

Abstract
Microdeletion of the human 16p11.2 gene locus has been linked to autism spectrum disorder (ASD) and intellectual disability and confers risk for a number of other neurodevelopmental deficits. Transgenic mice carrying 16p11.2 deletion (16p11+/−) display phenotypes reminiscent of those in human patients with 16p11.2 deletion syndrome, but the molecular mechanisms and treatment strategies for these phenotypes remain unknown. In this study, we have found that both male and female 16p11+/− mice exhibit deficient NMDA receptor (NMDAR) function in the medial prefrontal cortex (mPFC), a brain region critical for high-level “executive” functions. Elevating the activity of mPFC pyramidal neurons with a CaMKII-driven Gq-DREADD (Gq-coupled designer receptors exclusively activated by designer drugs) led to the significant increase of NR2B subunit phosphorylation and the restoration of NMDAR function, as well as the amelioration of cognitive and social impairments in 16p11+/− mice. These results suggest that NMDAR hypofunction in PFC may contribute to the pathophysiology of 16p11.2 deletion syndrome and that restoring PFC activity is sufficient to rescue the behavioral deficits.
SIGNIFICANCE STATEMENT The 16p11.2 deletion syndrome is strongly associated with autism spectrum disorder and intellectual disability. Using a mouse model carrying the 16p11.2 deletion, 16p11+/−, we identified NMDA receptor hypofunction in the prefrontal cortex (PFC). Elevating the activity of PFC pyramidal neurons with a chemogenetic tool, Gq-DREADD, led to the restoration of NMDA receptor function and the amelioration of cognitive and social impairments in 16p11+/− mice. These results have revealed a novel route for potential therapeutic intervention of 16p11.2 deletion syndrome.
Keywords: 16p11.2 deletion, autism spectrum disorder, DREADD, NMDA, prefrontal cortex
Introduction
The 16p11.2 human gene locus, which spans ∼550 kb and encompasses 26 genes (Portmann et al., 2014), has been identified as a hotspot for copy number variations that confer risk for autism spectrum disorder (ASD; Kumar et al., 2008; Weiss et al., 2008; McCarthy et al., 2009; Fernandez et al., 2010; Hanson et al., 2010; Rosenfeld et al., 2010; Shinawi et al., 2010; Zufferey et al., 2012; D'Angelo et al., 2016), intellectual disability (Hanson et al., 2010, 2015; Walters et al., 2010; Zufferey et al., 2012; Maillard et al., 2015; D'Angelo et al., 2016), schizophrenia (McCarthy et al., 2009), and obesity (McCarthy et al., 2009; Rosenfeld et al., 2010; Walters et al., 2010; Zufferey et al., 2012). ASD, a group of neurodevelopmental disorders characterized by social deficits, repetitive behaviors, and intellectual dysfunction, has been reported in both 16p11.2 deletions and 16p11.2 duplications but appears to be more strongly associated with 16p11.2 deletions (Kumar et al., 2008; Rosenfeld et al., 2010). Approximately 19–27% of 16p11.2 deletion carriers were found to meet criteria for ASD diagnosis (Hanson et al., 2010, 2015; Rosenfeld et al., 2010). Furthermore, genetic screenings suggest that 16p11.2 deletions account for ∼0.5% of all ASD cases, positioning it among the strongest known genetic predictors of ASD (Christian et al., 2008; Kumar et al., 2008; Bijlsma et al., 2009; Rosenfeld et al., 2010).
Human patients with 16p11.2 deletions commonly exhibit motor and developmental delays, below-average intelligence, speech and language impairments, attention deficits, autism-related symptoms, macrocephaly, and dysmorphic features (Hanson et al., 2010, 2015; Rosenfeld et al., 2010; Shinawi et al., 2010). Transgenic mouse models carrying 16p11.2 deletions (16p11+/−) have been developed, which display similar phenotypes, including impairments in novel object recognition memory (Portmann et al., 2014; Yang et al., 2015b; Arbogast et al., 2016), delayed learning in multiple paradigms (Tian et al., 2015; Yang et al., 2015b), loss of social novelty recognition, and disrupted ultrasonic vocalizations (Yang et al., 2015a,b; Arbogast et al., 2016). Molecular and cellular mechanisms underlying the autism-like behavioral abnormalities in 16p11+/− mice are primarily unknown.
