0.01) following UniProt database searching using SEQUEST. DIA data files were analyzed using Pinnacle software (v. 1.0.42.0, OPTYS) to perform spectral matching and quantification.
DDA LC-MS/MS analysis was carried out using Andromeda37 and MaxQuant38 for peptide identification and quantification searching data against human entries in UniProt (downloaded December 2015) with the following dynamic modifications: methionine oxidation, cysteine carbamidomethylation, and lysine desthiobiotinylation (+196.121178 Da). DDA and DIA datasets were searched again with Mascot using similar parameters to be compatible with PRIDE requirements.
Skyline was used for peak integration of all PRM and MRM data as well as the DIA data from the analysis of the dilution series.31 Spiked PRTC peptides and peptides from two metabolic proteins, ENOA_HUMAN and G3P_HUMAN, were assessed for retention time stability (see Tables S4-S7 in Supporting Information for average retention times) and ion signal consistency, respectively (see Supplemental Figure 2A,B). ENOA and G3P peptides were also used as a quality control for the sample processing and peptide enrichment. Peptides lacking peaks in the control samples (DMSO) were removed from the dataset. Following manual evaluation of peak picking to eliminate incorrect assignments or transitions with interference, MRM data were exported from Skyline, with quantification by the sum of all transitions. The PRM Skyline document initially scanned for all b and y fragment ions from ion 3 (i.e. b3 and y3) to the second to last ion in both series (i.e. b(n-2) and y(n-2), where n is the number of amino acids in the peptide). The document was then automatically refined to include only the top 5 ions for each peptide, favoring higher m/z fragment ions. Each peak was manually assessed and all transitions with an error greater than 10 ppm were removed.
Raw peak area values for DDA and DIA datasets were processed to remove minor variability using iterative rank order normalization (IRON)39 implemented in Galaxy.40 Statistical analysis and data visual generation was performed using R programming in R Studio.41–42 The following R packages were used for data processing and visualization: ggplot2,43 dplyr,44 reshape2,45 LSD,46 VennDiagram,47 and stringr.48
In DIA and DDA datasets, all peptides with less than 3 values across the dataset (n = 18) or that were not seen in the control (DMSO) sample were removed. Peptides with a Pinnacle score less than 3, meaning inadequate MS/MS data (less than 4 fragment ions matched and ≤ 0.65 MS/MS dot product score) for identification or poor MS1 ion signal insufficient for quantification (≤ 0.94 MS1 dot product score), were removed from DIA datasets. Retention time correlations were also used to remove outliers from the datasets. Concordance across quantitative analysis methods was assessed between peak areas for each data point (protein, peptide, treatment) computing Pearson’s correlation coefficient (R).
Relative quantification (without stable-isotope labeled standards) was performed by log2 transforming the peak areas and the calculating fold changes of inhibitor treatment compared to DMSO vehicle controls. If a peak area was missing in controls or deviated by more than 4x the average peak area across the control replicates, the DMSO peak area for a different technical replicate was used to calculate the fold change (5-15% of peptides, depending on the dataset). Standard error (SE), 95% confidence intervals, and boxplot statistics were calculated for each drug treatment MRM, PRM, and DIA datasets. Significance between SE boxplots was determined using the Wilcoxon Rank-Sum Test for data that did not fit to a normal distribution.
Significance of log2-fold change values was determined using the student’s one-tailed t-test when identifying proteins or peptides that are down-regulated in response to treatment (p < 0.05). To identify peptides that were differentially regulated (up or down), a two-tailed t-test was used. Forest plots show 95% confidence intervals for all peptides observed across MRM, PRM, and DIA that had a log2 fold change less than −1 and a p-value ≤ 0.05 in at least one MS quantification platform.
The dilution series dataset was filtered using a mean bias cutoff (observed versus expected peak area ratio compared to highest average peak area per peptide) of 100% and peak areas of < 1,500 were removed.
Raw data and log transformed fold change calculations are included in the Supporting Information for DDA (Tables S8-S12), DIA (Tables S13-S17), MRM (Tables S18-S22), and PRM (Tables S23-S27).
Pathway Enrichment Analysis
To determine which biological processes were altered following 24 hour BEZ-235 treatment, enrichment analysis using MetaCore GeneGO (Thomson Reuters) gene ontology49 was performed using all protein/peptide pairs with a mean positive or negative fold change larger than 1.8 and a p-value ≤ 0.05 in each platform, analyzed separately or together. This information identified the top ten cellular processes impacted by BEZ-235 treatment, which could indicate potential biomarkers or drug resistance mechanisms. Proteins with changing ATP-uptake computed by both PRM and MRM datasets were used to generate a schematic representation of the altered signaling pathway, using MetaCore GeneGO pathway map creator to identify protein-protein interaction mechanisms.
RESULTS & DISCUSSION
To elucidate the strengths and weaknesses of different quantitative proteomics approaches in the context of low abundance peptides in a complex mixture, the MET amplified lung adenocarcinoma cell line, H1993, was used as a model system due to identification of many kinases and pY sites in a preliminary lung cancer cell line LC-MS/MS (DDA) screening. This study (Supp. Fig. 1) examined the impact of each MS acquisition method from the raw data level to the biological insights drawn from downstream interpretation. Specifically, the global data metrics (correlations, coverage, missingness, and precision), identification of potential drug targets, and elucidation of signaling changes in response to therapy are explored.
Global Comparison of ATP-Uptake in Lung Cancer Cells in Response to Drug Treatment
To ensure datasets were similar to one another, concordance was examined between DDA, DIA, PRM, and MRM (Fig. 1, Supp. Fig. 3). Following normalization (IRON), log2 transformation and filtering for kinase peptides, a Pearson’s product-moment correlation (R) was calculated to confirm the consistency across datasets. Each data point in the correlation plots (Fig. 1A,B, Supp. Fig. 3A,B) are specific to a peptide and treatment, resulting in up to 18 measurements for each peptide. The 903 measurements for kinase peptides observed by both DDA and DIA (Fig. 1a) had a strong correlation (R = 0.92, p-value < 0.001). PRM and MRM peak areas (Fig. 1b) also strongly correlate (R = 0.8, p-value < 0.001). Correlations between MS1 and MS/MS quantification methods are lower due to the evaluations of the overall dataset, rather than peptide-by-peptide comparisons. Differences in the number of transitions selected per peptide and their percentage of the total fragment ion signal contribute to the lower correlation values. Therefore, we will primarily (but not exclusively) focus on the two individual comparisons: DDA vs. DIA and MRM vs. PRM.
Figure 1. Global Comparison of Detection of Desthiobiotinylated Peptides from Kinases Across Data Acquisition Approaches.
