

 We report the orthogonal assembly of two structurally dissimilar catalytic hydrogelators with mutually incompatible functional groups able to retain their individual catalytic activity and thus catalyse tandem reactions in one pot.
We report the orthogonal assembly of two structurally dissimilar catalytic hydrogelators with mutually incompatible functional groups able to retain their individual catalytic activity and thus catalyse tandem reactions in one pot.
Abstract
By equipping mutually incompatible carboxylic acid and proline catalytic groups with different self-assembling motives we have achieved self-sorting of the resulting catalytic gelators, namely SucVal8 and ProValDoc, into different supramolecular fibers, thus preventing the acidic and basic catalytic groups from interfering with each other. The resulting spatial separation of the incompatible catalytic functions is found to be essential to achieve one-pot deacetalization–aldol tandem reactions with up to 85% efficiency and 90% enantioselectivity. On the contrary, when SucVal8 was co-assembled with a structurally similar catalytically active hydrogelator (ProVal8), self-sorting was precluded and no tandem catalysis was observed.
Introduction
Tandem catalysis has received increasing attention in recent years due to the process simplicity, economy of time and energy and waste reduction, among other practical advantages, associated with one-pot procedures. In particular, orthogonal tandem catalysis is a one-pot reaction in which sequential catalytic processes occur through two or more functionally distinct and non-interfering catalytic cycles.1 Excellent examples are found in multi-enzymatic complexes present in biosynthetic pathways, in which a substrate is sequentially transformed towards a product in an effective and selective manner.2 Multifunctional catalysts have been prepared based on the spatial separation of catalytic groups in nanostructured materials such as star polymers, silicas, zeolites and MOFs, among others, quite often requiring tedious preparation procedures.3 Some of these systems are based on the controlled organization of incompatible acidic and basic catalytic groups into non-interacting locations. In this context, self-assembly of discrete functional components into multifunctional self-organized materials by bottom-up non-covalent synthesis could represent a powerful strategy incorporating both functional diversity as well as synthetic economy. Catalytic supramolecular nanomaterials can be prepared by self-assembly of tailor-made low molecular weight functional building blocks.4 In this respect, we and others have reported several examples of catalytic molecular gels in which catalytic activity has emerged as a consequence of the supramolecular organization of functional groups.5
These examples were based on a single catalytic component. However, supramolecular tandem catalysis requires the spatial separation of two incompatible catalytic moieties. One way to achieve spatial separation by self-assembly is self-sorting, a relevant phenomenon in complex supramolecular systems that can be defined as a high fidelity recognition between molecules based on non-covalent interactions and structural complementarity.6 Seminal work in this field was reported by Menger et al. showing self-sorting of fibers and microcrystallites based upon side chain differences of related compounds, as well as by van Esch et al. who reported orthogonal self-assembly of surfactants and hydrogelators into co-existing fibers and vesicles.7 Since then, several groups have studied this phenomenon in depth in order to prepare spatially and temporally resolved multicomponent nanoscale materials.8,9
In this communication we present for the first time the use of two orthogonally self-assembled catalytic molecular hydrogel networks for performing one-pot tandem catalysis. In contrast, an analogous co-assembled network is shown to be catalytically inactive (Scheme 1a). For this purpose, we selected three low-molecular weight hydrogelators with different catalytic groups and different self-assembly motives, which were previously described in our group (Scheme 1b). Compounds SucVal8 and ProVal8 present a bolaamphiphilic structure of similar molecular dimensions bearing carboxyl groups and l-proline residues, respectively, as end-groups.10 Compound ProValDoc is an amphiphilic dipeptide with an l-proline residue at the N-terminus.5b
Scheme 1. (a) Reaction scheme and self-sorting vs. co-assembly of different self-assembling components based on the degree of interaction and recognition between them; (b) structures of hydrogelators SucVal8, ProValDoc and ProVal8.