Prefrontal cortex (PFC), a brain region critical for high-level executive functions, plays an essential role in mediating social cognition (Amodio and Frith, 2006). Disrupted NMDA receptor (NMDAR) function in PFC has been found to underlie the autism-like social deficits in the Shank3-deficient model of autism (Duffney et al., 2015). In this study, we sought to find out whether 16p11+/− mice also exhibit NMDAR hypofunction in PFC and whether elevating PFC neuronal activity is able to rescue the synaptic and behavioral deficits in 16p11+/− mice.
One way to manipulate the activity of specific neuronal populations in vivo is to use the chemogenetic tool designer receptors exclusively activated by designer drugs (DREADDs) (Roth, 2016). Gq-coupled DREADDs use a modified form of the human M3 muscarinic receptor (hM3Dq) to induce an excitatory cellular response in the presence of their ligand, clozapine-N-oxide (CNO; Armbruster et al., 2007; Alexander et al., 2009). Activating hM3Dq DREADDs with CNO increases neuron excitability by mobilizing intracellular calcium (Armbruster et al., 2007). Here we investigated the rescuing effect of Gq-DREADD-induced activation of PFC pyramidal neurons in 16p11.2 microdeletion mice.
Materials and Methods
Animals
The 16p11.2 deletion mice (The Jackson Laboratory) with one copy (heterozygous for a deletion or deficiency allele) of the chromosomal region corresponding to 16p11.2 were generated using chromosome engineering as described previously (Horev et al., 2011). Heterozygous 16p11.2 deletion female mice and wild-type (WT; C57BL/6N129Sv) male mice were bred together to generate heterozygous offspring for experimental use. Wild-type littermates were used as controls. Mice (8–12 weeks old, both males and females) of different genotypes were randomly assigned to treatment groups. Researchers were blind to both genotypes and treatments during experiments. All animal studies were performed with the approval of the Institutional Animal Care and Use Committee of the State University of New York at Buffalo.
Behavioral testing
Barnes maze.
Animals were placed on a round platform with eight equally spaced holes at the edge, one of which was attached with an escape box. A weak aversive stimulus, light, was applied to increase the motivation to escape from the circular platform. During the two learning phases (information acquisition), the animal was allowed to explore the platform for 3 min until finding the correct hole using distal visual cues and entering the escape box. The interval between each learning phase was 5 min. After two learning sessions, the animal was placed in its home cage to rest for 15 min. In the memory phase (information retention and retrieval), the escape box was removed, and the animal was put back on the platform to explore for 3 min. The time spent on the correct hole (T1) and the other seven incorrect holes (T2) were counted. The spatial memory index was calculated by T1/T2.
Social approach.
The test animal was habituated in an apparatus (length, 67.7 cm; width, 50.8 cm; height, 50.8 cm) containing a capsule (an inverted pencil cup, placed in the center area) for 10 min and was returned to the home cage. The apparatus was cleaned, and a social stimulus (an age- and sex-matched mouse) was placed inside the capsule. The test animal was put back into the apparatus to explore for 10 min. The time spent on interacting with the social stimulus and the number of social approaching were measured.