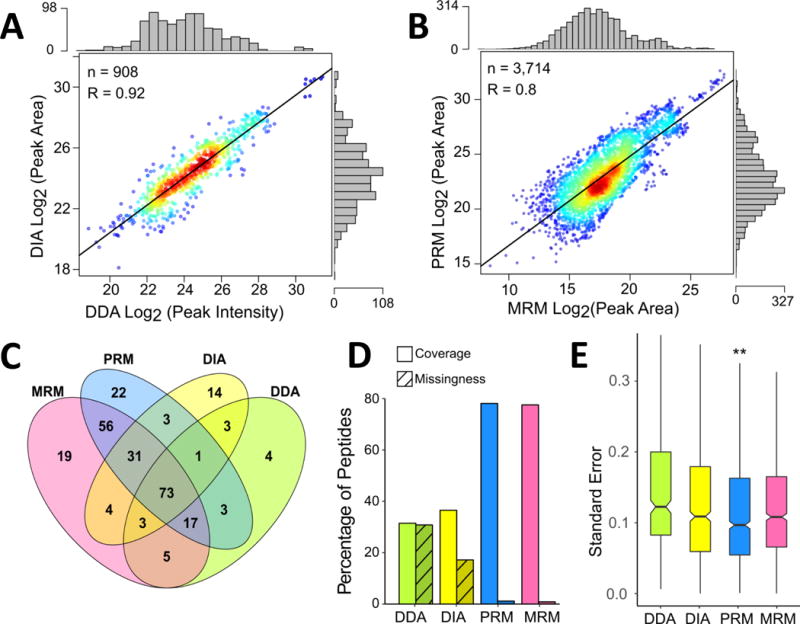
Correlation between log2 transformed peak area or intensity values following IRON normalization for desthiobiotinylated kinase peptides detected in DDA and DIA LC-MS/MS experiments (A). Comparison of peak areas for detected kinase peptides in LC-MRM and LC-PRM datasets (B). In both panels, each point represents a specific peptide measurement in a single replicate and the marginal distribution of measurements across the log2 transformed peak areas or intensities is shown as histograms on the opposite axis. R values are calculated for Pearson correlation and p-values < 0.001. Venn diagram (C) comparing the numbers of kinases detected and quantified by each method. Assessment of kinase peptide detection and data missingness across platforms (D). Coverage is defined as the percentage of peptides quantified in each method compared to the total identified across all MS methods. Missingness is the percentage of missing values for peptides detected with each method. Boxplots showing the distribution of standard errors for means of log2 fold-change ratios following drug treatment (E). Outliers were removed from the boxplots to retain scale. (**: p-value < 0.01, calculated using a paired t-test, compared to each other method).
Distance from the regression line correlating LC retention times in the different experiments was used to eliminate outliers for data analysis moving forward. Outliers generally presented themselves as a cluster of points, which would correspond to the same peptide, resulting in removal of data for 6 peptides from the MRM and PRM datasets due to poor data quality (based on poor LC elution profiles and peak shapes, or fluctuating retention time). Reduced correlation in quantification using PRM and MRM datasets may be attributed to the use of different instrumentation (quadrupole-orbital ion trap vs. triple-quadrupole) and quantification was performed using potentially different sets and numbers of MS2 fragment ions.
Across all four platforms, peptides from a total of 260 kinases were observed. The MRM and PRM panels consist of 264 kinases, with a large portion of those identifiable in the H1993 cell line. DDA, DIA, PRM, and MRM were able to quantify 109, 132, 206, and 208 kinases, respectively, across the whole dataset (Fig. 1C). A total of 73 kinases (28% of the total) were quantified across all four platforms. The DDA dataset had the fewest quantifiable kinases; although more were identified, not all were observed consistently enough for quantification. The 21 kinases quantified by only DDA and DIA represent unique information gained from including the “discovery” aspect of an LC-MS/MS experiment. MRM and PRM acquisition methods, however, were able to quantify 97 unique kinases, which is an improvement on the discovery acquisition methods (DDA/DIA). Next, the coverage and consistency of measurement at the peptide level was assessed (Fig. 1D, Supp. Fig. 3C). Out of the 409 peptides included in the MRM/PRM panels, 356 peptides were observed between MRM and PRM datasets. About 78% were observed in both (276 with MRM and 277 with PRM), each with their own set of unique peptides. Of the peptides in the targeted panels, 130 and 112 were quantifiable by DDA and DIA, respectively (Supp. Fig. 3C). 286 peptides total were quantified by DIA, however, indicating that the DIA dataset included a higher number of peptides per protein. To assess reproducibility of peptide detection, missingness across samples for the set of observed peptides specific to each method was assessed. MRM and PRM both had less than 100 missing values for the whole set of kinase peptides (43 missing from MRM and 58 missing from PRM), representing less than 2% of the observed values. DIA dataset resulted in 884 missing values (17%), while DDA contained the most missing values: 987 (31%).
The distribution of standard errors (SE) following log2 transformation and fold-change calculation (the final datasets for biological interpretation) were compared (Fig. 1E). The SE was used to evaluate the precision of measurement in each dataset. PRM had the lowest average standard error, 0.122, which was significantly lower than the other three methods (p-values ≤ 0.01). MRM acquisition resulted in an average SE of 0.132 and DIA’s was 0.143, although the difference was not significant. DDA was the poorest performer in this analysis, which was expected due to the larger number of missing data points for statistical calculations. Both DIA and DDA had increased variability of standard error values and more outliers, (removed from Fig. 1E). MRM acquisition resulted in the lowest variability and fewest outliers. Larger SEs often resulted for peptides that were either increasing or decreasing in the dataset due to variability from biology and experimental preparation.
Results indicate strong agreement across datasets, showing the quality of all data acquisition and processing methods is sufficient for quantification and downstream analyses for a selected subset of labeled kinase peptides. Global results demonstrate that both MRM and PRM acquisition allow quantification of the largest numbers of peptides. DIA and DDA are still able to provide additional value through the identification of new kinases or desthiobiotinylation sites, with DIA showing improved coverage and fewer missing values than DDA. The kinases quantified by the discovery acquisition methods could be added to the MRM/PRM kinase panel. However, this leads to the main limitation to PRM and MRM: targeted MS/MS panels can only include a few hundred peptides due to MS acquisition speed limitations and the resolution of the chromatography. While data quality is improved by PRM and MRM, clear upper limits on the number of peptides and kinases should be considered when deciding which quantitative method to use. DIA can be used to maximize the number of proteins and peptides quantified, while reducing missingness and improving coverage compared to DDA. Even in the context of low abundance peptides amongst great complexity, its superiority to label-free DDA quantification is demonstrated. For targeted quantification, average and median SEs calculated from the MRM dataset were higher compared to PRM. This difference could result from noise and interfering ions due to the wider mass filtering compared to PRM high resolution data analysis. Additionally, PRM data from the orbital ion trap does not include the baseline, which could also impact measurement precision.
