

Abstract
Small molecule inhibitors of indoleamine 2,3-dioxygenase-1 (IDO1) are emerging at the vanguard of experimental agents in oncology. Here pioneers of this new drug class provide a bench-to-bedside review on preclinical validation of IDO1 as a cancer therapeutic target and on the discovery and development of a set of mechanistically distinct compounds – indoximod, epacadostat and navoximod – that were first to be evaluated as IDO inhibitors in clinical trials. As ‘immunometabolic adjuvants’ to widen therapeutic windows, IDO inhibitors may leverage not only immuno-oncology modalities but also chemotherapy and radiotherapy as standards of care in the oncology clinic.
Keywords: indoleamine 2,3-dioxygenase; tryptophan dioxygenase; IDO1; IDO2; TDO; TDO2; tryptophan catabolism; immunometabolism; immune escape; immune surveillance; angiogenesis; vasculogenesis; inflammatory programming; cancer-associated inflammation
Introduction
Present therapies fail many patients with metastatic cancer, generally a terminal stage after the relapse of drug-resistant disease. Tumors display abundant immunogenic antigens yet ultimately escape immune rejection through the evolution of various tactics to evade, subvert and reprogram innate and adaptive immunity. Over the past decade, the molecular mechanisms enabling immune escape have been unraveled to a sufficient extent that mainstream acceptance of immunotherapy in the field of cancer research has been restored after an historical divorce occurring nearly half a century ago (1, 2). Perhaps the most impactful achievement of work in this area has been to resolve the historical failure of active immunotherapy to treat solid tumors, by revealing that such approaches are generally ineffective due to the dominance in most patients of tumor-mediated immune suppression (1, 3). One such immune ‘braking’ mechanism that has emerged with broadly useful appeal is mediated by the cytosolic enzyme indoleamine 2,3-dioxygenase-1 (IDO1), which acts in immunometabolism and inflammatory programming through its biochemical function in tryptophan catabolism (4, 5). Indeed, as demonstrated in preclinical models starting over a decade ago, inhibiting the IDO1 enzyme can empower the efficacy of cytotoxic chemotherapy, radiotherapy and immune checkpoint therapy without increasing their side effects (6–9). In this review of the foundations of IDO1 inhibitor validation and development, we survey key observations which have catapulted the relatively obscure area of tryptophan catabolism to the forefront of experimental cancer therapy.
The IDO1 enzyme is activated in many human cancers in tumor, stromal and innate immune cells where its expression tends to be associated with poor prognosis (10). Its role in immunosuppression is multifaceted, involving the suppression of CD8+ T effector cells and natural killer cells as well as increased activity of CD4+ T regulatory cells (Treg) and myeloid-derived suppressor cells (MDSC) (4). In tumor neovascularization, IDO1 acts as a key node at the regulatory interface between IFN-γ and IL-6 that shifts the inlammatory milieu towards promoting new blood vessel development (11, 12). As considered below, pharmacological and genetic studies in autochthonous transgenic and graft mouse models point to a root function for IDO1 in programming an inflammatory tumor microenvironment characterized by a tolerizing immune attitude and a permissive angiogenic context. Accordingly, there are preclinical proofs that IDO1 inhibitors may be able to safely empower the efficacy of cytotoxic or targeted chemotherapy, radiotherapy, immune checkpoint therapy and cancer vaccines. Further, there is emerging interest in two other tryptophan catabolic enzymes, TDO and IDO2, which also appear to influence inflammatory programs, with ongoing exploration of IDO1 inhibitors that also block TDO and IDO2 to deepen efficacy, limit inherent or acquired resistance to IDO1 blockade, and reduce autoimmune side-effects of immune therapy.
IDO1 and immune suppression
The discovery of the IDO1 enzyme was rooted in initial observations made in the 1950s in cancer patients where tryptophan catabolism was found to be elevated (13). In the 1970s, the gene encoding IDO1 was identified as the first interferon-activated gene to be described (14), but the precise meaning of this connection was obscure. In 1998 a pivotal conceptual breakthrough emerged from the work of Munn, Mellor and their colleagues suggesting that IDO1 could mediate immunosuppression based on the preferential sensitivity of T cells to tryptophan deprivation (15, 16). Briefly, they proposed that tryptophan deprivation would impair antigen-dependent T cell activation in microenvironments where IDO1 was active. Initial evidence supporting this concept was offered by studies of how immune tolerance to ‘foreign’ paternal antigens in pregnant mice could be reversed by a simple tryptophan mimetic that disrupts IDO signaling, 1-methyl-tryptophan (1MT), the administration of which was sufficient to elicit MHC-restricted T cell-mediated rejection of hemiallogeneic concepti presenting ‘foreign’ paternal antigens (17, 18). Subsequent work developed this concept as a mechanism to defeat immune surveillance in cancer (as reviewed initially in (19–22). Two pivotal connections were the following. First, IDO1 was found to be broadly expressed in human tumors (23, 24). Second, IDO1 expression in tumor cells was discovered to be linked to the status of Bin1 (25), a tumor suppressor gene among the most frequently attenuated genes in human cancer (26), due either to aberrant RNA splicing patterns that abolish its tumor suppressor function (27–31), or to altered gene methylation patterns that extinguish its expression (32–36). Bin1 deficiency in an oncogenically transformed cell was sufficient to facilitate IDO1-mediated immune escape by a cell-intrinsic mechanism (25). Notably, subsequent bone marrow transplant experiments argued that IDO1 acts in non-hematopoietic cells to support inflammatory skin carcinogenesis (37). Together, these observations support the concept that IDO1 can act solely in tumor cells and that its overexpression there is sufficient to drive immune escape. The discovery of a link between IDO1 expression and Bin1 status provided the first sound genetic connection of IDO1 to cancer pathophysiology. As surveyed below, Figure 1 provides a cartoon summary of the biological impact of IDO1 expression in cancer, whereas Figure 2 provides an overview of its regulation in expressing cells and the effector signals it generates in downstream responding cells.
Figure 1. Impact of IDO1 immunometablism in cancer.
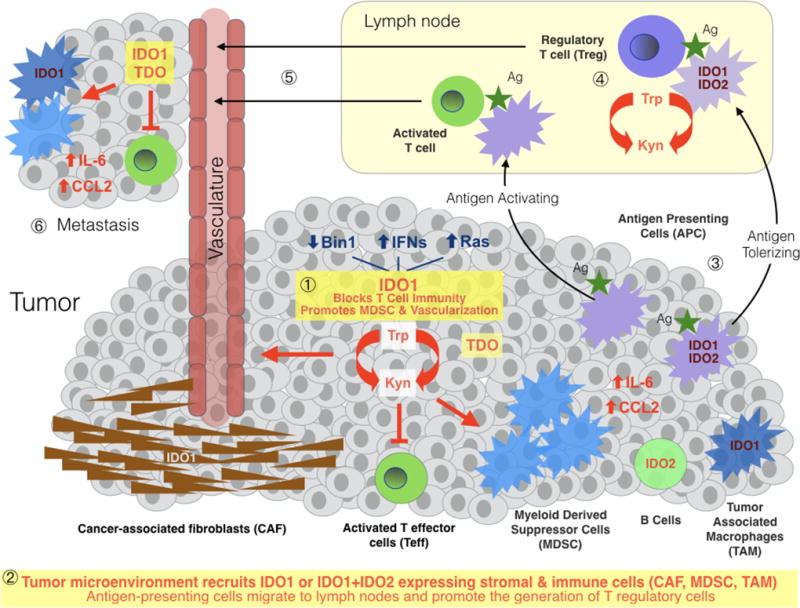
IDO1 expression patterns in human cancer are complex, occurring heterogeneously in malignant, immune, stromal and vascular cells within the tumor microenvironment and in antigen-presenting cells (APC) within tumor-draining lymph nodes. TDO and IDO2 are more narrowly expressed than IDO1 in human cancers, with TDO mainly in malignant cells and IDO2 mainly in immune cells. TDO is highly expressed in tumors independently or in parallel with IDO1; it has been ascribed both similar and distinct functions contributing to metastatic progression. IDO2 is expressed in antigen-presenting cells including B cells where it may influence IDO1 function (88); IDO2 is infrequently overexpressed in tumor cells. Tryptophan catabolism in tumor cells leads to local kynurenine generation and tryptophan depletion in the tumor microenvironment, enabling local suppression of T effector cells (Teff), functional licensing of myeloid-derived suppressor cells and recruitment of the tumor vasculature ❶. As conditioned by tumor cells, the tumor microenvironment recruits stromal cells expressing IDO1 and innate immune cells expressing IDO1 and IDO2, including cancer-associated fibroblasts, myeloid-derived suppressor cells and tumor-associated macrophages, the latter of which generate IL-6 and CCL2 in a manner dependent on local IDO1 activity, positively reinforcing the function of these cells and regulatory T cells that arrive ❷. Tumor antigens absorbed and presented to T cells by antigen-presenting cells which have roved away to a local draining lymph node ❸ promote the formation of activated T cells or tolerizing T cells (i.e. regulatory T cells), depending on whether the APC expresses IDO1 and perhaps IDO2 ❹. Antigen-specific T cells leave the lymph node and enter the vasculature ❺ where they can engage the primary tumor and contribute to the immune attitude of a latent metastatic niche ❻. APC, antigen-presenting cell; CAF, cancer-associated fibroblast; CCL2, a potent myeloid cell attractant and pro-differentiation agent, including for MDSC and TAM; IL-6, the master pro-inflammatory cytokine interleukin-6, which in tumors helps sustain myeloid-based and lymphoid-based immunosuppression and promotes neovascularization; MDSC, myeloid-derived suppressor cell; TAM, tumor-associated macrophage; Teff, activated effector T cell; Treg, regulatory T cell.
Figure 2. Sites of IDO1 expression and effector function in tumors.
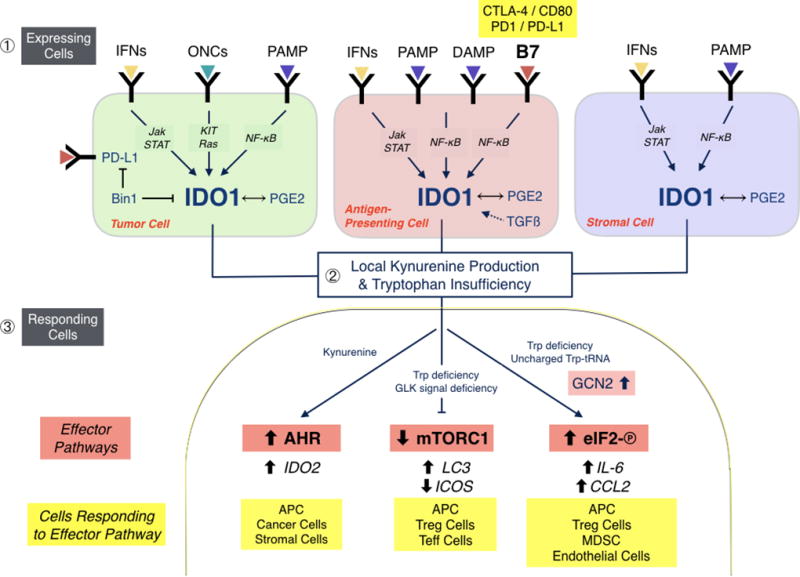
IDO1 is expressed in tumor cells, inflammatory/antigen-presenting cells and stromal cells under the diverse controls indicated in different tumor types ❶. In tumor cells, Bin1 attenuation and PGE2 production are key modifiers of IDO1 expression, which is transcriptionally controlled in different tumor settings by the interferon/Jak/STAT, ONC and PAMP signaling pathways. In inflammatory/antigen-presenting cells, B7 ligand reverse signaling is a major driver of IDO1 expression, most notably by CTLA-4 binding to CD80/CD86 or PD-1 binding to PD-L1 on the cell surface. Thus, tolerance mediated by PD-1 and CTLA-4 from regulatory T cells is intertwined with IDO1 upregulation, engendering a feed-forward loop to suppress adaptive immunity. In stromal cells, IDO1 can also be upregulated variably by interferon and PAMP signaling and PGE2 production. Altogether, IDO1 upregulation in tumor cells and the tumor microenvironment leads to locoregional deprivation of tryptophan and production of its catabolite kynurenine ❷. Responding cells interpret Trp insufficiency through the mTORC1 and GCN2/eIF-2 pathways, whereas kynurenine acts as a native ligand for the xenobiotic receptor AHR ❸. Downstream effector pathways with responsive target regulators are shown along with the different types of responding cells. AHR, arylhydrocarbon receptor; APC, antigen-presenting cell; B7, T cell co-receptor-ligand complexes (e.g. CTLA4-CD80/86 or PD1-PDL1) which stimulate signals into T cells and ‘reverse signals’ into antigen-presenting cells; IFN, interferon; DAMP, damage-associated molecular pattern (e.g. extracellular HMGB1 or ATP ligands for TLR4 or A2A receptors, respectively); eIF-2, a key regulatory factor for mRNA translation initiation; GCN2, a stress response kinase that is activated by binding uncharged tRNA, indicative of amino acid starvation; ONC, oncogenic ligand-receptor signaling complex (e.g. EGFR); mTORC1, a master metabolic regulatory complex that monitors amino acid pools for cell growth or autophagic decisions; PAMP, pathogen-associated molecular pattern (e.g. LPS or CpG ligands of Toll-like receptors [TLR]); Teff, activated effector T cells; Treg, regulatory T cells.