Results and discussion
Two different hydrogels were prepared by mixing SucVal8 and the two l-proline derivatives (ProValDoc and ProVal8). We envisaged that the structural differences between SucVal8 and ProValDoc would lead to their orthogonal self-assembly into two self-sorted networks, whereas the similar backbone structure of SucVal8 and ProVal8 would render a single co-assembled network. Then, these two mixed networks were studied for the one-pot deacetalization–direct aldol tandem reaction (Scheme 1a). All the individual gels and systems of two hydrogels were prepared by gentle heating to completely dissolve the compounds in water followed by sonication for 1 min, and left to stand overnight at 25 °C. For catalysis, the reactants were added to the gels and vortexed. Upon reaction, the products were extracted at the desired time by extracting twice with 1 mL CDCl3, dried over MgSO4, and observed by 1H NMR.
The hydrogels of ProValDoc, on the one hand, are white turbid looking gels with a minimum gel concentration (m.g.c) of 5.7 mM and temperature for the gel to solution transition, Tgel = 40 °C (see ESI† for gel preparation methods; Tgel was determined by inverted vial test). The catalytic activity of ProValDoc for the direct aldol condensation of aldehydes and ketones has already been reported by our group.5b,d On the other hand, SucVal8 is a bolaamphiphilic hydrogelator with carboxylic acid groups at the two ends, and is able to form transparent self-standing hydrogels showing a m.g.c of 7 mM with Tgel = 70 °C.10b The use of Brønsted acids for deprotection of acetal groups is well known.11 First, we checked the catalytic activity of SucVal8 fibers for the deacetalisation reaction. Complete conversion of 35 mM benzaldehyde dimethyl acetal into benzaldehyde was observed in 10 hours when 20 mol% (7 mM gel) of catalyst was used, while a lower Kobs was observed in samples of SucVal8 in solution (Table S1 in ESI†). ProValDoc did not catalyse this reaction, either in solution or as a gel.
Subsequently, a mixture of both SucVal8 + ProValDoc at their respective m.g.c values was used to obtain a strong turbid translucent gel. The AFM (Fig. 1) and TEM (Fig. S1–S5 in ESI†) images of this gel revealed the presence of two different kinds of fibers (Fig. 1c and S8c†). The larger, more crystalline fibers were attributed to ProValDoc, whereas the coexisting twisted thinner fibers were assigned to SucVal8, based on their similarity with the fiber morphologies observed for the single component gels (Fig. 1a and b). The height and width profiles provided in the ESI (Fig. S7 and S8†) show the presence of two different kinds of fibers with profiles similar to the individual gelators.
Fig. 1. AFM images of hydrogels of (A) SucVal8, (B) ProValDoc, (C) ProValDoc + SucVal8 and (D) ProVal8 + SucVal8.

To further confirm the orthogonal self-assembly of the catalytic network components, we performed wide angle powder X-ray diffraction (WAXD) of the xerogels of the two different gelators at their m.g.c and of their mixture. The WAXD data for SucVal8 reflected an amorphous structure with no crystallinity (Fig. 2a), whereas that for ProValDoc alone showed more crystallinity, which can be attributed to one of the previously described polymorphs of this compound, the most thermostable one (Fig. 2b).12 Interestingly, the WAXD of the mixture showed an overlap of the patterns of amorphous SucVal8 and a different polymorph of ProValDoc, which is dominant when gels are formed by pH change from acidic solutions (Fig. 2c and d). Even though gels of this compound alone and in the mixture of two hydrogelators were both prepared by heating followed by sonication, we obtained different polymorphs. This result suggests, as already mentioned in our previous report, a seeding effect caused by a small amount of protonated ProValDoc crystals which could be formed in the presence of the acidic counterpart. Nevertheless, as we reported before, all the polymorphs are catalytically active with only slight differences in the respective reaction rates.12 To further confirm the self-sorting of the two fibers, rheological experiments were done, which revealed the G′max for the mixed system (SucVal8 + ProValDoc) to be 11 430 Pa, which is one order of magnitude higher than the individual gelators SucVal8 (G′max = 1578 Pa) and ProValDoc (G′max = 650 Pa) (Fig. 3 and S10†).