Electrophysiological recordings
The whole-cell voltage-clamp recording technique was used to measure synaptic currents in prelimbic and infralimbic layer V pyramidal neurons from slices (1.70–2.00 mm anterior to bregma) as described previously (Yuen et al., 2010, 2012). Mouse slices (300 μm) were positioned in a perfusion chamber attached to the fixed stage of an upright microscope (Olympus) and submerged in continuously flowing oxygenated ACSF (in mm: 130 NaCl, 26 NaHCO3, 1 CaCl2, 5 MgCl2, 3 KCl, 1.25 NaH2PO4, 10 glucose, pH 7.4, 300 mOsm). Bicuculline (20 μm) and CNQX (20 μm) were added in NMDAR–EPSC recordings. Bicuculline and d-APV (50 μm) were added in AMPA receptor (AMPAR)–EPSC recordings. Patch electrodes contained internal solution (in mm): 130 Cs-methanesulfonate, 10 CsCl, 4 NaCl, 10 HEPES, 1 MgCl2, 5 EGTA, 2 QX-314, 12 phosphocreatine, 5 MgATP, 0.2 Na3GTP, 0.1 leupeptin, pH 7.2–7.3, 265–270 mOsm. Layer V medial PFC (mPFC) pyramidal neurons were visualized with a 40× water-immersion lens and recorded with the Multiclamp 700A amplifier (Molecular Devices). Evoked EPSCs were generated with a pulse from a stimulation isolation unit controlled by a S48 pulse generator (Grass Technologies). A bipolar stimulating electrode (FHC) was placed ∼100 μm from the neuron under recording. For NMDAR–EPSC, the cell (clamped at −70 mV) was depolarized to +40 mV for 3 s before stimulation to fully relieve the voltage-dependent Mg2+ block. Membrane potential was maintained at −70 mV for AMPAR–EPSC recordings. For input–output responses, the EPSC was elicited by a series of pulses with different stimulation intensities (20–100 μA) delivered at 0.033 Hz. To obtain the NMDAR–EPSC-to-AMPAR–EPSC ratio, AMPAR–EPSC was first recorded at −70 mV in ACSF solution (containing bicuculline). Then the mixture of AMPAR–EPSC and NMDAR–EPSC was recorded at +40 mV with the same stimulation pulse (0.5 ms, 50 μA). The peak of NMDAR–EPSC was calculated at 50 ms from the onset of the EPSC mixture. Spontaneous EPSCs (sEPSCs) were recorded in neurons (clamped at −70 mV) with the same external and internal solutions.
To record the spontaneous action potential (sAP), slices were bathed in a modified ACSF (in mm: 130 NaCl, 26 NaHCO3, 1 CaCl2, 0.5 MgCl2, 3.5 KCl, 10 glucose, 1.25 NaH2PO4) to slightly elevate basal neuronal activity (Zhong and Yan, 2016; Wei et al., 2018), which more closely mimics the ionic composition of brain interstitial fluid in situ (1.0–1.2 mm Ca2+, 1 mm Mg2+, and 3.5 mm K+; Sanchez-Vives and McCormick, 2000). Whole-cell current-clamp techniques were used to measure action potential firing with the internal solution containing (in mm) 20 KCl, 100 K-gluconate, 10 HEPES, 4 ATP, 0.5 GTP, and 10 phosphocreatine (Maffei and Turrigiano, 2008). A small depolarizing current was applied to adjust the interspike potential to −60 mV.
Data analyses were performed with Clampfit (Molecular Devices), Mini Analysis Program 6.0.3 (Synaptosoft), Kaleidagraph (Synergy Software), and GraphPad Prism 6 (GraphPad Software).
Biochemical measurement of total and synaptic proteins
Western blots were performed using antibodies against NR1 (1:1000, D65B7, Cell Signaling Technology), NR2A (1:500, 07-632, Millipore), NR2B (1:1000, 06-600, Millipore), GluR1 (1:500, SC-13152, Santa Cruz Biotechnology), GluR2 (1:500, Ab20673, Abcam), PSD-95 (1:1000, 75-028, Neuromab), Tubulin (1:10,000, T9026, Sigma), Synaptophysin (1:500, 611880, BD Biosciences), and phosphorylated-NR2B (S1303; 1:500, Ab81271, Abcam).
Subcellular fractions were prepared as described previously (Duffney et al., 2015). In brief, punches of frontal cortex were cut out, weighed, and homogenized in ice-cold lysis buffer (10 ml/g, 15 mm Tris, pH 7.6, 0.25 m sucrose, 1 mm PMSF, 2 mm EDTA, 1 mm EGTA, 10 mm sodium pyrophosphate, protease inhibitor tablet). After centrifugation at 800 × g for 5 min to remove nuclei and large debris, the remaining supernatant was subjected to 10,000 × g centrifugation for 10 min. The crude synaptosome fraction (pellet) was suspended in lysis buffer containing 1% Triton X-100 and 300 mm NaCl, homogenized again, and centrifuged at 16,000 × g for 15 min. Triton-insoluble fraction, which mainly includes membrane-associated proteins from synaptosomes, was dissolved in 1% SDS. Samples were boiled in 2× SDS loading buffer for 5 min and separated on 7.5% SDS-PAGE.