IDO1 is an inducible enzyme that is produced widely in response to the pivotal immune regulatory Th1 cytokine IFN-γ in various myeloid lineage-derived cells including dendritic cells and macrophages as well as endothelial cells, mesenchymal stromal cells and fibroblasts (38). Early studies accumulated evidence that IDO1 activity in cellular or tissue environments could suppress the function of T cells (39–43) and natural killer cells (44), and also that IDO1 was critical for the production and function of T regulatory cells (Tregs) (45) and myeloid derived suppressor cells (MDSCs) (11). Effector functions for both tryptophan depletion and catabolite production by IDO1 have been described, many focusing on antigen-presenting dendritic cells (DC) where IDO1 is upregulated by interferons, TLR ligands and other important immune signals (46). Notably, IDO1 expression in a small minority population of dendritic cells (DC) enables them to dominantly suppress effector T cell responses (47, 48). The anergizing effect of IDO1-mediated tryptophan depletion on T cells requires the stress response kinase GCN2, which is also required for IDO1-induced activation of Tregs (21, 43). Likewise, tryptophan catabolites block T cell activation and trigger T cell apoptosis while also promoting IDO1-induced differentiation of CD4+ cells into Tregs (through a TGF-β dependent mechanism), with apparent synergistic effects of this effector arm with tryptophan deprivation (49). While the role of kynurenine and other tryptophan catabolites has been less studied than the effects of tryptophan deprivation, a key advance reported by Platten and colleagues was the discovery that kynurenine acts as a native ligand for the aryl hydrocarbon receptor (AHR), a transcription factor that contributes to pro-inflammatory programs and xenobiotic responses of great relevance to cancer and inflammatory carcinogenesis (50).
Inflammatory programming by IDO1 in cancer
Mouse genetic studies indicate that IDO1 exerts a proximal influence on inflammation that can not be understood as simply immunosuppressive. It is notable that IDO1 deficiency in mice does not produce rampant inflammation in contrast to deletion of true immunosuppressive functions like CTLA-4 or PD-1. Even when mice were exposed to topical applications of the pro-inflammatory agent TPA (12-O-tetradecanoylphorbol-13-acetate), no discernable exacerbation in the severity of resulting contact dermatitis occurred (37). In contrast, genetic ablation of IDO1 markedly attenuated the ability of this inflammatory regimen to promote the development of premalignant papillomas following mutagenic initiation with the chemical carcinogen DMBA (7,12-dimethylbenz[a]anthracene) (51), while in the absence of inflammatory tumor promotion, the loss of IDO1 had no effect on papilloma development (37). These results suggest that IDO1 functions as a pivotal driver of ‘tumor-promoting’ inflammation, raising the possibility of using IDO1 inhibitors as chemopreventive agents to broaden their utility.
A key aspect of IDO1’s ability to establish a tumor-promoting inflammatory milieu is its status as a nodal interface beteween the inflammatory cytokines IFNγ and IL-6. The importance of IFN-γ in anti-tumor immune responses is well established (52) while IL-6 is recognized as an important pro-tumorigenic cytokine (53, 54). IFN-γ has also been long known to be a primary driver of IDO1 induction (14) while more recent data have shown IDO1 to be involved in the induction of IL-6 (11, 12). These findings have suggested a broad conceptual framework in which IDO1 acts as a negative feedback check on IFN-γ, mediated at least in part through upregulation of IL-6.
Further study indicates that this cytokine connection is particularly relevant to the recently recognized involvement of IDO1 in supporting neovascularzation – the abnormal development of new blood vessels associated with tumors as well as ischemia in general. Indeed, IDO1 was shown to be required for neovascularization not only in a mouse model of pulmonary breast cancer metastasis but also oxygen-induced retinopathy, and this was found to be the case for IL-6 as well (11, 12). IFN-γ can act to eliminate neovascularization as an important aspect of its antitumor activity (55, 56). As predicted by the negative feedback model, the reductions in neovascularization associated with losses of either IDO1 or IL-6 were completely reversed by the concomitant loss of IFN-γ, while pathophysiologically, the loss of IFN-γ correspondingly negated the increased resistance to pulmonary metastasis development associated with losses of either IDO1 or IL-6 (11, 12). Such are the hallmarks of a disease modifier, the function of which is contingent on influencing disease context. This distinction has important ramifications for IDO1 as a therapeutic target in contrast to general regulators of immunity (e.g. CTLA-4 or PD-1) or cell growth (e.g. EGFR, BRAF or TGF-β), which are not disease context limited and therefore fraught with therapeutic risk including major side-effects. Here it is notable that IDO expression is associated with other immune checkpoints, most notably PD-L1 and CTLA-4 supporting the concept of their joint targeting (57). While preclinical studies have suggested IDO1 blockade may not be entirely without risk (58), they have been pivotal in establishing that IDO1 provides integral support for tumor-promoting inflammation, thereby helping validate its status as a disease modifier and consequently the rationale for its development as a therapeutic target.
Discovery and preclinical development of IDO1 inhibitors
From a pharmacological standpoint IDO1 is very appealing for small molecule drug development. It is a single-chain catalytic enzyme with a well-defined biochemistry, one of only a small number of structurally distinct tryptophan catabolizing enzymes (TDO2, IDO2, TPH) with more restricted patterns of expression and substrate specificity compared to IDO1. Pharmacodynamic measurements of tryptophan and kynurenine, the chief substrate and downstream product of the IDO/TDO reaction, are readily obtained from blood specimens. Bolstering the rationale to develop IDO1 inhibitors, it was discovered that the anticancer effects of certain targeted drugs can be traced to IDO1 blockade (e.g. imatinib [Gleevec] in GIST treatment (59)). Several excellent comprehensive reviews have surveyed these medicinal chemistry, pharmacological and biological considerations in a detailed manner (5, 60–66).
Here we survey the preclinical work leading to clinical translation of the IDO pathway inhibitor indoximod and the mechanistically distinct IDO1 enzyme inhibitors epacadostat, navoximod and BMS-986205. Figure 3 presents a summary of the mechanisms through which these different agents affect tryptophan catabolism by IDO1 and other tryptophan catabolic enzymes. Figure 4A–D introduces the preclinical discovery and development teams at Lankenau, the Medical College of Georgia, NewLink Genetics and Incyte responsible for the compounds indoximod, navoximod and epacadostat, the first to enter clinical trials, and reflects an historiography of common roots among founders of the IDO programs. Figure 5 presents a timeline of IDO1 preclinical proof of concept and bench-to-bedside milestones from these teams. Figure 6A–F presents key chemical structures as discussed in the text.
Figure 3. IDO1 inhibitors and their impact on tryptophan immunocatabolism.
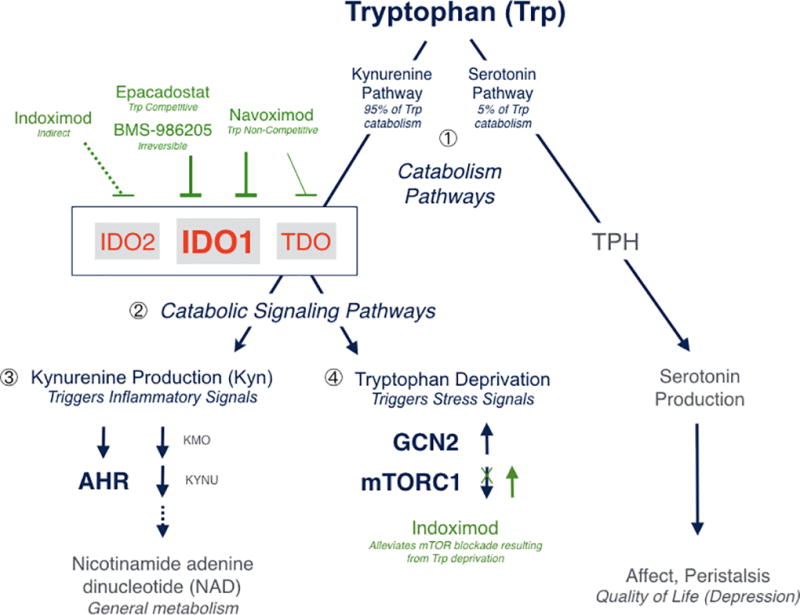
Tryptophan (Trp) catabolism proceeds through one of two pathways in mammals, leading to production of nicotinamide adenine dinucleotide or serotonin. The kynurenine pathway accounts for ~95% and the serotonin pathway for 5% of tryptophan catabolism ❶, with possible implications on affect and quality of life in cancer patients where the kynurenine pathway is driven by IDO/TDO dysregulation. Epacadostat is >100-fold selective for IDO1 against TDO and represents a highly specific agent with competitive inhibitory kinetics for tryptophan binding. Navoximod is ~20-fold selective for IDO1 against TDO and exhibits non-competitive inhibitory kinetics for tryptophan binding. BMS-986205 is an irreversible inhibitor of IDO1 that is highly specific for that enzyme. None of these agents inhibit IDO2 appreciably. Indoximod is not a direct enzyme inhibitor and its action is complex; it has been reported to indirectly inhibit IDO2 and/or IDO1 in some settings. Its primary mechanism of action appear to be downstream, in its high potency as a tryptophan mimetic interpreted by mTORC1 as L-tryptophan under conditions of high tryptophan catabolism and autophagy due to tryptophan deprivation by any catabolic enzyme. The targeted enzyme inhibitors affect both catabolic effector signaling pathways ❷. Kynurenine functions as a native ligand for the pro-inflammatory receptor AHR, which activates downstream gene expression ❸. Tryptophan deprivation triggers starvation-induced signals mediated by upregulation of the general stress kinase GCN2 and downregulation of the mTORC1 complex (which monitors amino acid pools for growth versus autophagy decisions, which are critical for T cell function) ❹.
Figure 4. Discovery teams in preclinical proof of concept.