Fig. 2. WAXD patterns of the xerogels of (a) SucVal8, (b) ProValDoc and (c) SucVal8 + ProValDoc. (d) ProValDoc gel formed by pH change.

Fig. 3. Comparison of G′max of different gels. The plateau region of the graph was taken as the G′max of each system (Fig. S9†).

This falls in line with the previous reports of orthogonal systems being mechanically stronger than the individual hydrogelators owing to the higher density of fibers.13 Additionally, DSC experiments revealed the thermal imprints of both SucVal8 and ProValDoc in the mixed system, suggesting the self-sorted assembly of both the hydrogelators (Fig. S11†).
From the Tgel experiments we know that SucVal8 forms more thermally stable gels than ProValDoc. Therefore, we formed different gels by mixing the two gelators and varying the concentration of ProValDoc to see if this induced an effect on the overall thermal stability of the gelator (Fig. 4). The gels of the mixture proved to be slightly more (∼5 °C higher) thermally stable than SucVal8, probably due to the higher density of fibers in the system owing to the presence of two different self-sorted fiber networks, making the gel stiffer and more stable.13,9b Thus, the self-sorting of molecules in a gel system consisting of two structurally different hydrogelators was suggested by the AFM, TEM, rheology, Tgel, WAXD and DSC experiments.
Fig. 4. T gel of the different gels of SucVal8 + ProValDoc and SucVal8 + ProVal8 formed by varying the concentration of the proline component. SucVal8 was present in the mixtures at its m.g.c.

The orthogonally assembled catalytic fibers were then examined for catalytic activity for the two previously mentioned reactions in tandem and in one pot. The SucVal8 + ProValDoc gel was first tested for deprotection of the acetal group. A quantitative yield of benzaldehyde from benzaldehyde dimethyl acetal (35 mM) was obtained in 10 h using 7 mM (20 mol%) of the catalyst. To examine the viability of the SucVal8 + ProValDoc gel for the tandem reaction, the two substrates, benzaldehyde dimethyl acetal and cyclohexanone, were simultaneously introduced to the system at t = 0. In 72 hours more than 85% of the aldehyde obtained by deacetalisation was converted to the final aldol product with an anti : syn ratio of 84 : 16 (Fig. 5a, 90% ee). Kobs = (2.1 ± 0.4) × 10–5 s–1 for the tandem reaction. No final product was observed when both the gelators were mixed when completely soluble (see ESI† for solubility procedure). These results confirm that both networks maintain their catalytic activity without interfering with each other. Moreover, when the direct aldol reaction between benzaldehyde and cyclohexanone was performed in the SucVal8 + ProValDoc gel, a 91% yield was obtained in 72 hours (anti : syn ratio of 90 : 10; 91% ee). Kobs = (2.2 ± 0.3) × 10–5 s–1, suggesting that the acetal deprotection was not the rate limiting step.
Fig. 5. (a) Kinetics of the tandem ( ) and direct aldol reaction (
) and direct aldol reaction ( ) in the mixture of SucVal8 + ProValDoc at their respective m.g.c values; (b) velocity of product formation versus substrate concentration for the tandem and direct aldol reaction.
) in the mixture of SucVal8 + ProValDoc at their respective m.g.c values; (b) velocity of product formation versus substrate concentration for the tandem and direct aldol reaction.