Viral vectors and animal surgery
Adeno-associated virus (AAV)8/CaMKIIα-hM3Dq(Gq)-mCherry (4.1 × 1012 vp/ml) was obtained from Addgene. AAV8-CaMKIIα-GFP (5.1 × 1012 vp/ml) was obtained from the Vector Core at the University of North Carolina at Chapel Hill. Stereotaxic injection of the virus (1 μl) to the medial PFC (prelimbic and infralimbic regions) was performed as described previously (Duffney et al., 2015). In brief, mice were anesthetized and placed on a stereotaxic apparatus (David Kopf Instruments). The injection was performed with a Hamilton syringe (needle gauge 31) at a speed of ∼0.2 μl/min, and the needle was kept in place for an additional 5 min. The virus was delivered bilaterally to the target area using the following coordinates: 1.98 mm anterior to bregma; 0.25 mm lateral; and 2.2 mm dorsal to ventral. All behavioral and electrophysiological tests were conducted 2 weeks after surgery. Behavioral testing or electrophysiological recordings were performed at 1–6 h after intraperitoneal injection of CNO or saline in viral-infected mice. Fluorescence images were taken on a Zeiss Axio Observer inverted microscope.
Immunohistochemistry
Mice were anesthetized and transcardially perfused with PBS, followed by 4% paraformaldehyde (PFA) before brain removal. Brains were postfixed in 4% PFA for 2 d and cut into 100 μm slices. Slices were cut coronally and permeabilized using 0.5% Triton in PBS for 1 h, washed, and blocked for 1 h in PBS containing 3% BSA and 0.3% Triton. After washing, slices were incubated with the primary antibody against CaMKII (ab134041, 1:500, Abcam) for 48 h at 4°C. After washing three times (30 min, 1 h, and 1 h with gentle shaking) in PBS, slices were incubated with the secondary antibody (Alex488, A21202, 1:1000, Invitrogen) for 1 h at room temperature, followed by three washes with PBS. Slices were mounted on slides with VECTASHIELD mounting medium (Vector Laboratories). Images were acquired using a 40× objective on a Zeiss LSM 510 confocal microscope.
Statistical analyses
All data are presented as the mean ± SEM. No samples were excluded from the analysis. The sample size was based on power analyses and was similar to those reported in previous work (Yuen et al., 2012; Wei et al., 2018). The variance between groups being statistically compared was similar. Experiments with two groups were analyzed statistically using unpaired Student's t tests. Experiments with more than two groups were subjected to one-way ANOVA, two-way ANOVA, or two-way repeated-measures ANOVA, followed by post hoc Bonferroni's tests.
Results
16p11+/− mice exhibit NMDA receptor hypofunction in PFC
To determine whether deletion of the 16p11.2 region affects glutamatergic transmission in the PFC, we performed whole-cell electrophysiological recordings in PFC pyramidal neurons from 16p11+/− (8- to 10-week-old) and age-matched WT mice. NMDAR–EPSCs evoked by multiple stimulation intensities were significantly reduced in PFC neurons of 16p11+/− mice (Fig. 1A; 39–52% decrease; WT, n = 21 cells/4 mice; 16p11+/−, n = 26 cells/5 mice; F(1,45) (genotype) = 22.5, p < 0.001, two-way ANOVA). The ratio of NMDAR–EPSC to AMPAR–EPSC was also significantly lower in 16p11+/− mice compared with WT mice (Fig. 1B; WT, 0.62 ± 0.02, n = 13 cells/4 mice; 16p11+/−, 0.47 ± 0.05, n = 10 cells/3 mice; p < 0.01, t test). However, the paired-pulse ratio of NMDAR–EPSCs, a measurement of presynaptic vesicle release (Zucker, 1989), did not differ between WT and 16p11+/− mice (Fig. 1C; WT, n = 23 cells/5 mice; 16p11+/−, n = 24 cells/4 mice; F(1,45) (genotype) = 0.20, p > 0.05, two-way ANOVA), suggesting a lack of presynaptic changes.
Figure 1.

16p11+/− mice exhibit diminished NMDAR-mediated synaptic responses in PFC. A, Summarized input–output curves of NMDAR–EPSC in PFC pyramidal neurons from WT and 16p11+/− mice. Inset, Representative NMDAR–EPSC traces. *p < 0.05, ***p < 0.001, two-way ANOVA. B, Bar graphs (mean + SEM) of NMDAR–EPSC-to-AMPAR–EPSC ratio in PFC pyramidal neurons from WT and 16p11+/− mice. Inset, Representative traces. **p < 0.01, t test. C, Plot of paired-pulse ratio (PPR) of NMDAR–EPSC evoked by double pulses with various intervals in PFC pyramidal neurons from WT or 16p11+/− mice. Inset, Representative traces.