(A.) NewLink Genetics team with academic collaborators. Left to right, Alexander Muller (Lankenau), Nicholas Vahanian (New Link), Andrew Mellor (Georgia), David Munn (Georgia), Charles Link (New Link), Mario Mautino (New Link), George Prendergast (Lankenau). The New Link IDO1 program was initiated by Mario Mautino after the company in-licensed founding intellectual property from the Munn/Mellor team at the Medical College of Georgia (now Augusta University) and the Prendergast/Muller team at the Lankenau Institute for Medical Research (LIMR). (B.) Lankenau team. Left to right, George Prendergast, Alexander Muller, James DuHadaway, William Malachowski (Bryn Mawr College). (C.) Incyte and Lankenau. Left to right, Peggy Scherle, a key proponent of the IDO1 program at Incyte, with spouse Alexander Muller, who initiated the IDO1 program at Lankenau with George Prendergast. (D.) Incyte medicinal chemistry team contributing to the development of epacadostat (99). Left to right, Andrew Combs, Dilip Modi, Joe Glenn, Brent Douty, Padmaja Polam, Brian Wayland, Rick Sparks, Wenyu Zhu, and Eddy Yue.
Figure 5. Discovery timelines in preclinical proof of concept.
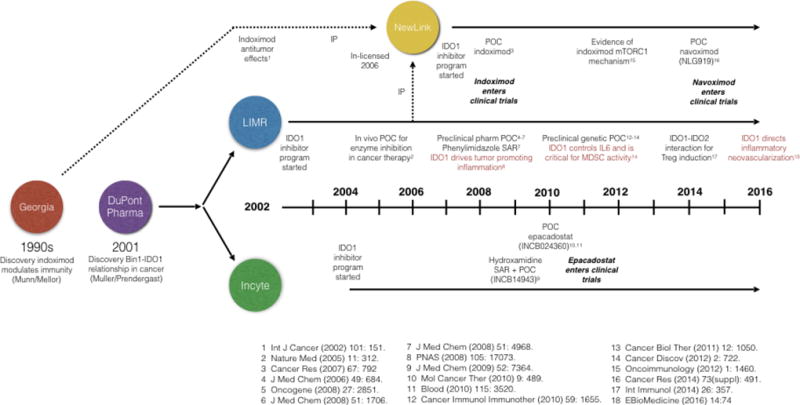
Key milestones are cited in the discovery and preclinical validation of IDO1 as a cancer therapeutic target as referenced in the Figure. IP, intellectual property; MDSC, myeloid-derived suppressor cells; POC, proof of concept; SAR, structure-activity relationship analysis (for inhibitor class noted).
Figure 6. IDO1 inhibitor structures and intermediates.
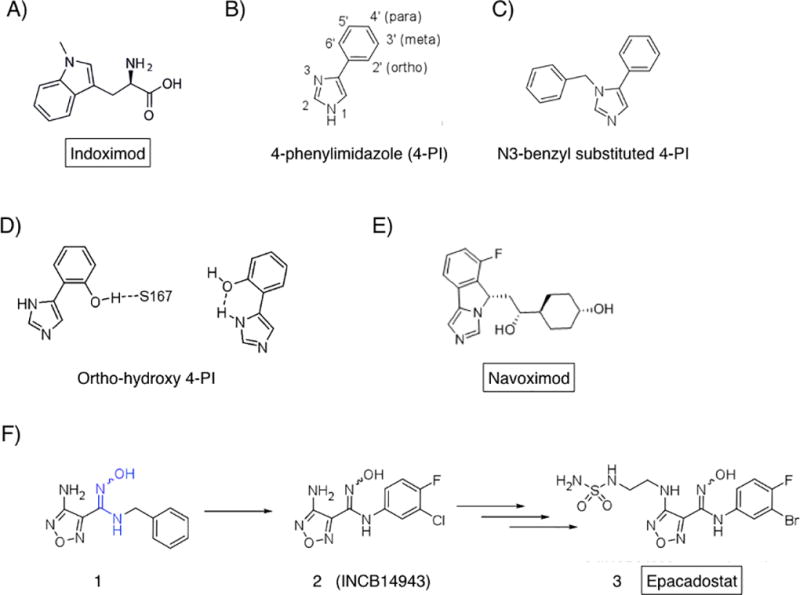
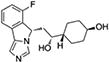
(A.) Indoximod. A 1-methyl derivative of D-tryptophan interpreted by mTORC1 in cells as L-tryptophan. (B.) 4-PI. Founding compound of the phenylimidazole chemotype series. (C.) N3-benzyl derivative elaborating this site. (D.) Ortho-hydroxyl modifications which elaborate potency. (E.) Navoximod (NLG-919), clinical lead from the imidazoisoindole series. (F.) Epacadstat discovery milestones. 1, original hit including critical hydroxyamidine (blue). 2, preclinical proof of concept lead compound (INCB14943). 3, epacadostat (INCB024360) as final clinical lead.
Indoximod (D-1MT; NLG-8189)
By far the IDO probe most employed in the preclinical literature is the simple racemic compound 1-methyl-D,L-tryptophan (1MT) with a reported Ki for IDO1 of 34 µM (67, 68). The L isomer acts as a weak substrate for IDO1 and is ascribed the weak inhibitory activity observed with the racemate, as the D isomer neither binds nor inhibits the purified IDO1 enzyme (4). Evidence offered in the early 2000s showed that 1MT could weakly retard the growth of cancer cells engrafted into syngeneic hosts (23, 69). However, as it soon became clear to molecular pharmacologists that 1MT is not a valid inhibitor of IDO1 enzyme activity, and that the experiments with 1MT could not address the pivotal question of whether IDO1 enzyme blockade could exert a significant therapeutic effect. Nevertheless, racemic 1MT has continued to serve as a common probe of the IDO pathway in preclinical studies, and further investigations of its antitumor activity led to the eventual clinical translation of the D racemer, now known as indoximod, as a drug candidate with potentially unique effects (Figure 6A).
In detailed studies of antitumor efficacy in a transgenic model of HER2-driven breast cancer, 1MT exerted little single agent effect on tumor outgrowth but was found to dramatically empower the efficacy of a variety of chemotherapeutic agents, especially DNA damaging drugs or taxol, triggering regressions of otherwise recalcitrant tumors (25). This was a pivotal observation in establishing how 1MT may offer a useful tool in cancer therapy. Regressions seen did not reflect drug-drug interaction, that is, by acting to raise the effective dose of the cytotoxic agent, because efficacy was increased without increased side effects. Significantly, depleting CD4+ or CD8+ T cells abolished the effects of not only 1MT but also of another true enzymatic inhibitor of IDO1, consistent with the observed efficacy being dependent on T cell-mediated immunity (25). A later study attributed the majority of the antitumor activity of 1MT to the D racemer, which has no inhibitory effect on IDO1 enzyme activity (7). However, factors in choosing to clinically translate it ultimately included (i.) its potency in relieving T cell suppression by mouse and human IDO1+ plasmacytoid DCs; (ii.) favorable pharmacology and toxicology; (iii.) superior preclinical antitumor activity relative to L-1MT in side-by-side comparisons, singly or in combination with chemotherapy; and (iv.) its genetic validation based on loss of activity in IDO1-deficient mice (7). The preclinical efficacy of D-1MT/indoximod in combination with chemotherapy led to its inclusion on a selective list of key agents for clinical assessment in 2008 by an NCI immunotherapy workshop (70).
A number of studies have addressed the mechanism of action of indoximod. However, in considering an evaluation of human pharmacokinetics where clinical responses have been noted (71), only one study has provided an explanative mechanism consistent with blood serum levels achieved in clinical trials (72). Specifically, this study revealed that indoximod can resuscitate cellular mTORC1 activity inhibited by tryptophan depletion with an IC50 of ~70 nM (72). Thus, indoximod acts as a high-potency tryptophan mimetic in reversing mTORC1 inhibition and the accompanying autophagy that is induced by tryptophan depletion in cells. As a central integrator of cell growth signals, mTORC1 receives signals that monitor levels of essential amino acids needed to activate cell growth, including in T cells. Interestingly, indoximod was able to relieve mTORC1 suppression created by tryptophan deprivation with a higher potency than L-tryptophan itself (i.e. at lower concentrations) (72). More recent findings confirm this activity in human T cell cultures, including for indoximod derivatives with superior pharmacokinetic qualities, establishing a unique mechanism of action for this compound acting directly on T cells (73).
The surprising finding that mTORC1 interprets indoximod – essentially a D-tryptophan analog – as a mimetic of L-tryptophan is a pivotal observation. IDO1 is not expressed in T cells but exerts its effects on them indirectly from a neighboring cellular milieu (Figures 1, 2). In acting directly upon T cells, indoximod acts differently from other IDO1 enzyme inhibitors which act outside T cells. Therefore, indoximod use is rationalized in tumors driven by any tryptophan catabolizing enzyme, and it might be combined productively with frank IDO1 enzyme inhibitors to enhance activity. Recent clinical evidence of the ability to indoximod to safely heighten the efficacy of anti-PD1 in melanoma patients (74), consistent with preclinical results (8), suggests that mTORC1 restoration may be a sufficient cause of the antitumor effects of IDO1 enzyme inhibitors (74). These findings are interpretable in light of evidence that the T cell co-regulatory receptor ICOS is elevated in T effector cells which infiltrate melanomas with favorable survival outcomes (75), insofar as ICOS levels are elevated in such cells by mTORC1 activation (76). On the basis of this information, one would predict that therapeutic responses to indoximod occur through mTORC1-mediated ICOS upregulation in tumor-infilitrating T effector cells. In summary, indoximod targets IDO signaling indirectly as an IDO pathway inhibitor that appears to act by derepressing mTORC1 in T cells, and perhaps other cells, to produce its antitumor effects.
Phase 1 studies of indoximod suggest it is well tolerated (71, 77). In a dose escalation study of 22 evaluable advanced cancer patients receiving taxotere, the co-administration of indoximod was found to be well tolerated to the maximum delivered dose of 1200 mg twice daily. In this set of patients, 4 partial responses were observed (2 breast cancer, 1 lung adenocarcinoma, 1 thymic tumor) and no drug-drug interactions were noted (71). In a second dose escalation study in 48 advanced cancer patients, a maximum tolerated dose was not reached at the top dose of 2000 mg twice daily (77). Notably, three patients previously treated with anti-CTLA-4 (ipilimumab) who received 200 mg once daily developed hypophysitis, an autoimmune signature of these inhibitors. In this set of patients, 5 cases of stable disease >6 months were observed. Pharmacokinetic analysis showed a plateau in plasma AUC and Cmax for indoximod at doses beyond 1200 mg twice daily. Cmax occurred at 2.9 hr and the halflife was 10.5 hr. An increase in levels of C reactive protein (CRP) was noted across multiple dose levels with additional evidence of an increase in tumor antigen autoantibodies (77). Overall, these studies supported the initiation of multiple Phase 2 studies at the dose of 1200 mg twice daily.
Early reports of Phase 2 trials with indoximod combinations been provocative. Perhaps most strikingly, administering indoximod with the PD1 antibody pembrolizumab has been reported to achieve disease control rates in melanoma patients that parallel those produced by the approved combination of nivolumab with the CTLA-4 antibody ipilimumab, but without the high-grade autoimmune side-effects seen with this therapy (78, 79). In another Phase 2 trial, in this case in metastatic prostate cancer patients treated with the dendritic cell vaccine sipuleucel-T (Provenge®), co-administration of indoximod post-vaccination led to a >2-fold extension in patient survival (80). In a Phase 2 study of 169 first-line metastatic breast cancer patients treated with taxotere, co-administration of indoximod did not produce a statistically significant difference in progression-free survival, overall survival or objective response rate. However, early promising results have been reported from Phase 1B combination trials where indoximod was combined with standard of care chemotherapy in patients with acute myeloid leukemia or pancreatic adenocarcinoma (81, 82), with ongoing Phase 2 trials now proceeding in these disease settings.