Comparison of MS Quantification Methods for Determining Kinase Inhibitor Targets
Kinome profiling using chemical proteomics has identified numerous kinases as off-targets for selective KIs.50–51 Identification of all inhibitor-kinase interactions can identify drug repurposing opportunities in different cancer types and explain unintended side effects in patients.52 Therefore, we have selected a few KIs as a test case to compare the different data acquisition strategies. H1993 cell lysates were treated with 10 μM Erlotinib, an EGFR inhibitor, or Dasatinib, a SRC family-kinase inhibitor, prior to labeling with the desthiobiotinylating ATP-probe (see workflow diagram, Supp. Fig. 1). Erlotinib and Dasatinib molecular targets should have reduced ATP-probe uptake in treatment samples compared to lysates treated with vehicle (DMSO). Erlotinib and Dasatinib were selected as the model for method comparison because they are well characterized KIs.53 Because many kinase targets have been identified,54–56 the two KIs provide a known system to tease apart the differences between quantitative platforms. Only down-regulated peptides were included in the analysis because lysates, not live cells, were treated, resulting in inhibitors competing with the ATP-probe for the kinase domains. Any peptides that were increased upon lysate treatment may be artifacts, rather than biological feedback or therapeutic escape mechanisms. Artifacts may include ion suppression or a false increase in ATP-uptake by non-targeted kinases as a result of inhibitor treatment with a high concentration (10 μM) of promiscuous inhibitors, thus increasing the amount of unbound ATP probe.
MRM, PRM, and DIA identified a number of peptides corresponding to known kinase targets57 upon Erlotinib treatment, including EGFR (Fig. 2A-C). PRM and MRM exhibited similar distribution of fold-changes and p-values, while down-regulated peptides quantified by DIA resulted in increased p-values because of higher variability in the measurements (Fig. 2A, Supp. Fig. 4A-B). MRM analysis (Supp. Fig. 4A) resulted in 15 peptides corresponding to 12 kinases (log2-Fold-change ≤ −1, p-value ≤ 0.05) while PRM (Supp. Fig. 4B) yielded 23 peptides with decreasing signal corresponding to 18 kinases; the majority (11) of the kinases were shared between the two platforms (Fig. 2B, Supp. Fig. 4). While quantification using DIA (Supp. Fig. 4C) resulted in fewer peptides (17) and kinases (11) with decreased ATP-uptake, the number of peptides per kinase was higher than either PRM or MRM, increasing confidence in these identifications and perhaps improving the confidence in the quantitative differences despite lower p-values at the peptide level. All kinases with significantly reduced ATP-uptake upon Erlotinib treatment (EGFR, EPHB4, ILK, MET, SLK, STK10, ULK3) have been previously identified as Erlotinib targets.50, 56
Figure 2. Kinases downregulated by Erlotinib and Dasatinib across three quantitative platforms.
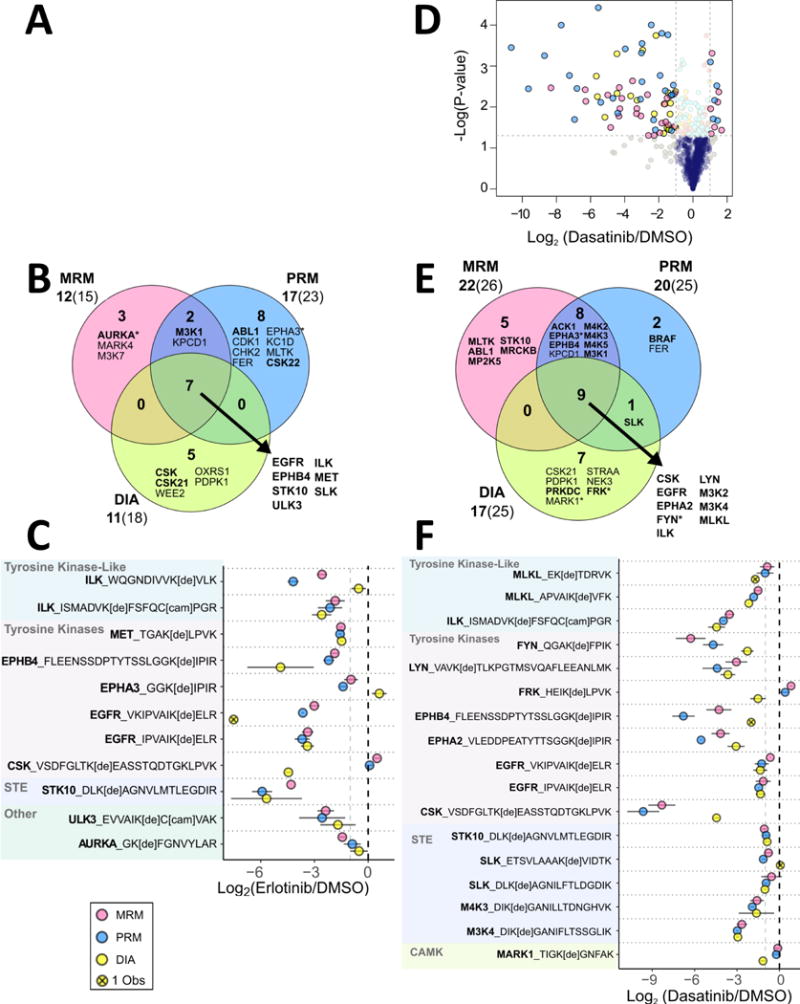
Volcano plot (A) of average desthiobiotin-labeled peptide fold changes (n = 2-3) with statistical significance in H1993 cell lysate treatment with 10 μM Erlotinib. P-values were calculated using a two-tailed T-test. The cut off lines are at a p-value of 0.05 and a log2(Fold Change) of ± 1. Overlap between the platforms is demonstrated (B), with the list of proteins down-regulated by all platforms (in the center of each Venn diagram) listed on the lower right. Proteins in bold have been previously identified as Erlotinib molecular targets. The asterisk indicates that the peptide(s) identified are not unique to that protein. Proteins that had a log2(Fold Change) ≤ −1 and a p-value ≤ 0.05 were included. Beneath each quantitative method name is the number of down-regulated proteins (bold) followed by the number of peptides. Desthiobiotin-labeled peptides down-regulated by Erlotinib treatment in at least two MS platforms (C) are shown grouped by kinase family. Points represent the average a log2(Fold Change) with bars indicating 95% confidence intervals. Panels D-F show similar analyses for H1993 cell lysates treated with 10 μM Dasatinib.
Dasatinib treatment resulted in a greater number of kinases (32 compared to 25 after Erlotinib treatment) with reduced ATP-uptake, which is consistent with expectation due to the larger number of known Dasatinib targets, shown in Figure 2d-f.53, 55, 58 Significantly decreased peptides from PRM analysis had lower p-values and higher negative fold-change values compared to MRM and DIA (Fig. 2D, Supp. Fig. 5). Both MRM and PRM identified CSK as the kinase with the largest reduction in ATP-uptake (Fig. 2E-F, Supp. Fig. 5). Quantification in response to Dasatinib by MRM, PRM, and DIA showed a greater degree of correlation, although the DIA dataset had more missing values in peptides observed across all platforms. Nine kinases that are known Dasatinib targets (CSK, EGFR, EPHA2, FYN, ILK, LYN, M3K2, M3K4, and MLKL) were reduced in all analyses. Seventeen out of 32 kinases were common between PRM and MRM results. Kinases only reduced with statistical significance in PRM (BRAF and FER) had either poor peak quality in MRM or the p-value was not significant. Of note, 5 peptides that were observed to be significantly reduced in MRM quantification (Supp. Fig. 5a) were completely missing from the Dasatinib post-treatment dataset in PRM analysis, even though they displayed strong signals in control samples. These included peptides from known Dasatinib targets: ABL1, CSK, ILK, and LYN. For the purpose of this analysis, imputation was not performed. When using PRM quantification, imputation could be necessary or peptides that are strongly differentially expressed in datasets (or not detected in one sample group) in order to fully evaluate the relevant biology.