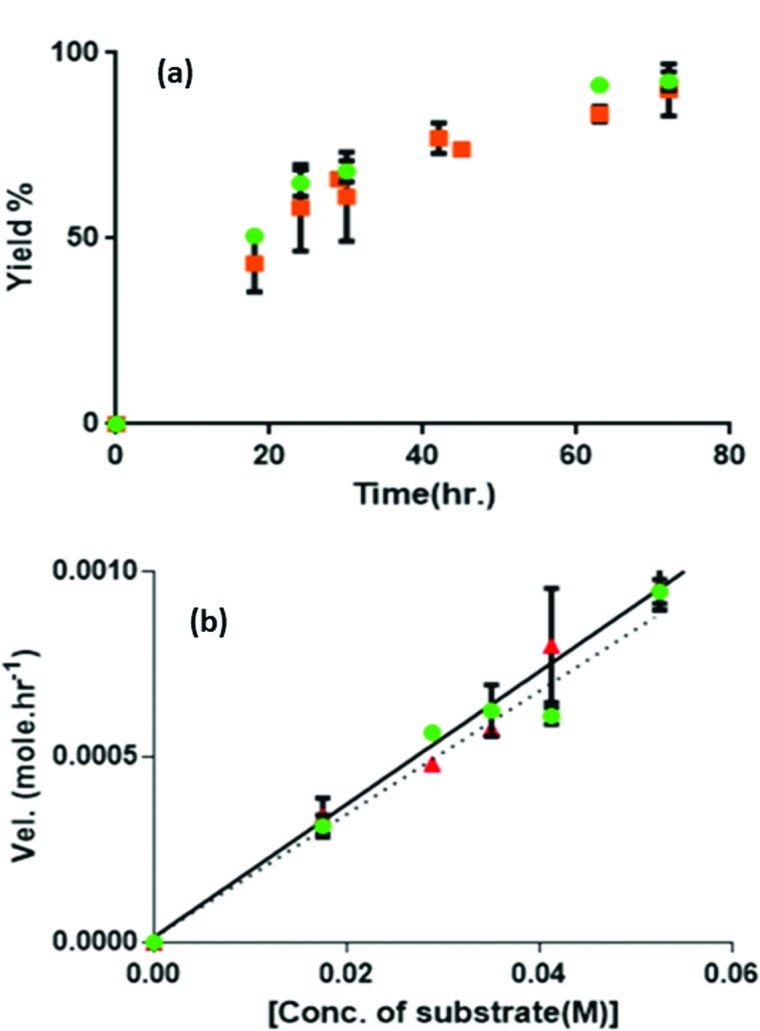
Orthogonal assembly is mandatory for the successful isolation of catalytic groups with opposite acid–base characteristics for dual catalysis in one pot. To highlight this fact, we performed similar experiments to those described above but this time using compound ProVal8, which is structurally very similar to SucVal8. ProVal8 is a bolamphiphilic hydrogelator like SucVal8, but with a proline group at each end (Scheme 1), that forms a white turbid weak gel at an m.g.c of 6 mM with Tgel = 37 °C. As we have reported before for other bolaamphiphilic analogues,14 we expected ProVal8 to co-assemble with SucVal8. ProVal8 forms large crystalline fibers with a width varying from 40–50 nm (see Fig. S3 and S6 in ESI†). However, the TEM images of the mixed gel SucVal8 + ProVal8 revealed a homogeneous system of only one kind of fiber with a width of 20–30 nm (Fig. S5†). On the other hand, AFM of the mixture (Fig. 1d) showed the presence of twisted thin fibrils (Fig. 1a). A closer analysis of the AFM images revealed that the fibrils in the mixture SucVal8 + ProVal8 had a pitch value of 70 nm, which is almost twice the value of the pure SucVal8 (45 nm) (see Fig. S7 in ESI†). Therefore, fibers of the mixture are different from fibers constituting the individual gels, alluding towards the co-assembly of the two molecules to form a different kind of homogenous network.