Learning more about the distinct mechanism of action of indoximod may yield significant insights. Since it relieves a common downstream IDO/TDO effector signal in mTORC1, indoximod is rationalized to attack tumors that overexpresses any tryptophan catabolizing enzyme. Further, its more narrow mechanism of action opens the possibility that relieving mTORC1 blockade in T cells is sufficient for anticancer efficacy by an IDO/TDO enzyme inhibitor, meaning that inhibiting all IDO/TDO function is unnecessary and may even heighten side-effects. Further studies of the connections between tryptophan catabolism and mTORC1 control may yield an important new crop of useful immunoregulatory principles. In developing the intriguing research directions opened by indoximod, a pro-drug form termed NLG-802 with superior pharmacokinetics has been reported recently that has entered Phase 1 testing (73).
Navoximod (NLG-919)
Building on the discovery of the Bin1-IDO1 connection in tumoral immune escape (25), the Prendergast/Muller group embarked in 2002 at the Lankenau Institute for Medical Research (LIMR) on a project to genetically and pharmacologically validate IDO1 as a therapeutic target in preclinical models of cancer. This group initiated an IDO1 inhibitor discovery program by screening publically available indoleamines and indoleamine mimetics and then, in collaboration with Malachowski and colleagues at Bryn Mawr College, synthesizing sets of chemical derivatives for hits of interest. Bioactive inhibitors identified in several structural classes in this manner displayed similar biological properties, namely, antitumor effects which relied upon T cell function and IDO1 targeting (6, 7, 83–86). Among these bioactive compounds, MTH-trp was the first to clearly demonstrate that impeding IDO1 enzyme activity could elicit antitumor effects (25). This pivotal observation was extended by studies of additional structural classes of bioactive inhibitors in establishing a pharmacological proof of concept for IDO1 as a cancer drug target (7, 83, 84, 86). In one important direction influencing subsequent drug discovery, their investigation of structure-activity relationships for a phenylimidazole chemotype founded in an early crystallographic study provided initial clues to key features of a bioactive IDO1 inhibitor (85, 87). Genetic proof of concept was subsequently achieved in mice lacking IDO1, corroborating evidence of a therapeutic effect for IDO1 blockade in cancer as mediated by effects on inflammation, adaptive immunity and neovascularization (11, 12, 51). Interestingly, later work also implicated IDO2 in Treg control by IDO1 (88). Together these pharmacological and genetic projects were pivotal in preclinical validation of IDO1 as a therapeutic target in cancer.
Emerging from this proof of concept project, the phenylimidazole chemotype appeared the most promising with a drug-like profile (85). This series had its roots in 4-phenylimidazole (4-PI) identified originally in 1989 as a weak non-competitive inhibitor of IDO1 enzyme activity (89). Despite showing non-competitive kinetics, early spectroscopic studies confirmed by later crystallographic analyses showed that 4-PI bound the heme iron at the IDO1 active site (89, 90). This work seeded initial structure-based drug design from Malachowski and colleagues (85), as an informative step in later discovery of the imidazoisoindole chemotype and the clinical lead navoximod, as developed independently by a medicinal chemistry team at NewLink Genetics Corporation.
In exploring chemical derivatives of 4-PI, Malachowski and colleagues probed the active site of IDO1 with structural modifications to explore binding interactions within it, namely, (i.) the active site entrance region decorated with heme 7-propionic acid, (ii.) the active site interior, especially interactions with C129 and S167; and (iii.) the heme iron binding group. Substitutions at the active site entrance region focused on the N-1, C-2, and N-3 positions of the imidazole ring (Figure 6B). In the 4-PI-bound crystal structure, the entrance region is occupied by an N-cyclohexyl-2-aminoethanesulfonic acid (CHES) buffer molecule, with its alkyl portion making hydrophobic interactions with F163 and F226, and its amino group forming an ion pair with the heme 7-propionic acid. N-1 derivatives lost inhibitory activity, confirming binding of the N-1 nitrogen to the heme iron, and, more importantly, demonstrating that the N-3 nitrogen of the imidazole cannot substitute to bind at the heme iron. However, the N-3 benzyl-substituted derivatives generated were observed to be equipotent to 4-PI, showing that imidazole ring substitutions might be tolerable (Figure 6C) to situate a hydrocarbon moiety at the active site entrance. The N-3 benzyl-substituted compound suggested the correct imidazole ring location and spatial tolerance, likely occupying the active site entrance where the CHES buffer molecule sits in the IDO1-4-PI crystal structure (90). This insight was supported by studies of brassinin derivatives as IDO1 inhibitors, i.e. a heme iron-binding group flanked by two large aromatic or hydrocarbon structures (83).
In probing the active site interior, S167 and C129 were in close proximity to the phenyl ring of 4-PI bound to IDO1 (90). Ortho, meta and para substitutions of the phenyl ring revealed that ortho-hydroxy (2’-OH) modifications had the greatest impact on potency (Figure 6D). In exploring this effect, it was found that a 2’,6’-dihydroxy-phenyl derivative presenting a hydroxy group to S167 or to N-3 imidazole was roughly equipotent to the 2’-hydroxy derivative, indicating no additional benefit from both events. Retrospectively, this observation may explain the benefits of planarization via a hydrocarbon bridge to replicate the H-bond to the N-3 imidazole. In evaluating the heme iron-binding group, substituting alternative aromatic rings for the imidazole in 4-PI always yielded less potent compounds. Later work illustrated that triazole can substitute for imidazole for presumptive histidine binding (91). In summary, these early studies of the phenylimidazole series of IDO1 inhibitors yielded three insights, namely, that (i.) N-3 substitutions suggested the active site could accommodate large hydrocarbon moieties, (ii.) an ortho-hydroxyl group was beneficial, and (iii.) the imidazole was optimal for binding to the heme iron.
Later development of an imidazoisoindole series incorporated some of these insights but also yielded novel information incorporated into the clinical lead navoximod (92, 93) (Figure 6E). X-ray structural information shows that the ring planes of the PI and imidazoisoindole series assume similar but not identical positions in the active site (94).
Navoximod has several features of interest defined in preclinical studies. Its potency as an IDO1 inhibitor is EC50=75 nM in cell-based assays, with only 10- to 20-fold selectivity against TDO (92). Thus, navoximod may exhibit unique activity in tumors expressing both IDO1 and TDO, the latter of potential relevance to intrinsic or acquired resistance to IDO1 blockade (see below). Like earlier compounds in the phenylimidizaole series, navoximod exhibits non-competitive inhibition kinetics. These features contrast with the greater IDO1 specificity and competitive inhibition kinetics displayed by epacadostat, with which a clinical comparison may be informative. Navoximod is orally bioavailable with a superior pharmacokinetic and toxicity profile in the imidazoisoindole series (92). Its oral administration reduced plasma kynurenine levels by approximately 50% in mice and it relieved IDO1-induced T cell suppression in human cell cultures. In preclinical models, navoximod greatly enhanced vaccine responses against B16 melanoma, reducing tumor size ~95% within 4 days of vaccination, and improved the efficacy of anti-PD-1 against EMT6 mammary carcinoma (95), where increased CD8+ T/Treg ratioes, plasma interferon-γ levels and activated intratumoral macrophages and DC were noted.
There has been limited clinical study of navoximod as yet. In patients with recurrent/advanced solid tumors and the safety, pharmacokinetic and pharmacodynamic results from a Phase 1A study have been reported (93). As monotherapy, it was well tolerated at up to 600 mg twice daily on a 21/28 day cycle, with stable disease observed in 7/17 patients. Safety, pharmacokinetics and pharmacodynamics have been evaluated on a continuous dosing schedule (twice daily, 28/28 days), also in combination with the anti-PD-L1 antibody atezolizumab (96).
Epacadostat (INCB024360)
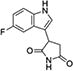
Building on evidence of immunomodulatory effects of 1MT in preclinical graft models (97, 98), Incyte Corporation initiated a project in 2004 to discover small molecule inhibitors of the IDO1 enzyme, focusing ultimately on a hydroxylamidine chemotype series from which the tryptophan competitive inhibitor epacadostat (INCB024360) was developed as a lead clinical agent. Details regarding the chemistry program that developed epacadostat from earlier compounds in this series have been reported elsewhere (99, 100).
This project was initiated by a high throughput screen of a proprietary collection of ~300,000 compounds which identified a single viable hit exhibiting selective micromolar-level activity as a tryptophan competitive inhibitor against the purified IDO1 enzyme and in cell culture (1, Figure 6F) (101). This novel chemotype contained hydroxyamidine as a unique functional group that structure-activity relationship studies revealed was essential for IDO1 enzyme inhibitory activity. Adsorption spectroscopy supported the hypothesis that the hydroxyamidine moiety bound directly to the iron of the heme in the IDO1 active site. Parallel synthesis and a proprietary preparative LC/MS purification technology provided rapid access to hundreds of analogs and afforded a proof-of-concept compound INCB14943 (2, Figure 6F). Preclinical efficacy was demonstrated by dosing 2 subcutaneously in a B16 melanoma graft model. Importantly, no tumor growth inhibition was achieved by dosing in immunodeficient nu/nu B16 mice, consistent with on-target activity. A full program of medicinal chemistry was engaged to overcome the low oral bioavailability of the lead (100). ADME investigations identified glucuronidation of the hydroxyamidine pharmacophore as the main metabolic liability and established in vitro phase two assays to allow medicinal chemistry to test the propensity of subsequent compounds to undergo phase two metabolism.
In analyzing thousands of synthetic derivatives, the most potent compounds with the best oral bioavailabilities unexpectedly required a larger number of hydrogen bond donors (HBD) and hydrogen bond acceptors (HBA) and higher polar surface area (PSA) than one would predict to afford high Caco2 permeability and oral bioavailability. The unique chemotype was shown to form several internal hydrogen bonds, thus shielding the polar functionality to allow good membrane permeability. Through a data-centric medicinal chemistry approach focused on improving potency and pharmacokinetics rather than “drug-likeness”, INCB24360/epacadostat was ultimately developed as a clinical lead agent (3, Figure 6F). As a result of this approach, the calculated properties of epacadostat actually fall outside of the classic rules of drug discovery (Lipinski’s Rule of 5 and Veber’s PSA Rule) (102, 103), but nevertheless good oral bioavailabilities were observed across all species tested (rodent, canine, primate). In profiling the compound against a panel of enzymes, receptors, ion channels, along with related or unrelated iron-containing heme enzymes (IDO2, TDO2, Cyp P450 enzymes), epacadostat was shown to be a highly selective inhibitor for the IDO1 enzyme.
Detailed preclinical studies showed that epacadostat acts as a tryptophan competitive inhibitor of the catabolic activity of human IDO1 in cell-based assays (IC50=12 nM) with >100-fold selectivity exhibited against IDO2 and TDO2. In co-cultures of human allogeneic lymphocytes with DCs or tumor cells, epacadostat promoted the growth of effector T cells and NK cells, reduced conversion of naïve T cells to Tregs and increased the number of CD86high DC (104). Consistent with these effects, administration of epacadostat to tumor-bearing syngeneic mice inhibited kynurenine levels ~90% in both plasma and tumor and reduced tumor growth in immunocompetent but not immunocompromised mice, confirming that drug efficacy relies upon functional immunity. Further, in the B16 melanoma model, epacadstat was found to enhance the antitumor effects of anti-CTLA4 or anti-PD-L1 antibodies, where increased IL-2 production and CD8+ T cell proliferation were suggestive of more pronounced T cell activity (105).