The peptides that were observed across all platforms and significantly decreased in at least one dataset demonstrate the consistency in quantification (Fig. 2C, F). There was a greater degree of agreement in MRM and PRM results, with occasional conflicting data from DIA. The DIA dataset had the largest number of kinases that had not been previously identified as drug targets, perhaps indicating novel information, but also raising potential concerns related to false discovery rate. One advantage of DIA is that method scheduling is not required, which can be challenging with PRM and MRM quantification of large panels. For example, a DBT-peptide from STRAA_HUMAN indicated reduced ATP-probe uptake (~50 fold decrease) in the DIA dataset, but not in MRM and PRM results (Supp. Fig. 5). The peak quality and low mass measurement error of the DIA identification indicate that this is the correct peptide, revealing an incorrect retention time window in PRM and MRM methods that led to data loss for this peptide (Supp. Fig. 6). DIA requires less upfront work for acquisition method programming and testing compared to PRM and MRM, but more effort on subsequent data analysis.
PRM and MRM each exhibited advantages and disadvantages in a peptide-specific manner. PRM provides improved sensitivity in quantification of peptides that have interfering ion signals in MRM due to comparatively low resolution of quadrupole mass selection (0.4 or 0.7 m/z resolution for the mass selection windows in Q1 and Q3). Larger peptides with more opportunity for fragment ion signal dispersion (i.e. signal splitting) do not perform as well in MRM when compared to PRM. With proper peptide and fragment ion selection for peptides 7 to 15 a.a. in length, results indicate that MRM has a superior lower limit of detection. Following lysate incubation with Erlotinib and Dasatinib, known targets were sometimes missing all together in post-treatment PRM datasets, while detectable in MRM. Figure 3 shows representative peaks for WQGNDIVVKVLK from ILK, a known target of both Erlotinib and Dasatinib. Patricelli et al.12 reported ILK IC50 values of 1.1 μM and 0.24 μM for Erlotinib and Dasatinib, respectively. ABPP-LC-MRM quantification recapitulates that Dasatinib has increased potency against ILK compared to Erlotinib. MRM quantification showed a 6-fold decrease in ILK ATP-uptake upon Erlotinib treatment and an 11-fold decrease when treated with Dasatinib. PRM analysis of this ILK peptide had a quantifiable peak in the untreated sample, but no peak present following treatment with either inhibitor, indicating that ILK is a target for both, though the extent of inhibition cannot be determined.
Figure 3. Comparison of LC-MRM and LC-PRM Quantification of Differences in a Desthiobiotinylated Peptide Ion Signal due to Target Inhibition by Dasatinib and Erlotinib.
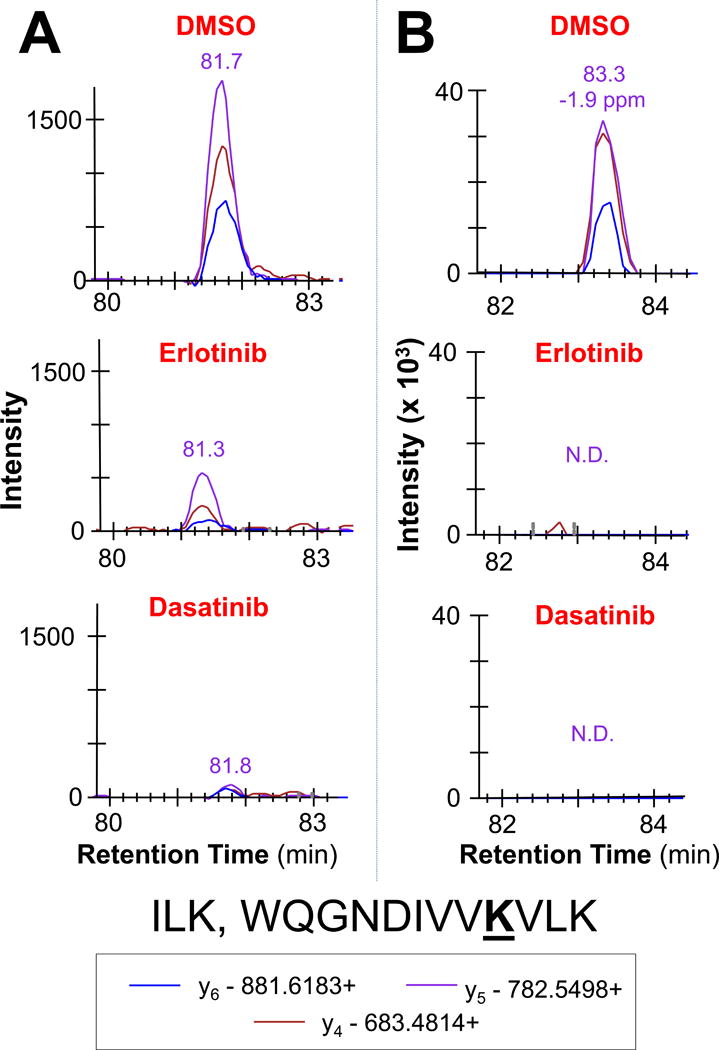
Example peaks for a peptide from ILK are shown, which is a stronger target for Dasatinib compared to Erlotinib. Quantifiable peaks are present in LC-MRM data (A), but not LC-PRM (B). N.D. indicates not detected.
To further investigate the relative values for lower limits of detection across quantitative platforms, reproducibility was assessed for a dilution series spanning 2 orders of magnitude (Supp. Fig. 7A-B). When examining the relationship between the coefficient of variation (CV) versus peptide response (peak area), MRM quantification maintained the majority (≥ 75%) of measurements below 20% CV across the largest range of analyte input and corresponding ion signal intensities, indicating superior linear range for quantification compared to PRM and DIA (Supp. Fig. 7B). A similar trend of increasing CVs as response decreases can be seen qualitatively for both MRM and PRM (Supp. Fig. 8A), although less obvious for DIA quantification. MRM was able to detect 40 peptides, 24 of which were reproducibly quantified (CV < 20%) in a sample with a dilution factor of 108 (Supporting Information, Table S28. Supporting previous results, PRM acquisition outperformed DIA, resulting in reproducible measurements for >50% down to a 1/36 dilution, compared to <25% of DIA measurements.). While additional analytical experiments are necessary to confirm performance for each peptide panel, these results indicate that MRM has superior sensitivity compared to PRM and DIA.