The WAXD of the xerogel of this mixture also showed a different pattern of packing to the individual hydrogelators (Fig. 2a and 6a, b). Moreover, the gel of this mixture seemed to be weaker than SucVal8 alone. This was further confirmed by Tgel experiments with a varying concentration of ProVal8 in the mixture. With an increase in the concentration of ProVal8, a decrease in Tgel was observed. On increasing the concentration of ProVal8 above 7 mM in the mixture, we could no longer obtain self-standing gels but only suspended aggregates (Fig. 4). The disruptive co-assembly of ProVal8 and SucVal8 was further vindicated by rheology and DSC experiments. G′max for ProVal8 + SucVal8 was measured to be 290 Pa, which was lower than that of SucVal8 (1578 Pa) and slightly higher than ProVal8 (138 Pa) (Fig. 3 and S10†). The thermal imprint of the mixed system was also different from the individual systems (Fig. S11†). All of the above experiments suggested that the molecules of ProVal8 and SucVal8 showed a disruptive co-assembly that interferes with the gelation ability of each hydrogelator.15 Subsequently, catalytic studies were performed for this co-assembled mixture. Single component gels of ProVal8 were able to catalyze the aldol condensation of benzaldehyde and cyclohexanone in water with a yield of 55% in 96 hours. However, when the tandem deacetalization/aldol reaction was tested with the mixture of SucVal8 + ProVal8, only 58% conversion of benzaldehyde dimethyl acetal to benzaldehyde was observed in 96 hours, and no final aldol product was obtained even after much longer times. These results suggest a specific interaction between the acidic and proline groups of the two molecules while co-assembling, presumably involving a salt bridge, and thus rendering them catalytically inactive for the enamine-based aldol reaction, which is hampered if the proline amino group is protonated. FTIR did not show significant differences in the position of bands between the different samples, however, broad bands were obtained for the xerogel of the mixture SucVal8 + ProVal8, suggesting the formation of ammonium salts to a greater extent (see Fig. S14†).
Fig. 6. WAXD patterns of the xerogels of (a) ProVal8 and (b) SucVal8 + ProVal8.

Conclusions
In conclusion, we have reported for the first time two catalytic molecular hydrogelators that, owing to their structural differences, are able to orthogonally self-assemble into a two-network gel and perform two reactions in one pot which would be otherwise incompatible, as exemplified by a related co-assembled network. We have shown that it is possible, by the sole use of molecularly instructed non-covalent interactions, to construct spatially separated catalytic sites able to work in tandem, mimicking multi-enzymatic complexes found in cells. This work is an example of a tailored design of self-assembled supramolecular catalysts for one-pot tandem reactions, which is often not easy to achieve when working in homogeneous catalysis, and it could be extended to other subsets of incompatible reactions.16
Supplementary Material
Acknowledgments