In a Phase I human study to investigate safety and maximum-tolerated dose, pharmacokinetics, pharmacodynamics and antitumor activity (106), epacadostat was generally well tolerated, effectively normalizing plasma kynurenine levels with maximal inhibition of IDO1 activity at doses of ≥100 mg twice daily. No objective responses were detected, although stable disease for ≥16 weeks was observed in 7/52 patients (106). A randomized Phase 2 study compared epacadostat versus tamoxifen treatment in 42 patients with biochemically recurrent-only epithelial ovarian cancer, primary peritoneal carcinoma or fallopian tube cancer, where epacadostat was found to be well tolerated but showed no difference in efficacy (107).
Far more exciting are early reports of the results of combining epacadostat with PD1 antibodies in melanoma, head and neck cancer and lung, renal and urothelial cancers (108). In melanoma, anti-PD-1 combinations (either pembrolizumab or nivolumab) show rates of overall response and disease control similar to those produced by the approved combination of PD-1 and CTLA-4 antibodies (ipilimumab), but without the high-grade autoimmune side-effects of combining those antibodies (109, 110). In head and neck cancer, an interim report of 38 heavily pretreated patients suggested that epacadostat could increase rates of overall response and disease control when administered with anti-PD1, in a manner independent of PD-L1 levels or HPV status, also without any notable increase in side-effects. On the basis of such results, the clinical development of epacadostat is currently being advanced aggressively in a total of 14 tumor types (including the above cancers) where it is being co-administered with anti-PD-1 antibodies (nivolumab or pembrolizumab) or anti-PD-L1 antibodies (atezolizumab and duvalumab). A definitive Phase 3 trial in melanoma for the combination with pembrolizumab is reported to be completely enrolled. The similar observations produced in melanoma patients so far by epacadostat or indoximod in combination with anti-PD-1 is extremely encouraging given the different mechanisms of action of these two agents. In summary, clinical validation is emerging for IDO1 and tryptophan catabolic pathways as therapeutic targets to improve cancer management.
BMS-986205 and other IDO1 inhibitors
An increasing number of other IDO1 enzyme inhibitors are being registered in the patent literature, as listed elsewhere (61), however, information on these compounds in the peer-reviewed biomedical literature remains limited. Several IDO1 inhibitory compounds developed in the pharmaceutical industry have been reported to be in later stage preclinical development or early clinical testing (see Table I for summary). BMS-986205 is an IDO1 inhibitor licensed by Flexus Inc. to Bristol-Myers Squibb for clinical development. This compound appears to act as an irreversible inhibitor of high potency (~2 nM) with superior pharmacokinetics relative to epacadostat and navoximod. In 2015, BMS-986205 entered a Phase 1/2 study in melanoma patients as monotherapy and in combination with nivolumab (NCT02658890). An interim report noted the compound was well tolerated in patients (111). A second Phase 1 study of the same combination is being conducted in various other types of advanced cancer patients (NCT03192943). PF-06840003 is a tryptophan non-competitive, non-heme binding IDO1 inhibitor licensed by iTeos SA to Pfizer for clinical development (Wythes et al, SITC 2016, poster 253). This compound is predicted to have favorable human PK characteristics, a prolonged human half-life that may allow single dose daily administration, and CNS penetration properties that may enable efficient access to brain metastases. In a preclinical study, PF-06840003 enhanced the antitumor efficacy of anti-PD1/PDL1 axis blockade. A first-in-patient study was initiated in 2016 in malignant gliomas (NCT02764151). Other IDO1 inhibitors developed by pharmaceutical groups in late preclinical development, including IOM2983 (Merck/IO-Met) and RG-70099 (Roche/CuraDev), have little information disclosed in the biomedical literature as yet (Table I). Preclinically, a number of new IDO1 inhibitor scaffolds have been reported, many of which are covered in a recent review (61). Of particular recent note, identification of a benzenesulfonylhydrazide series of inhibitors (112) yielded compound 40 as a potent IDO1 inhibitor in cells with EC50=68 nM, 59% oral bioavailability, the administration of which produced significant tumor growth delay without body weight loss in mouse tumor models (113).
Table I.
IDO1 inhibitors reported in development from pharmaceutical industry.
| NewLink | NewLink | Incyte | BMS Flexus |
Pfizer iTeos |
Merck IO-Met |
Roche Curadev |
|
|---|---|---|---|---|---|---|---|
| Name | Indoximod NLG-8186 | Navoximod NLG-919 | Epacadostat INCB024360 | BMS-986205 F001287 | PF-06840003 | IOM2983 | RG-70099 |
| Structure |
|
|

|

|
|
Unknown Patent Published | Unknown Patent Published |
| Mechanism | Stimulates mTORC1 downstream of IDO1/TDO | Catalytic inhibitor | Catalytic inhibitor | Suicide inhibitor | Catalytic inhibitor | Unknown | Unknown |
| Inhibitory Kinetics | NA | Tryptophan non-competitive | Tryptophan competitive | Irreversible | Tryptophan non-competitive | Unknown | Unknown |
| Cell-Based Potency | NA | 75 nM | 12 nM | 2 nM | 1100 nM | Unknown | Unknown |
| TDO Selectivity | Non-selective | 10–20 fold | >100-fold | >100-fold | >100-fold | >100-fold | ~5-fold |
| Clinical Entry | 2008 | 2015 | 2012 | 2015 | 2016 | NA | NA |
| Program Status | Phase 3 | Phase 1B | Phase 3 | Phase 2 | Phase 1 | Preclinical | Preclinical |
On the horizon: TDO and IDO2 in inflammatory programming and therapeutic utility
Accumulating evidence points to the distinct tryptophan catabolic enzyme TDO as another means of immune escape (64, 114, 115). Thus, TDO inhibition has emerged as a parallel immunomodulatory strategy to attack tumors (116–121), the rationale for which has been reviewed recently by pioneers in this area (64, 115). An early bioactive inhibitor termed 68OC91 (116) has been used for mouse studies, but more potent and pharmacologically favorable compounds have been reported (117–120). Deleting the TDO gene in mice (Tdo2) causes L-tryptophan to accumulate and these mice show neurologic alterations, possibly due to serotonin elevation (122). Treating mice with 680C91 phenocopies Tdo2 deletion and increases sensitivity to endotoxin-induced shock, implicating TDO, like IDO1, in inflammatory programming (123). However, there are differences in the inflammatory characteristics conferred by TDO despite a common role of these enzymes in tryptophan catabolism (124). While enzymological differences may help explain these different roles, they may also reflect differences in locoregional control of kynurenine production or in the availability or efficiency of kynurenine effector mechanisms (e.g. AhR binding, kynurenine pathway catabolic enzymes, etc.). While there is evidence of a contribution to tumoral immune escape using bioactive inhibitors (50, 118), no genetic proof in mice exists as yet, nor an understanding of the nature or extent of TDO expression in tumor cells or the tumor microenvironment. In preclinical studies, TDO inhibitors have not exhibited major safety concerns, although the neurological effects produced by genetic deletion of TDO in the mouse suggest an area of concern in their clinical evaluation (115, 122). With the generation of TDO-deficient mice (122), these animals should be available to investigators for in depth analysis of this issue of critical importance to consideration of TDO inhibitor development. Nevertheless, despite the current lack of a genetic preclinical proof of concept, there exists a pharmacological rationale to explore TDO inhibitors along with IDO1/TDO combined inhibitors as next-generation modalities in the field, for which navoximod and RG-70099 represent initial steps from pharmaceutical industry (Table I).
Beyond its role in immune escape, upregulation of TDO in cancer cells has been found to contribute to tumor cell survival and metastatic prowess as well (125). Anoikis resistance is a key step in metastatic progression (126). In a seminal study of aggressive ‘triple-negative’ breast cancer (TNBC), Richer and colleagues showed how TDO upregulation in cancer cell suspension culture was essential for resistance to anoikis and for metastatic capacity (125). Furthermore, TDO-induced kynurenine production was sufficient to activate the AhR signaling pathway, as is also the case with IDO1, and pharmacological inhibition or genetic attenuation of either TDO or AhR was sufficient to restore anoikis sensitivity and reduce invasive character. Lastly, 680C91 treatment was sufficient to reduce pulmonary metastasis in tumor-bearing mice, and TDO status in clinical TNBC specimens associated with increased grade, ER-negative status and shorter overall survival (125). These findings extend the concept that TDO can act like an oncogene in cancer cells to directly promote malignant outgrowth, suggesting that upregulation of TDO may provide a selective advantage beyond simply enabling peripheral immune tolerance.
Although less studied, IDO2 is a structural relative of IDO1 also implicated in immunomodulation. The evidence for this function is mainly through studies of autoimmunity to date (127), but IDO2 is anticipated to make contributions to cancer given its overexpression in some solid tumors (128, 129). In mice, Ido2 gene deletion does not affect embryonic development, hematopoiesis or immune character, nor does it affect blood levels of tryptophan or kynurenine (88). The tryptophan catalytic activity of IDO2 is weaker than that of IDO1 or TDO, especially for the human enzyme, but it is clear that IDO2 biochemistry also relies upon a different co-reductant system in cells (130). Indeed, earlier characterizations of human IDO2 as ‘inactive’ are incorrect, reflecting only non-optimal biochemical conditions that have been recently improved (131) (S.-R. Yeh, pers. communication). The fact that IDO2 deletion does not affect systemic levels of tryptophan likely reflects its far narrower range of expression, relative to IDO1 and TDO which are expressed both more broadly and more strongly.
Mouse genetic studies offer early evidence of a role for IDO2 in immune tolerance. Ido2−/− mice exhibit deficiency in their ability to support IDO1-activated T regulatory cells (88). A role in tolerance has also been suggested by work in human dendritic cells (132). Reciprocally, hematopoietic cells from Ido1−/− mice are mosaically deficient for IDO2 function, reinforcing the idea of IDO1-IDO2 interaction in immune control (88). Interestingly, in a model of autoimmune arthritis the administration of indoximod is therapeutic, phenocopying the amelioration of this chronic inflammatory disease achieved by Ido2 deletion (133). Combining indoximod treatment with Ido2 deletion provided no added benefit, consistent with IDO2 inibition being the mechanism of action as suggested by earlier evidence that indoximod blunts IDO2 catalytic activity (134). Even more strikingly, the therapeutic effect of indoximod is abolished by Ido1 deletion consistent with genetic evidence of IDO1-IDO2 interaction (133). Accordingly, common genetic variations in human IDO2 which reduce its catalytic activity may be relevant to clinical responses to indoximod (134). Reduced inflammatory disease in Ido2−/− mice is associated with diminished levels of auto-antibody and has been traced specifically to the lack of IDO2 in B cells, suggesting that IDO2 acts to support B cell involvement in eliciting inflammation (133, 135, 136). These findings are interesting in light of evidence that certain cancers rely upon B cell dependent inflamed states for their development (137, 138). While IDO2-deficient mice are unchanged with regard to their susceptibility to inflammatory skin carcinogenesis (88), they resist the development of K-Ras-induced pancreatic cancers (G.C.P. and A.J.M., unpublished observations) where B cells have been implicated (139, 140). Cancers do not tend to overexpress IDO2 although this has been observed in melanoma and gastric, brain and pancreatic cancers, in the latter case quite widely (129). The IDO2 gene is regulated by the aryl hydrocarbon receptor (AHR) (141, 142), which binds Kyn as a endogenous ligand produced by the more active IDO1 enzyme (50). Thus, given genetic evidence of IDO1-IDO2 interactions (88), it is conceivable that locoregional IDO1 activity may, by producing Kyn, increase levels of IDO2 in roving antigen-presenting cells in the tumor microenvironment, perhaps contributing to a tolerized state that engenders IDO1-mediated Treg formation in tumor-draining lymph nodes (4). Small molecule inhibitors of IDO2 have been reported (131, 143–146), but they are not bioactive and have yet to be studied in vivo. Interestingly, a B cell-penetrating bioactive antibody against IDO2 has been reported that can phenocopy the anti-arthritic effects of Ido2 genetic deficiency in the mouse (136), but this antibody has yet to be tested in cancer models.