Multiplexed MRM assays can be developed in silico using LC-MS/MS discovery datasets, but without standard peptides for assay development and collision energy optimization, the MRM ion signal may not be optimized, negatively impacting sensitivity. Stable isotope-labeled standard (SIS) peptides can be used to characterize the targets for assay development (e.g. determine lower limits of detection and quantification) and spiked into each sample for absolute quantification.59 However, the financial burden, reagent availability/stability (e.g. FMOC protected desthiobiotinyl-lysine for peptide synthesis is not available), and time requirements are not always feasible, especially when targeting a panel of 400+ labeled peptides. PRM target panels, on the other hand, require minimal a priori knowledge other than LC-MS/MS. There should be no difference in fragmentation or retention time, because analysis is performed on the same instrument; PRM fragment ion selection and collision energy optimization are unnecessary, stream-lining implementation. The high-resolution accurate mass measurements (<10 ppm) afford improved confidence in peptide identification without spiked SIS peptides. However, cycle time due to ion accumulation and Fourier Transform mass spectrometry is a significant limitation for PRM, decreasing the number of peptides that can be monitored simultaneously. In order to achieve a minimum of 7 points across a 25 second peak with 100 ms for each mass analysis, only 35 precursors can be monitored during the same period in the experiment. If SIS peptides are used for absolute quantification, then only 17 peptides can be analyzed by PRM in any given scheduled elution time window. Triple quadrupole transition monitoring is quicker with dwell times as low as 5 ms per transition, which leads to higher multiplexing capability. If four transitions, on average, are monitored per precursor with 10 ms dwell times, then about 90 peptides can be concurrently quantified. To compensate for this limitation, PRM scheduled time windows were reduced by ~50%. The ABPP PRM platform described here is at the limit of what can be monitored with this acquisition method during the most congested parts of the gradient (e.g. the elution periods with the highest peptide density); some peptide peaks found in the middle of the gradient contain fewer than 7 points. The MRM ABPP assay, on the other hand, can accommodate more than double the amount of peptides analyzed here. Additional assay content can then be included to increase kinome coverage as more kinases are observed in discovery datasets and determined to be relevant for cancer treatment.
PRM is a useful tool for MRM assay refinement, especially when developing targeted assay panels from discovery datasets acquired using linear ion-trap fragmentation typical of linear ion trap-orbital ion trap hybrid instruments (e.g. LTQ-Orbitrap). The LC-MRM ABPP peptide panel was initially created by selecting the top fragments from linear ion-trap data. Once incorporating high resolution and accurate mass (HRAM) data (acquired on the Q Exactive Plus) into the spectral libraries, the top fragment ions changed and were more consistent with QqQ fragmentation. For some peptides, the top ions selected from the PRM experiment were not the same as the MRM transitions chosen from linear ion trap fragmentation data. In addition, a PRM error cut-off can be used to eliminate interfering transitions that were indistinguishable by ion trap MS/MS and MRM, which relies on deviations in transition ratios to observe interferences. PRM data was used to refine MRM transition selection and improve the ABPP-LC-MRM panel; an example is shown in Supp. Fig. 8.
BEZ-235 Treatment Induces Signaling Changes Impacting Cell Cycle and Autophagy Regulation
Kinome profiling with ABPP is a useful tool to identify perturbed signaling pathways in response to drug treatment. In this study, H1993 cells, not lysates (see workflow diagram, Supp. Fig. 1), were treated with either Crizotinib, a Met inhibitor, or BEZ-235, which was originally marketed as a dual PI3K/MTOR inhibitor60 to improve MTOR inhibition by blocking AKT1 reactivation through a PI3K feedback mechanism61–62 and later shown to be a potent inhibitor of ATM and DNA-PKcs.63 Because BEZ-235 targets multiple known pathways, it is expected to show quantifiable changes for comparison of MS acquisition methods (see Supp. Fig. 8, and Supp. Tables for Crizotinib analysis).
To assess fold-changes quantified across MRM, PRM, and DIA, 95% confidence intervals were plotted for peptides shared across the three datasets (ERN1 and AKT1 peptides were included, although not observed in DIA analysis) that had an average fold change in either direction of at least 1.8 (Fig. 4A, Supp. Fig. 9A-C). Results indicate agreement between MRM and PRM identification of differentially regulated peptides, although the degree of change differed. DIA, on the other hand, showed little to no change or fold-change values in the opposite direction, compared to MRM and PRM for 7 peptides (46%). In addition, the top list of differentially regulated proteins identified by DIA differed from MRM and PRM (Supp. Fig. 10). Metacore GeneGO analysis was conducted to determine the biological significance of the disparate list of differentially regulated kinases following DIA quantification (Supporting Information, Tables S29-S32). Pathways regulated by BEZ-235 in H1993 cells are consistent between MRM and PRM results, but not DIA (Fig. 4B). MRM and PRM shared 3 out of 10 of the most statistically significant GeneGO process networks, although at least half of the top ten process networks for each method involved cell cycle or autophagy. DIA, on the other hand, only had 2 process networks in common with MRM and PRM, and no common pathway themes when examined alone.
Figure 4. ABPP Comparing BEZ-235 Treatment to Controls Shows Signaling Changes Impacting Cell Cycle Regulation in H1993 Lung Cancer Cells.
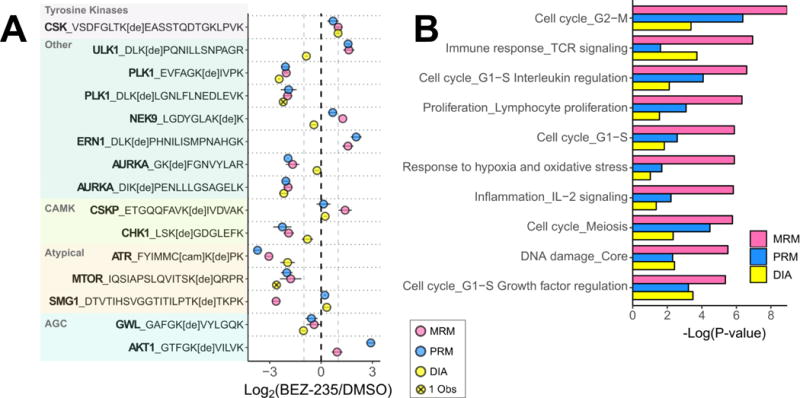
Differentially regulated desthiobiotinylated peptides (A) have been selected by |log2(fold change)| > 1 in at least two MS and grouped by kinase family. Data points represent the average log2(Fold Change) with bars indicating 95% confidence intervals. For pathway mapping, proteins in each dataset were selected based on peptide fold changes > 1.6 or <−1.6 between control and BEZ-235 treatment. The top ten statistically significant GeneGO process networks enriched across the three methods are shown for each MS dataset (B). Proteins used were filtered from each analysis, separately (fold changes greater than 1.6 or less than −1.6), then combined for MetaCore GeneGO network analysis.
The networks identified were driven by proteins unique to the DIA dataset, including MET and EGFR. Initial analysis was carried out at the peptide level, without review at the protein level. EGFR and MET had many peptides quantified in the DIA dataset, and the majority were not statistically different following treatment. EGFR, for example, had only one of eleven detected peptides significantly decreasing and that peptide contained a missed cleavage (~50% of total ion signal) and at least two modification sites (K716 and K737). If all peptides were considered for a protein level comparison, these two proteins would not be included. This observation underscores the importance of extensively reviewing the peptide level data and rolling them up to the protein level to fully understand the biological relevance of the data.