This work was supported by the EU (Marie Curie ITN–Smartnet). N. S. thanks EU for a Marie Curie ESR contract.
Footnotes
†Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, technical details and additional figures are provided. See DOI: 10.1039/c6sc01268j
References
- Lohr T. L., Marks T. J. Nat. Chem. 2015;7:477–482. doi: 10.1038/nchem.2262. [DOI] [PubMed] [Google Scholar]
- Voet D., Voet J. G. and Pratt C. W., Fundamentals of Biochemistry: Life at the Molecular Level, Wiley, 2nd Ed, 2006. [Google Scholar]
- (a) Helms B., Guillaudeu S. J., Xie Y., McMurdo M., Hawker C. J., Fréchet J. M. J. Angew. Chem., Int. Ed. 2005;44:6384–6387. doi: 10.1002/anie.200502095. [DOI] [PubMed] [Google Scholar]; (b) Li P., Cao C. –Y., Chen Z., Liu H., Yu Y., Song W. –G. Chem. Commun. 2012;48:10541–10543. doi: 10.1039/c2cc35718f. [DOI] [PubMed] [Google Scholar]; (c) Vernekar D., Jagadeesan D. Catal. Sci. Technol. 2015;5:4029–4038. [Google Scholar]; (d) Gao J., Zhang X., Lu Y., Liu S., Liu J. Chem.–Eur. J. 2015;21:7403–7407. doi: 10.1002/chem.201500532. [DOI] [PubMed] [Google Scholar]; (e) Aguirre-Díaz L. M., Gandara F., Iglesias M., Snejko N., Gutierrez-Puebla E., Monge M. A. J. Am. Chem. Soc. 2015;137:6132–6135. doi: 10.1021/jacs.5b02313. [DOI] [PubMed] [Google Scholar]
- (a) Raynal M., Ballester P., Vidal-Ferran A., van Leeuwen P. W. N. M. Chem. Soc. Rev. 2014;43:1660–1733. doi: 10.1039/c3cs60027k. [DOI] [PubMed] [Google Scholar]; (b) Raynal M., Ballester P., Vidal-Ferran A., van Leeuwen P. W. N. M. Chem. Soc. Rev. 2014;43:1734–1787. doi: 10.1039/c3cs60037h. [DOI] [PubMed] [Google Scholar]
- For instance: ; (a) Guler M. O., Stupp S. I. J. Am. Chem. Soc. 2007;129:12082. doi: 10.1021/ja075044n. [DOI] [PubMed] [Google Scholar]; (b) Rodríguez-Llansola F., Escuder B., Miravet J. F. J. Am. Chem. Soc. 2009;131:11478–11484. doi: 10.1021/ja902589f. [DOI] [PubMed] [Google Scholar]; (c) Rodríguez-Llansola F., Miravet J. F., Escuder B. Chem. Commun. 2009:7303–7305. doi: 10.1039/b916250j. [DOI] [PubMed] [Google Scholar]; (d) Berdugo C., Miravet J. F., Escuder B. Chem. Commun. 2013;49:10608–10610. doi: 10.1039/c3cc45623d. [DOI] [PubMed] [Google Scholar]; (e) Huang Z., Guan S., Wang Y., Shi G., Cao L., Gao Y., Dong Z., Xu J., Luo Q., Liu J. J. Mater. Chem. B. 2013;1:2297–2304. doi: 10.1039/c3tb20156b. [DOI] [PubMed] [Google Scholar]; (f) Zhang C., Xue X., Luo Q., Li Y., Yang K., Zhuang X., Jiang Y., Zhang J., Liu J., Zou G., Liang X.-J. ACS Nano. 2014;8:11715. doi: 10.1021/nn5051344. [DOI] [PubMed] [Google Scholar]; (g) Singh N., Conte M. P., Ulijn R. V., Miravet J. F., Escuder B. Chem. Commun. 2015;51:13213. doi: 10.1039/c5cc04281j. [DOI] [PubMed] [Google Scholar]; (h) Lee K. S., Parquette J. R. Chem. Commun. 2015;51:15653. doi: 10.1039/c5cc06142c. [DOI] [PubMed] [Google Scholar]; (i) Tena-Solsona M., Nanda J., Díaz-Oltra S., Chotera A., Ashkenasy G., Escuder B. Chem.–Eur. J. 2016;22:6687. doi: 10.1002/chem.201600344. [DOI] [PubMed] [Google Scholar]
- Safont-Sempere M. M., Fernández G., Würthner F. Chem. Rev. 2011;111:5784–5814. doi: 10.1021/cr100357h. [DOI] [PubMed] [Google Scholar]