In contrast to the reduction of NMDAR–EPSC, PFC pyramidal neurons from 16p11+/− mice had unchanged AMPAR–EPSC amplitudes (Fig. 2A; WT, n = 17 cells/5 mice; 16p11+/−, n = 14 cells/4 mice; F(1,29) (genotype) = 2.43, p > 0.05, two-way ANOVA). sEPSCs also had no difference between 16p11+/− and WT mice (Fig. 2B–D; WT, 2.89 ± 0.26 Hz, 12.2 ± 0.42 pA, n = 14 cells/4 mice; 16p11+/−, 2.98 ± 0.24 Hz, 12.5 ± 0.29 pA, n = 11 cells/3 mice; p > 0.05, t test), indicating normal levels of presynaptic glutamate release and postsynaptic AMPAR expression. Together, these results indicate that 16p11.2 deletion specifically induces NMDAR hypofunction in PFC.
Figure 2.

AMPAR-mediated synaptic transmission is unchanged in PFC of 16p11+/− mice. A, Summarized input–output curves of AMPAR–EPSC in PFC pyramidal neurons from WT and 16p11+/− mice. Inset, Representative AMPAR–EPSC traces. B, C, Bar graphs (mean ± SEM) showing sEPSC frequency (B) and amplitude (C) in PFC pyramidal neurons from WT and 16p11+/− mice. D, Representative sEPSC traces.
Given the observed NMDAR hypofunction and evidence implicating a postsynaptic mechanism, we examined whether the synaptic expression of NMDAR subunits was reduced in PFC neurons from 16p11+/− mice. Subcellular fractions containing only synaptic membrane proteins were isolated from PFC of 16p11+/− mice and WT controls. Protein expression levels of NMDAR and AMPAR subunits were measured in total lysates and synaptic membrane fractions through Western blotting. No difference was found between groups in total expression levels of NMDAR or AMPAR subunits (Fig. 3A,C; n = 5–6 mice per group, p > 0.05, t test). Normal levels of NMDAR and AMPAR subunits were also detected in the synaptic fraction (Fig. 3B,C; n = 5–6 mice per group, p > 0.05, t test). Western blots for Synaptophysin (a presynaptic vesicle protein) showed no differences between groups (Fig. 3A,C; n = 5–6 mice per group, p > 0.05, t test), suggesting a lack of changes in presynaptic components. Because NMDARs can be regulated by GPCR signaling-mediated phosphorylation (Yang et al., 2014), it is likely that NMDAR hypofunction in 16p11+/− mice is due to NMDAR dysregulation.
Figure 3.

The total and synaptic levels of glutamate receptor subunits are unchanged in PFC of 16p11+/− mice. A, B, Immunoblots showing the expression of NMDAR and AMPAR subunits in the total lysates (A) or the synaptic fraction (B) of PFC from WT versus 16p11+/− mice. C, Quantification analysis of total and synaptic protein levels in PFC from WT and 16p11+/− mice. Total protein levels were normalized to tubulin. Synaptic protein levels were normalized to PSD-95.
Chemogenetic activation of PFC pyramidal neurons in 16p11+/− mice rescues NMDAR hypofunction
Because16p11+/− mice exhibited deficits in NMDAR-mediated synaptic responses in PFC, we next tested whether elevating PFC neuronal activity via chemogenetic stimulation of the Gq pathway could restore NMDAR function. The mCherry-tagged, CaMKII-driven, Gq-coupled DREADD AAV (hM3Dq) was injected bilaterally to the mPFC of 16p11+/− and WT mice (Fig. 4A). Immunohistochemical staining of CaMKII indicated that ∼70% of mPFC pyramidal neurons in the proximity of the injection sites were DREADD positive (Fig. 4B). Selective activation of mPFC pyramidal neurons was achieved by the systemic administration of CNO (3 mg/kg, i.p.). Considering that systemically injected CNO converts to clozapine (Gomez et al., 2017), which could occupy endogenous GPCRs in the brain, we also injected the GFP-tagged, CaMKII-driven control virus to mPFC to determine the requirement of DREADD for the effect of CNO.