Summary and Future Perspectives
As is the case for combating microbial and viral infections, it is clear that systemic immunity is required for effective immunotherapy in cancer (147). Vaccines against infectious pathogens include the use of adjuvants to program locoregional inflammatory signals needed for effective priming of systemic immunity. These vaccines have the advantage of being administered prophylactically so that systemic immunity can be primed and ready to respond to an infection, whereas cancer immunotherapy involves the treatment of established disease in which dominant toleragenic mechanisms have already been engaged. Thus, the reprogramming of the tumor promoting inflammatory state to one that supports the creation of systemic immunity is important to developing any effective immunotherapy regimen. In reprogramming inflammation, IDO1 inhibitors may offer broadly useful ‘immunometabolic adjuvants’ to empower systemic immune responses with immunotherapy, as recent clinical studies focused on immune checkpoint inhibitors suggest, as well as with chemotherapy or radiotherapy. From this perspective, they may be effective in helping convert immunologically ‘cold’ tumors where an inflammatory stimulus is desired, e.g. recalcitrant squamous or simple epithelial tumors (e.g. head and neck, prostate cancers).
Immune combinations with pembrolizumab or nivolumab are currently most intriguing. In human melanoma, there is strong evidence of an intertwined relationship between CTLA-4, PD-L1 and IDO1 suggestive of an interrelated signaling network (57). Notably, circulating levels of these markers on plasmacytoid dendritic cells are altered with prior invasion of a sentinel lymph node, where IDO1 expression in the sentinel node correlated to increased numbers of IDO1+ peripheral blood mononuclear cells (PBMC). In identifying the patients most likely to benefit from IDO1 inhibitors, it is tempting to speculate that these markers in the peripheral blood – associated with advanced disease and negative outcome in melanoma – create a rationale for combination treatments directed against these markers to create a desired synergistic response. Vaccine combinations are also appealing to consider (e.g. sipuleucel-T), especially in the context of a coordinate pro-inflammatory stimulus of the sort created by oncolytic viruses or PAMP/DAMP admixture. Anti-neovascular effects have yet to been explored in combination with agents that target angiogenesis (e.g. bevicizumab, bortezimib) or the hypoxic environment (e.g. hypoxia-activated prodrugs, HIF-targeting drugs, antimetabolites). In this light, IDO inhibitors may offer a unique tool in certain settings such as brain tumors or leukemia, where anti-angiogenic approaches in the cranium or bone marrow clearly pose interest. Lastly, combinations with other ‘immune adjuvant’ approaches – radiotherapy, STING, glutamine/glucose starvation signaling and adenosine signaling – can be envisioned based on emerging mechanistic interfaces yet to be therapeutically explored.
In considering TDO and IDO2, which are currently little understood in cancer, addressing to what extent blocking TDO and/or IDO2 may widen efficacy and reduce inherent resistance or the risk of acquired resistance to IDO1 blockade are additional key questions for the future. In its connections to cancer and autoimmune disease, IDO2 poses an especially fascinating subject to explore, particularly with regard to the use of IDO2 inhibitors to limit autoimmune-related adverse events that immune checkpoint therapies and other cancer therapies can produce, which are often severe and durable in survivors (e.g. arthritis-like joint inflammation which IDO2 blockade might relieve (133, 135, 136)). As IDO1 inhibitors continue to be evaluated in the clinic, combinatorial targeting of IDO1, IDO2 and TDO to various degrees in a single modality may afford a special opportunity to safely widen the therapeutic window of many regimens, not only for the new expensive immuno-oncology agents of great current interest, but also the less expensive generic standard of care modalities involving chemotherapy and radiotherapy, which are likely to remain workhorses of the cancer clinic for years to come.
Acknowledgments
We thank our many colleagues and collaborators and apologize for omitting citations to many primary research reports cited only through reviews due to space limitations. We are grateful to Peggy Scherle and Andrew Combs at Incyte Corporation and Mario Mautino at NewLink Genetics Corporation for their critical comments and contributions to the text. G.C.P. recognizes support from the National Cancer Institute (R01 CA109542, R01 CA191191), the Lankenau Medical Center Foundation and Main Line Health, along with previous support for IDO-related research from the Department of Defense Breast and Lung Cancer Research Programs, Dan Green Foundation, Sharpe-Strumia Research Foundation and New Link Genetics Corporation. G.C.P. holds The Havens Chair for Biomedical Research at the Lankenau Institute for Medical Research. A.J.M. recognizes support from I-O Biotech, Inc., the Lankenau Medical Center Foundation and Main Line Health, as well as previous support for IDO-related research from the Department of Defense Breast Cancer Research Program, Susan G. Komen for the Cure and the Pennsylvania Department of Health. W.P.M. recognizes the support of NSF grant CHE-0958996 and Bryn Mawr College.
Footnotes
Conflict of Interest Statement
The authors state a conflict of interest as shareholders, W.J.M. and G.C.P. as compensated scientific advisors, and G.C.P. as a former grant recipient of NewLink Genetics Corporation, based on their roles as inventors of IDO intellectual property licensed to NewLink as described in U.S. Patents Nos. 7705022, 7714139, 8008281, 8058416, 8383613, 8389568, 8436151, 8476454 and 8586636.
References
- 1.Prendergast GC, Jaffee EM. Cancer immunologists and cancer biologists: why we didn't talk then but need to now. Cancer Res. 2007;67:3500–4. doi: 10.1158/0008-5472.CAN-06-4626. [DOI] [PubMed] [Google Scholar]
- 2.Prendergast GC. Immunological thought in the mainstream of cancer research: Past divorce, recent remarriage and elective affinities of the future. Oncoimmunology. 2012;1:793–7. doi: 10.4161/onci.20909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prendergast GC, Jaffee EM. Cancer immunotherapy: Immune suppression and tumor growth. 2. New York: Academic Press; 2013. [Google Scholar]
- 4.Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014;63:721–35. doi: 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016;37:193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–9. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 7.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 8.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210:1389–402. doi: 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monjazeb AM, Kent MS, Grossenbacher SK, Mall C, Zamora AE, Mirsoian A, et al. Blocking Indolamine-2,3-Dioxygenase Rebound Immune Suppression Boosts Antitumor Effects of Radio-Immunotherapy in Murine Models and Spontaneous Canine Malignancies. Clin Cancer Res. 2016;22:4328–40. doi: 10.1158/1078-0432.CCR-15-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res. 2011;17:6985–91. doi: 10.1158/1078-0432.CCR-11-1331. [DOI] [PubMed] [Google Scholar]
- 11.Smith C, Chang MY, Parker KH, Beury DW, Duhadaway JB, Flick HE, et al. IDO Is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2012;2:722–35. doi: 10.1158/2159-8290.CD-12-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mondal A, Smith C, DuHadaway JB, Sutanto-Ward E, Prendergast GC, Bravo-Nuevo A, et al. IDO1 is an Integral Mediator of Inflammatory Neovascularization. EBioMedicine. 2016;14:74–82. doi: 10.1016/j.ebiom.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyland E, Williams DC. The metabolism of tryptophan. 2. The metabolism of tryptophan in patients suffering from cancer of the bladder. Biochem J. 1956;64:578–82. doi: 10.1042/bj0640578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshida R, Imanishi J, Oku T, Kishida T, Hayaishi O. Induction of pulmonary indoleamine 2,3-dioxygenase by interferon. Proc Natl Acad Sci U S A. 1981;78:129–32. doi: 10.1073/pnas.78.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 16.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–72. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 18.Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today. 1999;20:469–73. doi: 10.1016/s0167-5699(99)01520-0. [DOI] [PubMed] [Google Scholar]
- 19.Muller AJ, Prendergast GC. Marrying immunotherapy with chemotherapy: why say IDO? Cancer Res. 2005;65:8065–8. doi: 10.1158/0008-5472.CAN-05-2213. [DOI] [PubMed] [Google Scholar]
- 20.Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat Rev Cancer. 2006;6:613–25. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- 21.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–54. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prendergast GC. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene. 2008;27:3889–900. doi: 10.1038/onc.2008.35. [DOI] [PubMed] [Google Scholar]
- 23.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 24.Theate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld JC, et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer immunology research. 2015;3:161–72. doi: 10.1158/2326-6066.CIR-14-0137. [DOI] [PubMed] [Google Scholar]
- 25.Muller AJ, DuHadaway JB, Sutanto-Ward E, Donover PS, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunomodulatory target of the tumor suppressor gene Bin1, potentiates cancer chemotherapy. Nature Med. 2005;11:312–9. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 26.Prendergast GC, Muller AJ, Ramalingam A, Chang MY. BAR the door: cancer suppression by amphiphysin-like genes. Biochim Biophys Acta. 2009;1795:25–36. doi: 10.1016/j.bbcan.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ge K, DuHadaway J, Du W, Herlyn M, Rodeck U, Prendergast GC. Mechanism for elimination of a tumor suppressor: aberrant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proc Natl Acad Sci USA. 1999;96:9689–94. doi: 10.1073/pnas.96.17.9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pineda-Lucena A, Ho CS, Mao DY, Sheng Y, Laister RC, Muhandiram R, et al. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. Journal of molecular biology. 2005;351:182–94. doi: 10.1016/j.jmb.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 29.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14:185–93. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anczukow O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19:220–8. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakacs A, Coppola L, et al. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71:4464–72. doi: 10.1158/0008-5472.CAN-10-4410. [DOI] [PubMed] [Google Scholar]
- 32.Barekati Z, Radpour R, Lu Q, Bitzer J, Zheng H, Toniolo P, et al. Methylation signature of lymph node metastases in breast cancer patients. BMC Cancer. 2012;12:244. doi: 10.1186/1471-2407-12-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radpour R, Barekati Z, Kohler C, Lv Q, Burki N, Diesch C, et al. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One. 2011;6:e16080. doi: 10.1371/journal.pone.0016080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Radpour R, Kohler C, Haghighi MM, Fan AX, Holzgreve W, Zhong XY. Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array. Oncogene. 2009;28:2969–78. doi: 10.1038/onc.2009.149. [DOI] [PubMed] [Google Scholar]