- (a) Kölbel M., Menger F. M. Langmuir. 2001;17:4490–4492. [Google Scholar]; (b) Heeres A., van der Pol C., Stuart M., Friggeri A., Feringa B. L., van Esch J. J. Am. Chem. Soc. 2003;125:14252–14253. doi: 10.1021/ja036954h. [DOI] [PubMed] [Google Scholar]; (c) Brizard A., Stuart M., van Bommel K., Friggeri A., de Jong M., van Esch J. Angew. Chem., Int. Ed. 2008;47:2063–2066. doi: 10.1002/anie.200704609. [DOI] [PubMed] [Google Scholar]
- Kumar D. K., Steed J. W. Chem. Soc. Rev. 2014;43:2080–2088. doi: 10.1039/c3cs60224a. [DOI] [PubMed] [Google Scholar]
- (a) Moffat J. R., Smith D. K. Chem. Commun. 2009:316–318. doi: 10.1039/b818058j. [DOI] [PubMed] [Google Scholar]; (b) Smith M. M., Smith D. K. Soft Matter. 2011;7:4856–4860. [Google Scholar]; (c) Nagy K. J., Giano M. C., Jin A., Pochan D. J., Schneider J. P. J. Am. Chem. Soc. 2011;133:14975–14977. doi: 10.1021/ja206742m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Morris K. L., Chen L., Raeburn J., Sellick O. R., Cotanda P., Paul A., Griffiths P. C., King S. M., O'Reilly R. K., Serpell L. C., Adams D. J. Nat. Commun. 2013;4:1480. doi: 10.1038/ncomms2499. [DOI] [PubMed] [Google Scholar]; (e) Li D., Shi Y., Wang L. Chin. J. Chem. 2014;32:123–127. [Google Scholar]; (f) Raeburn J., Alston B., Kroeger J., McDonald T. O., Howse J. R., Cameron P. J., Adams D. J. Mater. Horiz. 2014;1:241–246. [Google Scholar]; (g) Draper E. R., Eden E. G. B., McDonald T. O., Adams D. J. Nat. Chem. 2015;7:848–852. doi: 10.1038/nchem.2347. [DOI] [PubMed] [Google Scholar]; (h) Cornwell D. J., Daubney O. J., Smith D. K. J. Am. Chem. Soc. 2015;137:15486–15492. doi: 10.1021/jacs.5b09691. [DOI] [PubMed] [Google Scholar]; (i) Horgan C. C., Rodriguez A. L., Li R., Bruggeman K. F., Stupka N., Raynes J. K., Day L., White J. W., Williams R. J., Nisbet D. R. Acta Biomater. 2016 doi: 10.1016/j.actbio.2016.04.038. [DOI] [PubMed] [Google Scholar]
- (a) Rodríguez-Llansola F., Escuder B., Miravet J. F. Org. Biomol. Chem. 2009;7:3091–3094. [Google Scholar]; (b) Fontanillo M., Angulo-Pachón C. A., Escuder B., Miravet J. F. J. Colloid Interface Sci. 2013;412:65–71. doi: 10.1016/j.jcis.2013.08.055. [DOI] [PubMed] [Google Scholar]
- Wuts P. G. M. and Greene T. W., Protection for the Carbonyl Group, in Greene's Protective Groups in Organic Synthesis, John Wiley & Sons: Hoboken, 2006, pp. 431–532. [Google Scholar]
- Díaz-Oltra S., Berdugo C., Miravet J. F., Escuder B. New J. Chem. 2015;39:3785–3791. [Google Scholar]
- Colquhoun C., Draper E. R., Eden E. G. B., Cattoz B. N., Morris K. L., Chen L., McDonald T. O., Terry A. E., Griffiths P. C., Serpell L. C., Adams D. J. Nanoscale. 2014;6:13719–13725. doi: 10.1039/c4nr04039b. [DOI] [PubMed] [Google Scholar]
- Escuder B., Martí S., Miravet J. F. Langmuir. 2005;21:6776–6787. doi: 10.1021/la050655j. [DOI] [PubMed] [Google Scholar]
- Fleming S., Debnath S., Frederix P. W. J. M., Hunt N. T., Ulijn R. V. Biomacromolecules. 2014;15:1171–1184. doi: 10.1021/bm401720z. [DOI] [PubMed] [Google Scholar]
- Lu J., Dimroth J., Weck M., J. Am. Chem. Soc., 2015, 137 , 12984 –12989 , , and references cited therein . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.