Figure 4.

Chemogenetic activation of PFC restores neuronal excitability in 16p11+/− mice. A, Immunofluorescent image of a brain slice from a mouse showing the expression of Gq-DREADD (mCherry tagged) in medial PFC. B, High-resolution confocal images of CaMKII staining (green) and Gq-DREADD (red) in the viral infected mPFC region. C, D, Bar graphs (mean ± SEM) showing the frequency of synaptic-driven sAP in PFC pyramidal neurons from saline- or CNO-injected WT or 16p11+/− mice with prior infection of Gq-DREADD AAV (C) or GFP-AAV (D). The inset in C shows representative sAP traces. *p < 0.05, ***p < 0.001, ns, not significant, two-way ANOVA.
In hM3Dq-DREADD-injected animals, the frequency of synaptic-driven sAPs was significantly lower in PFC pyramidal neurons from 16p11+/− mice than those from WT mice, and intraperitoneal CNO injection significantly increased sAP frequency in 16p11+/− mice, restoring PFC neuronal activity to the normal level (Fig. 4C; WT plus saline, 1.87 ± 0.09 Hz; 16p11+/− plus saline, 0.99 ± 0.06 Hz; 16p11+/− plus CNO, 1.93 ± 0.08 Hz; WT plus CNO, 2.66 ± 0.18 Hz; n = 18–32 cells/3–4 mice per group, F(1,93) (genotype) = 59.6, p < 0.0001, F(1,93) (treatment) = 68.9, p < 0.0001, two-way ANOVA). In contrast, in GFP-injected animals, systemic administration of CNO failed to restore sAP frequency in 16p11+/− mice (Fig. 4D; WT plus saline, 1.96 ± 0.25 Hz; 16p11+/− plus saline, 1.06 ± 0.17 Hz; 16p11+/− plus CNO, 0.94 ± 0.16 Hz; WT plus CNO, 1.89 ± 0.24 Hz; n = 7–9 cells/2 mice per group, F(1,28) (genotype) = 16.5, p < 0.001, F(1,28) (treatment) = 0.83, p > 0.05, two-way ANOVA).
Next, we examined the impact of chemogenetic elevation of PFC activity on NMDAR–EPSC in PFC pyramidal neurons from 16p11+/− mice. In hM3Dq-DREADD-injected 16p11+/− mice, CNO injection significantly increased NMDAR–EPSC amplitudes compared with saline injection (Fig. 5A; 45–106% increase, n = 15–17 cells/3 mice per group, F(1,59) (genotype) = 10.37, p < 0.01, F(1,59) (treatment) = 8.95, p < 0.01, two-way ANOVA), raising NMDAR–EPSC amplitudes to levels similar to WT mice. Such rescuing effect of CNO was not observed in GFP-injected 16p11+/− mice (Fig. 5B; n = 16–17 cells/3 mice per group, F(1,63) (genotype) = 35.89, p < 0.001, F(1,63) (treatment) = 0.17, p > 0.05, two-way ANOVA).
Figure 5.

Chemogenetic activation of PFC restores NMDAR function and elevates NR2B phosphorylation in 16p11+/− mice. A, B, Summarized input–output curves of NMDAR–EPSC in PFC pyramidal neurons from saline- or CNO-injected WT or 16p11+/− mice with prior infection of Gq-DREADD AAV (A) or GFP-AAV (B). ***p < 0.001, two-way ANOVA. Insets, Representative NMDAR–EPSC traces. C, Immunoblots of S1303phos-NR2B and total NR2B in the lysates of saline- or CNO-treated PFC slices from WT versus 16p11+/− mice with prior infection of Gq-DREADD AAV. D, Quantification analysis of S1303phos-NR2B (normalized to total NR2B) levels in different groups. **p < 0.01, two-way ANOVA.