After removing proteins that had peptides with conflicting fold changes and lowering the cut off (fold-change of at least ± 1.6), the resulting enriched process networks from the DIA dataset had improved agreement between MRM and PRM (5/10 processes). However, reduction in protein number pushed the FDR past the cutoff threshold of 0.05 (< 5% false positives), reducing confidence in the results. Overall, the lack of consistency compared to other methods and increased FDR indicate that gaining meaningful biological insights from kinome profiling using ABPP and DIA to elucidate signaling changes may be challenging, when compared to reaction monitoring strategies.
To examine the ability for each method to detect changes in probe uptake across a biological network, Figure 5 presents a schematic representation of kinases that had BEZ-235 induced differential kinase ATP-uptake by both PRM and MRM analysis and above the fold change cut-off in at least one of the two datasets (gray proteins were not observed). Relationships were curated using GeneGO Pathway Maps and interactions found in the literature.60, 63–65 Known targets, like MTOR and ATM, had decreased ATP-uptake when quantified by MRM; PLK1 and AKT1 also emerged as signaling hubs perturbed by BEZ-235 treatment. The signaling pathways perturbed by BEZ-235 inhibition block DNA damage response, proliferation, and cell cycle progression by disrupting regulation at multiple points, while increasing autophagy signaling through ULK1 and IRE1/ERN1. Detection of kinases was assessed in this diagram for all four MS acquisition methods. Only CSK, PLK1, and AURKA were significantly changing across all four datasets out of the 18 kinases in the diagram, representing the subset that was quantifiable by DDA. DIA analysis improved quantification with significant changes seen in ~48%, or 8, of the kinases. The majority of these were quantifiable by both MRM and PRM, with only 3 kinases calculated to have a significant change by only one quantitative method.
Figure 5. Pathway Map Annotated with Detection and Quantification of Changes in Desthiobiotinylated Kinase Peptides in Response to BEZ-235 Treatment.
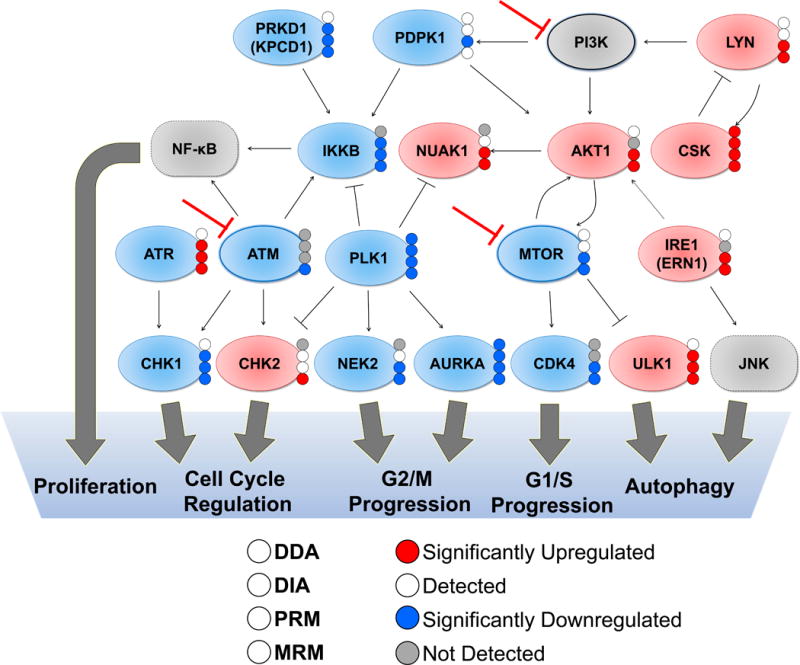
Schematic diagram of protein-protein interactions and downstream processes of a subset of kinases with differentially regulated probe labeling at 24 hours after BEZ-235 treatment. Proteins included had at least one peptide significantly increasing or decreasing (with an average fold-change > 1.6 or < −1.6 and p-values < 0.05 using student’s two tailed t-tests. Known BEZ-235 targets (ATM, MTOR, PI3K) are indicated with the red inhibition arrow. Proteins with red fill colors are increasing while those with blue fill colors are decreasing, and grey proteins are not detected in the dataset, but are included as known mediators of downstream signaling. The colored dots on the right of each protein indicate quantification results specific to each method, as indicated in the legend.
CONCLUSIONS
HRAM MS and MS/MS as well as improvements in bioinformatics have resulted in numerous possibilities for data acquisition. These platforms should be compared to evaluate their strengths and weaknesses for investigating biological questions. This study assessed the differences in relative quantification using four different MS/MS acquisition platforms: DDA, DIA, PRM, and MRM. Specifically, we examine the ability to reproducibly quantify low abundance peptides in a complex mixture. Activity-based protein profiling enrichment of kinases was used to examine drug target profiles and quantify downstream signaling events of clinically relevant KIs in human lung cancer cells. DIA, PRM, and MRM effectively identified inhibitor-protein interactions that were consistent with the literature. Pathway enrichment for kinases significantly changing after BEZ-235 treatment demonstrated that both PRM and MRM quantification led to similar profiles of differentially regulated biological processes. PRM quantification yielded a slight improvement on reproducibility, which could be explained in part by the reduction in noise and interference compared to MRM. MRM quantification demonstrated superior sensitivity with shorter, doubly-charged peptides. DIA reduced missingness across the dataset and increased quantifiable kinase identifications compared to DDA, but quantification was inferior, in these datasets, to both MRM and PRM. Both DDA and DIA identified many kinases that were eliminated throughout data analysis because there were not enough observations for quantification or statistical analysis. In the future, we would like to examine the capability of different types of DIA methods, including massively parallel PRM and SWATH, which use MS2 quantification instead of the MS1 quantification used in this study, and continue to improve data analysis workflows to quantify low abundance peptides, thus increasing our ability to gain biological insights into the mechanisms of cancer treatment.
In summary, DIA improves quantification compared to traditional DDA, while not limiting observations to an a priori target list. These discovery acquisition methods are ideal when asking the question, “What proteins can be observed in these biological samples?’ rather than “How much do these proteins change in different conditions?” For quantification, MRM should be selected when quantifying larger panels or low abundance peptides that require the most sensitivity, while PRM is an excellent choice for targeted quantification of peptide panels with limited size and where the sample complexity leads to interference that can only be eliminated with high resolution data acquisition. In addition, MRM reproducibility and sensitivity can be improved with the use of stable isotope labeled peptides and PRM can be used to direct and refine transition selection, supporting MRM assay development.