- 35.Kuznetsova EB, Kekeeva TV, Larin SS, Zemlyakova VV, Khomyakova AV, Babenko OV, et al. Methylation of the BIN1 gene promoter CpG island associated with breast and prostate cancer. Journal of carcinogenesis. 2007;6:9. doi: 10.1186/1477-3163-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenna ES, Tamayo P, Cho YJ, Tillman EJ, Mora-Blanco EL, Sansam CG, et al. Epigenetic inactivation of the tumor suppressor BIN1 drives proliferation of SNF5-deficient tumors. Cell Cycle. 2012;11:1956–65. doi: 10.4161/cc.20280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller AJ, DuHadaway JB, Chang MY, Ramalingam A, Sutanto-Ward E, Boulden J, et al. Non-hematopoietic expression of IDO is integrally required for inflammatory tumor promotion. Cancer Immunol Immunother. 2010;59:1655–63. doi: 10.1007/s00262-010-0891-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop LD, Metz R, et al. IDO in inflammatory programming and immune suppression in cancer. In: Gabrilovich DI, Hurwitz AA, editors. Tumor-Induced Immune Suppression. New York: Springer; 2014. [Google Scholar]
- 39.Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Diff. 2002;9:1069–77. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- 40.Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–68. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–57. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–70. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 43.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–42. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 44.Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. 2006;108:4118–25. doi: 10.1182/blood-2006-03-006700. [DOI] [PubMed] [Google Scholar]
- 45.Mellor AL, Chandler P, Baban B, Hansen AM, Marshall B, Pihkala J, et al. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. 2004;16:1391–401. doi: 10.1093/intimm/dxh140. [DOI] [PubMed] [Google Scholar]
- 46.Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008;8:74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- 47.Grohmann U, Fallarino F, Puccetti P. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol. 2003;24:242–8. doi: 10.1016/s1471-4906(03)00072-3. [DOI] [PubMed] [Google Scholar]
- 48.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–74. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 49.Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176:6752–61. doi: 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- 50.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 51.Muller AJ, Sharma MD, Chandler PR, Duhadaway JB, Everhart ME, Johnson BA, 3rd, et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci U S A. 2008;105:17073–8. doi: 10.1073/pnas.0806173105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beatty GL, Paterson Y. Regulation of tumor growth by IFN-gamma in cancer immunotherapy. Immunologic research. 2001;24:201–10. doi: 10.1385/IR:24:2:201. [DOI] [PubMed] [Google Scholar]
- 53.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–9. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dougan M, Li D, Neuberg D, Mihm M, Googe P, Wong KK, et al. A dual role for the immune response in a mouse model of inflammation-associated lung cancer. J Clin Invest. 2011;121:2436–46. doi: 10.1172/JCI44796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qin Z, Blankenstein T. CD4+ T cell--mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. 2000;12:677–86. doi: 10.1016/s1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- 56.Qin Z, Schwartzkopff J, Pradera F, Kammertoens T, Seliger B, Pircher H, et al. A critical requirement of interferon gamma-mediated angiostasis for tumor rejection by CD8+ T cells. Cancer Res. 2003;63:4095–100. [PubMed] [Google Scholar]
- 57.Chevolet I, Speeckaert R, Schreuer M, Neyns B, Krysko O, Bachert C, et al. Characterization of the in vivo immune network of IDO, tryptophan metabolism, PD-L1, and CTLA-4 in circulating immune cells in melanoma. Oncoimmunology. 2015;4:e982382. doi: 10.4161/2162402X.2014.982382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang MY, Smith C, Duhadaway JB, Pyle JR, Boulden J, Peralta Soler A, et al. Cardiac and gastrointestinal liabilities caused by deficiency in the immune modulatory enzyme indoleamine 2,3-dioxygenase. Cancer Biol Ther. 2011;12 doi: 10.4161/cbt.12.12.18142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17:1094–100. doi: 10.1038/nm.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeung AW, Terentis AC, King NJ, Thomas SR. Role of indoleamine 2,3-dioxygenase in health and disease. Clinical science. 2015;129:601–72. doi: 10.1042/CS20140392. [DOI] [PubMed] [Google Scholar]
- 61.Rohrig UF, Majjigapu SR, Vogel P, Zoete V, Michielin O. Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. J Med Chem. 2015;58:9421–37. doi: 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]
- 62.Brochez L, Chevolet I, Kruse V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur J Cancer. 2017;76:167–82. doi: 10.1016/j.ejca.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 63.Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular Pathways: Targeting IDO1 and Other Tryptophan Dioxygenases for Cancer Immunotherapy. Clin Cancer Res. 2015;21:5427–33. doi: 10.1158/1078-0432.CCR-15-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Baren N, Van den Eynde BJ. Tumoral Immune Resistance Mediated by Enzymes That Degrade Tryptophan. Cancer immunology research. 2015;3:978–85. doi: 10.1158/2326-6066.CIR-15-0095. [DOI] [PubMed] [Google Scholar]
- 65.Selvan SR, Dowling JP, Kelly WK, Lin J. Indoleamine 2,3-dioxygenase (IDO): Biology and Target in Cancer Immunotherapies. Current cancer drug targets. 2016;16:755–64. doi: 10.2174/1568009615666151030102250. [DOI] [PubMed] [Google Scholar]
- 66.Li F, Zhang R, Li S, Liu J. IDO1: An important immunotherapy target in cancer treatment. International immunopharmacology. 2017;47:70–7. doi: 10.1016/j.intimp.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 67.Cady SG, Sono M. 1-methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and beta-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch Biochem Biophys. 1991;291:326–33. doi: 10.1016/0003-9861(91)90142-6. [DOI] [PubMed] [Google Scholar]
- 68.Peterson AC, Migawa MT, Martin MJ, Hamaker LK, Czerwinski KM, Zhang W, et al. Evaluation of functionalized tryptophan derivatives and related compounds as competitive inhibitors of indoleamine 2,3-dioxygenase. Medicinal Chemistry Research. 1994;3:531–44. [Google Scholar]
- 69.Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. 2002;101:151–5. doi: 10.1002/ijc.10645. [DOI] [PubMed] [Google Scholar]
- 70.Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. 2008;222:357–68. doi: 10.1111/j.1600-065X.2008.00604.x. [DOI] [PubMed] [Google Scholar]
- 71.Soliman HH, Jackson E, Neuger T, Dees EC, Harvey RD, Han H, et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. 2014;5:8136–46. doi: 10.18632/oncotarget.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1:1460–8. doi: 10.4161/onci.21716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mautino MR, Kumar S, Zhuang H, Waldo J, Jaipuri F, Potturi H, et al. 2017 Annual Meeting of the American Association for Cancer Research. Washington D.C.: American Association for Cancer Research; 2017. A novel prodrug of indoximod with enhanced pharmacokinetic properties. [Google Scholar]
- 74.Colwell J. Indoximod combo triggers responses in melanoma. Cancer Discov. 2017;7:542–3. doi: 10.1158/2159-8290.CD-NB2017-056. [DOI] [PubMed] [Google Scholar]
- 75.Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011;71:5445–54. doi: 10.1158/0008-5472.CAN-11-1138. [DOI] [PubMed] [Google Scholar]
- 76.Xie DL, Wu J, Lou YL, Zhong XP. Tumor suppressor TSC1 is critical for T-cell anergy. Proc Natl Acad Sci U S A. 2012;109:14152–7. doi: 10.1073/pnas.1119744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soliman HH, Minton SE, Han HS, Ismail-Khan R, Neuger A, Khambati F, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget. 2016;7:22928–38. doi: 10.18632/oncotarget.8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zakharia Y, McWilliams R, Shaheen M, Grossman K, Drabick J, Milhem M, et al. Interim analysis of the Phase 2 clinical trial of the IDO pathway inhibitor indoximod in combination with pembrolizumab for patients with advanced melanoma. Cancer Res. 2017;77:CT117. [Google Scholar]
- 79.Zahkaria Y. Combined inhibition of the IDO and PD-1 pathways improves the response rate for patients with advanced melanoma; Third CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference; 2017; Mainz, Germay. [Google Scholar]
- 80.Jha GG, Gupta S, Tagawa ST, Koopmeiners JS, Vivek S, Dudek AZ, et al. A phase II randomized, double-blind study of sipuleucel-T followed by IDO pathway inhibitor, indoximod, or placebo in the treatment of patients with metastatic castration resistant prostate cancer (mCRPC) J Clin Oncol. 2017;35:3066. [Google Scholar]
- 81.Emadi A, Holtzman NG, Imran M, El-Chaer F, Koka M, Singh Z, et al. Indoximod in combination with idarubicin and cytarabine for upfront treatment of patients with newly diagnosed acute myeloid leukemia (AML): Phase 1 report; 22nd Congress of the European Hematology Association; 2017 22–25 June 2017; Madrid, Spain. [Google Scholar]
- 82.Bahary N, Garrido-Laguna I, Cinar P, Somer BG, Nayak-Kapoor A, Lee JS, et al. Phase 2 trial of the indoleamine 2,3-dioxygenase pathway (IDO) inhibitor indoximod plus gemcitabine/nab-paclitaxel for the treatment of metastastic pancreas cancer: interim analysis; 2016 Annual Meeting of the American Society of Clinical Oncology; 2017; Chicago. [Google Scholar]
- 83.Gaspari P, Banerjee T, Malachowski WP, Muller AJ, Prendergast GC, Duhadaway J, et al. Structure-activity study of brassinin derivatives as indoleamine 2,3-dioxygenase inhibitors. J Med Chem. 2006;49:684–92. doi: 10.1021/jm0508888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Banerjee T, DuHadaway JB, Gaspari PE, Sutanto-Ward E, Munn DH, Mellor AL, et al. A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene. 2008;27:2851–7. doi: 10.1038/sj.onc.1210939. [DOI] [PubMed] [Google Scholar]
- 85.Kumar S, Jaller D, Patel B, LaLonde JM, DuHadaway JB, Malachowski WP, et al. Structure based development of phenylimidazole-derived inhibitors of indoleamine 2,3-dioxygenase. J Med Chem. 2008;51:4968–77. doi: 10.1021/jm800512z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kumar S, Malachowski WP, DuHadaway JB, LaLonde JM, Carroll PJ, Jaller D, et al. Indoleamine 2,3-dioxygenase is the anticancer target for a novel series of potent naphthoquinone-based inhibitors. J Med Chem. 2008;51:1706–18. doi: 10.1021/jm7014155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sono M, Cady SG. Enzyme kinetic and spectroscopic studies of inhibitor and effector interactions with indoleamine 2,3-dioxygenase. 1. Norharman and 4-phenylimidazole binding to the enzyme as inhibitors and heme ligands. Biochemistry. 1989;28:5392–9. doi: 10.1021/bi00439a012. [DOI] [PubMed] [Google Scholar]
- 88.Metz R, Smith C, Duhadaway JB, Chandler P, Baban B, Merlo LM, et al. IDO2 is critical for IDO1-mediated T cell regulation and exerts a non-redundant function in inflammation. Int Immunol. 2014;26:357–67. doi: 10.1093/intimm/dxt073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sono M, Cady SG. Enzyme kinetic and spectroscopic studies of inhibitor and effector interactions with indoleamine 2,3-dioxygenase. 1. Norharman and 4-phenylimidazole binding to the enzyme as inhibitors and heme ligands. Biochemistry. 1989;28:5392–9. doi: 10.1021/bi00439a012. [DOI] [PubMed] [Google Scholar]
- 90.Sugimoto H, Oda S, Otsuki T, Hino T, Yoshida T, Shiro Y. Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc Natl Acad Sci U S A. 2006;103:2611–6. doi: 10.1073/pnas.0508996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rohrig UF, Majjigapu SR, Grosdidier A, Bron S, Stroobant V, Pilotte L, et al. Rational design of 4-aryl-1,2,3-triazoles for indoleamine 2,3-dioxygenase 1 inhibition. J Med Chem. 2012;55:5270–90. doi: 10.1021/jm300260v. [DOI] [PubMed] [Google Scholar]
- 92.Mautino MR, Jaipuri FA, Waldo J, Kumar S, Adams J, Van Allen C, et al. NLG919, a novel indoleamine-2,3-dioxygenase (IDO)-pathway inhibitor drug candidate for cancer therapy. Cancer Res. 2013;73(8 Suppl):491. [Google Scholar]
- 93.Nayak A, Hao Z, Sadek R, Dobbins R, Marshall L, Vahanian NN, et al. ECCO-ESMO 215. Vienna, Austria: 2015. Phase 1a study of the safety, pharmacokinetics, and pharmacodynamics of GDC-0919 in patients with recurrent/advanced solid tumors. [Google Scholar]