We next sought to determine the potential mechanism by which hM3Dq-DREADD activation restored NMDAR function in 16p11+/− mice. Because hM3Dq-DREADD uses Gq-coupled GPCR activity to induce an excitatory cellular response, we examined the link between Gq signaling and NMDA receptors. Protein kinase C (PKC) and CaMKII, which are activated downstream of Gq signaling (Lu et al., 1999), have been shown to phosphorylate NMDA receptors at multiple residues (Chen and Roche, 2007). Because NR2B phosphorylation at S1303 by PKC and CaMKII can potentiate NMDAR currents (Lu et al., 1999; Liao et al., 2001; Chen and Roche, 2007), we focused on this phosphorylation site. PFC slices collected from hM3Dq DREADD-injected WT or 16p11+/− mice were treated with either saline or CNO ex vivo for 20 min and probed for the level of phosphorylated NR2B at S1303 via Western blotting. As shown in Figure 5, C and D, the basal level of S1303phos-NR2B was lower in 16p11+/− mice compared with WT mice, and CNO treatment led to a significant elevation of S1303phos-NR2B in DREADD-injected 16p11+/− mice, whereas the effect of CNO in DREADD-injected WT mice was marginal (WT plus saline, 1.00 ± 0.12; 16p11+/− plus saline, 0.74 ± 0.14; 16p11+/− plus CNO, 1.30 ± 0.08; WT plus CNO, 1.14 ± 0.17; n = 6–10 slices/2–3 mice per group, F(1,25) (genotype) = 0.01, p = 0.72, F(1,25) (treatment) = 7.8, p < 0.01, two-way ANOVA). These data suggest that Gq-DREADD activation might restore NMDAR function in 16p11+/− mice via elevating the phosphorylation of NR2B subunits.
Chemogenetic activation of PFC pyramidal neurons in 16p11+/− mice ameliorates cognitive and social deficits
Considering the loss of NMDAR function and synaptic-driven excitability of PFC neurons in 16p11+/− mice, we next examined whether chemogenetic activation of PFC neurons could ameliorate the deficits in cognition and social behaviors exhibited in 16p11+/− mice (Portmann et al., 2014; Tian et al., 2015; Yang et al., 2015a,b; Arbogast et al., 2016). WT and 16p11+/− mice with PFC infection of the CaMKII-driven DREADD AAV (hM3Dq) were subject to a variety of behavioral assays. As controls, another cohort of WT and 16p11+/− mice received mPFC injection of the GFP virus before being subjected to the same behavioral assays. Both male and female WT and 16p11+/− mice were used in behavioral tests.
The Barnes maze, a cognitive test assessing spatial memory (Sharma et al., 2010), was first performed because NMDAR hypofunction in mPFC has been linked to impaired Barnes maze performance by a previous pharmacological study (Locklear et al., 2015). The Barnes maze tests the animal's spatial memory by recalling the location of one correct hole (where an escape box was attached before) from seven other incorrect holes on a round platform. The spatial memory index is calculated by the time spent exploring the correct hole divided by the time spent exploring all the other incorrect holes. We found that in Gq-DREADD-injected animals, 16p11+/− mice spent significantly less time exploring the correct hole and more time exploring the incorrect holes compared with WT mice, suggesting diminished spatial memory, and after CNO administration, 16p11+/− mice spent significantly more time exploring the correct hole and less time exploring the incorrect holes (Fig. 6A; correct hole: WT plus saline, 37.9 ± 1.7 s; 16p11+/− plus saline, 25.8 ± 2.1 s; 16p11+/− plus CNO, 35.9 ± 2.2 s; WT plus CNO, 36.0 ± 1.7 s; n = 6–8 per group, F(1,23) (genotype) = 9.9, p < 0.01, F(1,23) (treatment) = 4.5, p < 0.05, two-way ANOVA; incorrect holes: WT plus saline, 27 ± 1.0 s; 16p11+/− plus saline, 32.3 ± 1.4 s; 16p11+/− plus CNO, 25.3 ± 1.1 s; WT plus CNO, 24.6 ± 1.7; n = 6–8 per group, F(1,23) (genotype) = 4.9, p < 0.05, F(1,23) (treatment) = 11.9, p < 0.01, two-way ANOVA). Accordingly, the spatial memory index was significantly lower in 16p11+/− mice than WT animals, which was brought to the normal level by CNO (WT plus saline, 1.43 ± 0.12; 16p11+/− plus saline, 0.81 ± 0.1; 16p11+/− plus CNO, 1.43 ± 0.1; WT plus CNO, 1.49 ± 0.1; n = 6–8 each group, F(1,23) (genotype) = 10.61, p < 0.01, F(1,23) (treatment) = 10.48, p < 0.01, two-way ANOVA).
Figure 6.