Supplementary Material
Acknowledgments
All data analysis performed (by MAH) using R programming was learned in a short course taught at the Mass Spectrometry: Applications to the Clinical Lab annual meeting with attendance supported by the MSACL Young Investigator Travel Award. We’d like to thank Dr. Dan Holmes (University of British Columbia) and Dr. Stephen Master (Cornell University) for reference material and instruction provided in the R short course. We thank Brent Kuenzi (Moffitt Cancer Center) for providing advice with data analysis and visualization using R. We thank Dr. Amol Prakash (OPTYS) and Dr. Scott Peterman (Thermo Scientific) for providing technical support with DIA data analysis using Pinnacle. Fumi Kinose and Moffitt’s Lung Cancer Center of Excellence provided the lung cancer cell lines used in the study. This work has been supported in part by the Proteomics Core Facility at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated and funded Comprehensive Cancer Center (P30-CA076292).
Funding Sources
National Cancer Institute (P30-CA076292, R21-CA169980)
American Lung Association (Lung Cancer Discovery Award LCD-257857-N)
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website at DOI: xxxxxx 052317_Hoffman_etal_JPR_Supp_Figs.pdf. Figures S1-8, Includes workflow diagram, QC data, additional comparison figures, example chromatograms, and analyses of individual datasets.052317_Hoffman_etal_JPR_Supplemental_Tables.xlsx.
Tables S1-31, Excel spreadsheets containing DIA acquisition windows, MRM transition list, PRM isolation list, retention times, raw data for DDA, raw data and processed log2 fold-change spreadsheets with p-values for Erlotinib, Dasatinib, Crizotinib, and BEZ-235 treatment for each method, GeneGO process networks enrichment report for all methods and each method individually.
Notes
DDA and DIA datasets are publically available via PRIDE repository: PXD006095 and PXD006096, respectively.
MRM and PRM datasets are posted in Panorama: https://panoramaweb.org/labkey/quant_method_comparison.url
Author Contributions
JMK supervised all aspects of the project and led the study. BF, MAH, and JMK contributed to assay development and data acquisition. Experiments and data analysis were completed by MAH. The manuscript was written by MAH and JMK. All authors contributed to study design and provided input on the manuscript preparation. All authors have given approval to the final version of the manuscript.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61(2):203–12. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 4.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–65. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 5.Sholl LM, Aisner DL, Varella-Garcia M, Berry LD, Dias-Santagata D, Wistuba II, Chen H, Fujimoto J, Kugler K, Franklin WA, Iafrate AJ, Ladanyi M, Kris MG, Johnson BE, Bunn PA, Minna JD, Kwiatkowski DJ, Investigators, L Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2015;10(5):768–77. doi: 10.1097/JTO.0000000000000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Gramont A, Watson S, Ellis LM, Rodon J, Tabernero J, de Gramont A, Hamilton SR. Pragmatic issues in biomarker evaluation for targeted therapies in cancer. Nature reviews Clinical oncology. 2015;12(4):197–212. doi: 10.1038/nrclinonc.2014.202. [DOI] [PubMed] [Google Scholar]
- 7.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, Hu C, Luo Y, Chen L, Ye M, Huang J, Zhi X, Zhang Y, Xiu Q, Ma J, Zhang L, You C. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. The Lancet Oncology. 2011;12(8):735–42. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 8.Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(31):3987–96. doi: 10.1200/JCO.2012.45.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Fang B, Kinose F, Bai Y, Kim JY, Chen YA, Rix U, Koomen JM, Haura EB. Target Identification in Small Cell Lung Cancer via Integrated Phenotypic Screening and Activity-Based Protein Profiling. Mol Cancer Ther. 2016;15(2):334–42. doi: 10.1158/1535-7163.MCT-15-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao Y, Guo L, Wang Y. A targeted quantitative proteomics strategy for global kinome profiling of cancer cells and tissues. Molecular & cellular proteomics : MCP. 2014;13(4):1065–75. doi: 10.1074/mcp.M113.036905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee JD, Gray NS, Kozarich JW. In situ kinase profiling reveals functionally relevant properties of native kinases. Chemistry & biology. 2011;18(6):699–710. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang B, Hoffman MA, Mirza AS, Mishall KM, Li J, Peterman SM, Smalley KS, Shain KH, Weinberger PM, Wu J, Rix U, Haura EB, Koomen JM. Evaluating kinase ATP uptake and tyrosine phosphorylation using multiplexed quantification of chemically labeled and post-translationally modified peptides. Methods. 2015;81:41–9. doi: 10.1016/j.ymeth.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Worboys JD, Sinclair J, Yuan Y, Jorgensen C. Systematic evaluation of quantotypic peptides for targeted analysis of the human kinome. Nat Methods. 2014;11(10):1041–4. doi: 10.1038/nmeth.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rebecca VW, Wood E, Fedorenko IV, Paraiso KH, Haarberg HE, Chen Y, Xiang Y, Sarnaik A, Gibney GT, Sondak VK, Koomen JM, Smalley KS. Evaluating melanoma drug response and therapeutic escape with quantitative proteomics. Molecular & cellular proteomics : MCP. 2014;13(7):1844–54. doi: 10.1074/mcp.M113.037424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Gruidl M, Remily-Wood E, Liu RZ, Eschrich S, Lloyd M, Nasir A, Bui MM, Huang E, Shibata D, Yeatman T, Koomen JM. Quantification of beta-catenin signaling components in colon cancer cell lines, tissue sections, and microdissected tumor cells using reaction monitoring mass spectrometry. J Proteome Res. 2010;9(8):4215–27. doi: 10.1021/pr1005197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U S A. 2007;104(14):5860–5. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiang Y, Remily-Wood ER, Oliveira V, Yarde D, He L, Cheng JQ, Mathews L, Boucher K, Cubitt C, Perez L, Gauthier TJ, Eschrich SA, Shain KH, Dalton WS, Hazlehurst L, Koomen JM. Monitoring a nuclear factor-kappaB signature of drug resistance in multiple myeloma. Molecular & cellular proteomics : MCP. 2011;10(11):M110 005520. doi: 10.1074/mcp.M110.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng Y, Zhang C, Croucher DR, Soliman MA, St-Denis N, Pasculescu A, Taylor L, Tate SA, Hardy WR, Colwill K, Dai AY, Bagshaw R, Dennis JW, Gingras AC, Daly RJ, Pawson T. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature. 2013;499(7457):166–71. doi: 10.1038/nature12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim HJ, Lin D, Lee HJ, Li M, Liebler DC. Quantitative Profiling of Protein Tyrosine Kinases in Human Cancer Cell Lines by Multiplexed Parallel Reaction Monitoring Assays. Molecular & cellular proteomics : MCP. 2016;15(2):682–91. doi: 10.1074/mcp.O115.056713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Molecular & cellular proteomics : MCP. 2012;11(11):1475–88. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karakosta TD, Soosaipillai A, Diamandis EP, Batruch I, Drabovich AP. Quantification of Human Kallikrein-Related Peptidases in Biological Fluids by Multiplatform Targeted Mass Spectrometry Assays. Molecular & cellular proteomics : MCP. 2016;15(9):2863–76. doi: 10.1074/mcp.M115.057695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kockmann T, Trachsel C, Panse C, Wahlander A, Selevsek N, Grossmann J, Wolski WE, Schlapbach R. Targeted proteomics coming of age - SRM, PRM and DIA performance evaluated from a core facility perspective. Proteomics. 2016;16(15-16):2183–92. doi: 10.1002/pmic.201500502. [DOI] [PubMed] [Google Scholar]