- 94.Peng YH, Ueng SH, Tseng CT, Hung MS, Song JS, Wu JS, et al. Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives with IDO1. J Med Chem. 2016;59:282–93. doi: 10.1021/acs.jmedchem.5b01390. [DOI] [PubMed] [Google Scholar]
- 95.Spahn J, Peng J, Lorenzana E, Kan D, Hunsaker T, Segal E, et al. Improved anti-tumor immunity and efficacy upon combination of the IDO1 inhibitor GDC-0919 with anti-PD-L1 blockade versus anti-PD-L1 alone in preclinical tumor models. J Immunother Canc. 2015;3(Suppl 2):303. [Google Scholar]
- 96.Burris HA, Gordon MS, Hellmann MD, LoRusso P, Emens LA, Hodi FS, et al. A Phase 1b dose escalation study of combined inhibition of IDO1 (GDC-0919) and PD-L1 (atezolizumab) in patients with locally advanced or metastatic solid tumors. J Clin Oncol. 2017;35(suppl) abstr 105. [Google Scholar]
- 97.Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. 2002;101:151–5. doi: 10.1002/ijc.10645. [DOI] [PubMed] [Google Scholar]
- 98.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 99.Halford B. Drug Candidates Unveiled At ‘First-Time Disclosures’ Symposium In Boston. Chem Eng News. 2015;93:38–40. [Google Scholar]
- 100.Yue EW, Sparks R, Polam P, Modi D, Douty B, Wayland B, et al. INCB24360 (epacadostat), a highly potent and selective indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS medicinal chemistry letters. 2017;8:486–91. doi: 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yue EW, Douty B, Wayland B, Bower M, Liu X, Leffet L, et al. Discovery of Potent Competitive Inhibitors of Indoleamine 2,3-Dioxygenase with in Vivo Pharmacodynamic Activity and Efficacy in a Mouse Melanoma Model. Journal of Medicinal Chemistry. 2009;52:7364–7. doi: 10.1021/jm900518f. [DOI] [PubMed] [Google Scholar]
- 102.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 103.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–23. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 104.Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115:3520–30. doi: 10.1182/blood-2009-09-246124. [DOI] [PubMed] [Google Scholar]
- 105.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3. doi: 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beatty GL, O'Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle P, et al. First-in-Human Phase 1 Study of the Oral Inhibitor of Indoleamine 2,3-dioxygenase-1 Epacadostat (INCB024360) in Patients With Advanced Solid Malignancies. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kristeleit R, Davidenko I, Shirinkin V, El-Khouly F, Bondarenko I, Goodheart MJ, et al. A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)-only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol Oncol. 2017;146:484–90. doi: 10.1016/j.ygyno.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 108.Rose S. Epacadostat shows value in two SCCHN trials. Cancer Disc. 2017;7:OF2. doi: 10.1158/2159-8290.CD-NB2017-100. [DOI] [PubMed] [Google Scholar]
- 109.Gangadhar TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated Phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27:379–400. [Google Scholar]
- 110.Perez RP, Riese MJ, Lewis KD, Saleh MN, Daud A, Berlin J, et al. Epacadostat plus nivolumab in patients with advanced solid tumors: Preliminary phase I/II results of ECHO-204. J Clin Oncol. 2017;35:3003. [Google Scholar]
- 111.Siu LL. BMS-986205, an optimized indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor, is well tolerated with potent pharmacodynamic (PD) activity, alone and in combination with nivolumab (nivo) in advanced cancers in a Phase 1/2A trial; 2017 Annual Meeting of the American Association for Cancer Research; 2017 4 April 2017; Washington DO. [Google Scholar]
- 112.Cheng MF, Hung MS, Song JS, Lin SY, Liao FY, Wu MH, et al. Discovery and structure-activity relationships of phenyl benzenesulfonylhydrazides as novel indoleamine 2,3-dioxygenase inhibitors. Bioorganic & medicinal chemistry letters. 2014;24:3403–6. doi: 10.1016/j.bmcl.2014.05.084. [DOI] [PubMed] [Google Scholar]
- 113.Lin S-Y, Yeh T-K, Kuo C-C, Song J-S, Cheng M-F, Liao F-Y, et al. Phenyl Benzenesulfonylhydrazides Exhibit Selective Indoleamine 2,3-Dioxygenase Inhibition with Potent in Vivo Pharmacodynamic Activity and Antitumor Efficacy. Journal of Medicinal Chemistry. 2016;59:419–30. doi: 10.1021/acs.jmedchem.5b01640. [DOI] [PubMed] [Google Scholar]
- 114.Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 115.Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Frontiers in immunology. 2014;5:673. doi: 10.3389/fimmu.2014.00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Salter M, Hazelwood R, Pogson CI, Iyer R, Madge DJ. The effects of a novel and selective inhibitor of tryptophan 2,3-dioxygenase on tryptophan and serotonin metabolism in the rat. Biochem Pharmacol. 1995;49:1435–42. doi: 10.1016/0006-2952(95)00006-l. [DOI] [PubMed] [Google Scholar]
- 117.Dolusic E, Larrieu P, Moineaux L, Stroobant V, Pilotte L, Colau D, et al. Tryptophan 2,3-Dioxygenase (TDO) Inhibitors. 3-(2-(Pyridyl)ethenyl)indoles as Potential Anticancer Immunomodulators. J Med Chem. 2011;54:5320–34. doi: 10.1021/jm2006782. [DOI] [PubMed] [Google Scholar]
- 118.Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109:2497–502. doi: 10.1073/pnas.1113873109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pantouris G, Mowat CG. Antitumour agents as inhibitors of tryptophan 2,3-dioxygenase. Biochem Biophys Res Commun. 2014;443:28–31. doi: 10.1016/j.bbrc.2013.11.037. [DOI] [PubMed] [Google Scholar]
- 120.Wu JS, Lin SY, Liao FY, Hsiao WC, Lee LC, Peng YH, et al. Identification of Substituted Naphthotriazolediones as Novel Tryptophan 2,3-Dioxygenase (TDO) Inhibitors through Structure-Based Virtual Screening. J Med Chem. 2015;58:7807–19. doi: 10.1021/acs.jmedchem.5b00921. [DOI] [PubMed] [Google Scholar]
- 121.Abdel-Magid AF. Targeting the Inhibition of Tryptophan 2,3-Dioxygenase (TDO-2) for Cancer Treatment. ACS medicinal chemistry letters. 2017;8:11–3. doi: 10.1021/acsmedchemlett.6b00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kanai M, Funakoshi H, Takahashi H, Hayakawa T, Mizuno S, Matsumoto K, et al. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol Brain. 2009;2:8. doi: 10.1186/1756-6606-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature. 2014;511:184–90. doi: 10.1038/nature13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Larkin PB, Sathyasaikumar KV, Notarangelo FM, Funakoshi H, Nakamura T, Schwarcz R, et al. Tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase 1 make separate, tissue-specific contributions to basal and inflammation-induced kynurenine pathway metabolism in mice. Biochim Biophys Acta. 2016;1860:2345–54. doi: 10.1016/j.bbagen.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.D'Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, et al. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015;75:4651–64. doi: 10.1158/0008-5472.CAN-15-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 127.Prendergast GC, Metz R, Muller AJ, Merlo LM, Mandik-Nayak L. IDO2 in Immunomodulation and Autoimmune Disease. Frontiers in immunology. 2014;5:585. doi: 10.3389/fimmu.2014.00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lob S, Konigsrainer A, Zieker D, Brucher BL, Rammensee HG, Opelz G, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother. 2009;58:153–7. doi: 10.1007/s00262-008-0513-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Witkiewicz AK, Costantino CL, Metz R, Muller AJ, Prendergast GC, Yeo CJ, et al. Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. Journal of the American College of Surgeons. 2009;208:781–7. doi: 10.1016/j.jamcollsurg.2008.12.018. discussion 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Eldredge HB, Denittis A, Duhadaway JB, Chernick M, Metz R, Prendergast GC. Concurrent whole brain radiotherapy and short-course chloroquine in patients with brain metastases: a pilot trial. Journal of radiation oncology. 2013;2 doi: 10.1007/s13566-013-0111-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li J, Li Y, Yang D, Hu N, Guo Z, Kuang C, et al. Establishment of a human indoleamine 2, 3-dioxygenase 2 (hIDO2) bioassay system and discovery of tryptanthrin derivatives as potent hIDO2 inhibitors. Eur J Med Chem. 2016;123:171–9. doi: 10.1016/j.ejmech.2016.07.013. [DOI] [PubMed] [Google Scholar]
- 132.Trabanelli S, Ocadlikova D, Ciciarello M, Salvestrini V, Lecciso M, Jandus C, et al. The SOCS3-Independent Expression of IDO2 Supports the Homeostatic Generation of T Regulatory Cells by Human Dendritic Cells. J Immunol. 2014;192:1231–40. doi: 10.4049/jimmunol.1300720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Merlo LM, Pigott E, Duhadaway JB, Grabler S, Metz R, Prendergast GC, et al. IDO2 Is a critical mediator of autoantibody production and inflammatory pathogenesis in a mouse model of autoimmune arthritis. J Immunol. 2014;92:2082–90. doi: 10.4049/jimmunol.1303012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Metz R, Duhadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007;67:7082–7. doi: 10.1158/0008-5472.CAN-07-1872. [DOI] [PubMed] [Google Scholar]
- 135.Merlo LM, DuHadaway JB, Grabler S, Prendergast GC, Muller AJ, Mandik-Nayak L. IDO2 Modulates T Cell-Dependent Autoimmune Responses through a B Cell-Intrinsic Mechanism. J Immunol. 2016;196:4487–97. doi: 10.4049/jimmunol.1600141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Merlo LM, Grabler S, DuHadaway JB, Pigott E, Manley K, Prendergast GC, et al. Therapeutic antibody targeting of indoleamine-2,3-dioxygenase (IDO2) inhibits autoimmune arthritis. Clin Immunol. 2017;179:8–16. doi: 10.1016/j.clim.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Affara NI, Ruffell B, Medler TR, Gunderson AJ, Johansson M, Bornstein S, et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell. 2014;25:809–21. doi: 10.1016/j.ccr.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schioppa T, Moore R, Thompson RG, Rosser EC, Kulbe H, Nedospasov S, et al. B regulatory cells and the tumor-promoting actions of TNF-alpha during squamous carcinogenesis. Proc Natl Acad Sci U S A. 2011;108:10662–7. doi: 10.1073/pnas.1100994108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016;6:270–85. doi: 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D, et al. Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia. Cancer Discov. 2016;6:256–69. doi: 10.1158/2159-8290.CD-15-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bankoti J, Rase B, Simones T, Shepherd DM. Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicology and applied pharmacology. 2010;246:18–28. doi: 10.1016/j.taap.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Simones T, Shepherd DM. Consequences of AhR activation in steady-state dendritic cells. Toxicol Sci. 2011;119:293–307. doi: 10.1093/toxsci/kfq354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rohrig UF, Majjigapu SR, Caldelari D, Dilek N, Reichenbach P, Ascencao K, et al. 1,2,3-Triazoles as inhibitors of indoleamine 2,3-dioxygenase 2 (IDO2) Bioorganic & medicinal chemistry letters. 2016;26:4330–3. doi: 10.1016/j.bmcl.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 144.Austin CJ, Mailu BM, Maghzal GJ, Sanchez-Perez A, Rahlfs S, Zocher K, et al. Biochemical characteristics and inhibitor selectivity of mouse indoleamine 2,3-dioxygenase-2. Amino Acids. 2010;39:565–78. doi: 10.1007/s00726-010-0475-9. [DOI] [PubMed] [Google Scholar]
- 145.Bakmiwewa SM, Fatokun AA, Tran A, Payne RJ, Hunt NH, Ball HJ. Identification of selective inhibitors of indoleamine 2,3-dioxygenase 2. Bioorganic & medicinal chemistry letters. 2012;22:7641–6. doi: 10.1016/j.bmcl.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 146.Pantouris G, Serys M, Yuasa HJ, Ball HJ, Mowat CG. Human indoleamine 2,3-dioxygenase-2 has substrate specificity and inhibition characteristics distinct from those of indoleamine 2,3-dioxygenase-1. Amino Acids. 2014;46:2155–63. doi: 10.1007/s00726-014-1766-3. [DOI] [PubMed] [Google Scholar]
- 147.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell. 2017;168:487–502. e15. doi: 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]