- 24.Bilbao A, Varesio E, Luban J, Strambio-De-Castillia C, Hopfgartner G, Muller M, Lisacek F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics. 2015;15(5-6):964–80. doi: 10.1002/pmic.201400323. [DOI] [PubMed] [Google Scholar]
- 25.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods. 2004;1(1):39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 26.Panchaud A, Scherl A, Shaffer SA, von Haller PD, Kulasekara HD, Miller SI, Goodlett DR. Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Analytical chemistry. 2009;81(15):6481–8. doi: 10.1021/ac900888s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prakash A, Peterman S, Ahmad S, Sarracino D, Frewen B, Vogelsang M, Byram G, Krastins B, Vadali G, Lopez M. Hybrid data acquisition and processing strategies with increased throughput and selectivity: pSMART analysis for global qualitative and quantitative analysis. J Proteome Res. 2014;13(12):5415–30. doi: 10.1021/pr5003017. [DOI] [PubMed] [Google Scholar]
- 28.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Molecular & cellular proteomics : MCP. 2012;11(6):O111 016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muntel J, Xuan Y, Berger ST, Reiter L, Bachur R, Kentsis A, Steen H. Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer. J Proteome Res. 2015;14(11):4752–62. doi: 10.1021/acs.jproteome.5b00826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu A, Noble WS, Wolf-Yadlin A. Technical advances in proteomics: new developments in data-independent acquisition. F1000Research. 2016;5(419) doi: 10.12688/f1000research.7042.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–8. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Escher C, Reiter L, MacLean B, Ossola R, Herzog F, Chilton J, MacCoss MJ, Rinner O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12(8):1111–21. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maclean B, Tomazela DM, Abbatiello SE, Zhang S, Whiteaker JR, Paulovich AG, Carr SA, Maccoss MJ. Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Analytical chemistry. 2010;82(24):10116–24. doi: 10.1021/ac102179j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O’Kelly G, Schoenegger A, Ovelleiro D, Perez-Riverol Y, Reisinger F, Rios D, Wang R, Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic acids research. 2013;41:D1063–9. doi: 10.1093/nar/gks1262. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolome S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32(3):223–6. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma V, Eckels J, Taylor GK, Shulman NJ, Stergachis AB, Joyner SA, Yan P, Whiteaker JR, Halusa GN, Schilling B, Gibson BW, Colangelo CM, Paulovich AG, Carr SA, Jaffe JD, MacCoss MJ, MacLean B. Panorama: a targeted proteomics knowledge base. J Proteome Res. 2014;13(9):4205–10. doi: 10.1021/pr5006636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10(4):1794–805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 38.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 39.Welsh EA, Eschrich SA, Berglund AE, Fenstermacher DA. Iterative rank-order normalization of gene expression microarray data. BMC bioinformatics. 2013;14:153. doi: 10.1186/1471-2105-14-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Eberhard C, Gruning B, Guerler A, Hillman-Jackson J, Von Kuster G, Rasche E, Soranzo N, Turaga N, Taylor J, Nekrutenko A, Goecks J. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic acids research. 2016;44(W1):W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.RStudio Team. RStudio: Integrated Development Environment for R, 0.99.902. RStudio, Inc.; Boston, MA: 2015. [Google Scholar]
- 42.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2016. [Google Scholar]
- 43.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York: 2009. [Google Scholar]
- 44.Wickham H, Francois R. dplyr: A Grammar of Data Manipulation. 2016 R package version 0.5.0. [Google Scholar]
- 45.Wickham H. Reshaping Data with the reshape Package. Journal of Statistical Software. 2007;21(12):1–20. [Google Scholar]
- 46.Schwalb B, Tresch A, Torkler P, Duemcke S, Demel C. LSD: Lots of Superior Depictions. 2015 R package version 3.0. [Google Scholar]
- 47.Chen H. VennDiagram: Generate High-Resolution Venn and Euler Plots. 2016 R package version 1.6.17. [Google Scholar]
- 48.Wickham H. stringr: Simple, Consistent Wrappers for Common String Operations. 2016 R package version 1.1.0. [Google Scholar]
- 49.Bessarabova M, Ishkin A, JeBailey L, Nikolskaya T, Nikolsky Y. Knowledge-based analysis of proteomics data. BMC bioinformatics. 2012;13(Suppl 16):S13. doi: 10.1186/1471-2105-13-S16-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 51.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25(9):1035–44. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 52.Kerkela R, Woulfe KC, Durand JB, Vagnozzi R, Kramer D, Chu TF, Beahm C, Chen MH, Force T. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clinical and translational science. 2009;2(1):15–25. doi: 10.1111/j.1752-8062.2008.00090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J, Bennett KL, Gao J, Song L, Eschrich S, Superti-Furga G, Koomen J, Haura EB. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nature chemical biology. 2010;6(4):291–9. doi: 10.1038/nchembio.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hantschel O, Rix U, Superti-Furga G. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leukemia & lymphoma. 2008;49(4):615–9. doi: 10.1080/10428190801896103. [DOI] [PubMed] [Google Scholar]
- 55.Rix U, Hantschel O, Durnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV, Kaupe I, Bennett KL, Valent P, Colinge J, Kocher T, Superti-Furga G. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood. 2007;110(12):4055–63. doi: 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- 56.Conradt L, Godl K, Schaab C, Tebbe A, Eser S, Diersch S, Michalski CW, Kleeff J, Schnieke A, Schmid RM, Saur D, Schneider G. Disclosure of erlotinib as a multikinase inhibitor in pancreatic ductal adenocarcinoma. Neoplasia. 2011;13(11):1026–34. doi: 10.1593/neo.111016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kitagawa D, Yokota K, Gouda M, Narumi Y, Ohmoto H, Nishiwaki E, Akita K, Kirii Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes to cells : devoted to molecular & cellular mechanisms. 2013;18(2):110–22. doi: 10.1111/gtc.12022. [DOI] [PubMed] [Google Scholar]
- 58.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26(1):127–32. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 59.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100(12):6940–5. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 61.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer research. 2005;65(16):7052–8. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 62.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV, Pichamoorthy N, Burma S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012;14(1):34–43. doi: 10.1593/neo.111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karar J, Cerniglia GJ, Lindsten T, Koumenis C, Maity A. Dual PI3K/mTOR inhibitor NVP-BEZ235 suppresses hypoxia-inducible factor (HIF)-1alpha expression by blocking protein translation and increases cell death under hypoxia. Cancer biology & therapy. 2012;13(11):1102–11. doi: 10.4161/cbt.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Awasthi N, Yen PL, Schwarz MA, Schwarz RE. The efficacy of a novel, dual PI3K/mTOR inhibitor NVP-BEZ235 to enhance chemotherapy and antiangiogenic response in pancreatic cancer. Journal of cellular biochemistry. 2012;113(3):784–91. doi: 10.1002/jcb.23405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.