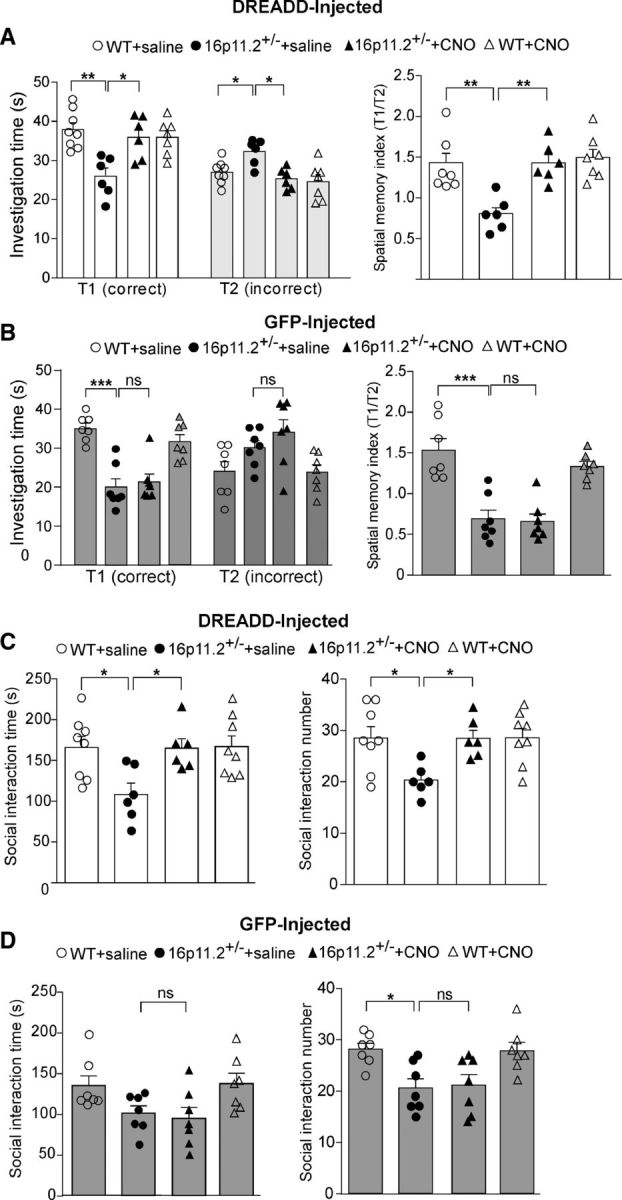
Chemogenetic activation of PFC ameliorates cognitive and social deficits in 16p11+/− mice. A, B, Bar graphs (mean ± SEM) showing the time spent exploring the correct hole (T1) versus the seven incorrect holes (T2) in the memory phase of Barnes maze tests (left) and the spatial memory index (T1/T2) in Barnes maze tests (right) of saline- or CNO-injected WT or 16p11+/− mice with prior infection of Gq-DREADD AAV (A) or GFP-AAV (B). *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, two-way ANOVA. C, D, Bar graphs (mean ± SEM) showing the time (left) and number (right) of social interactions in social approach tests of saline- or CNO-injected WT or 16p11+/− mice with prior infection of Gq-DREADD AAV (C) or GFP-AAV (D). *p < 0.05, ns, not significant, two-way ANOVA.
In GFP-injected animals (Fig. 6B), CNO administration did not affect the amount of time that 16p11+/− mice spent in exploring the correct hole (WT plus saline, 35.2 ± 1.3 s; 16p11+/− plus saline, 20.0 ± 2.2 s; 16p11+/− plus CNO, 21.4 ± 2.0 s; WT plus CNO, 31.7 ± 1.8; n = 7 per group, F(1,24) (genotype) = 48.53, p < 0.0001, F(1,24) (treatment) = 0.32, p > 0.05, two-way ANOVA) or incorrect holes (WT plus saline, 24 ± 2.5 s; 16p11+/− plus saline, 30.1 ± 1.8 s; 16p11+/− plus CNO, 34.1 ± 3.2 s; WT plus CNO, 23.0 ± 1.9; n = 7 each group, F(1,24) (genotype) = 11.2, p < 0.01, F(1,24) (treatment) = 0.64, p > 0.05, two-way ANOVA). The spatial memory index of 16p11+/− mice was also not restored after CNO injection (WT plus saline, 1.55 ± 0.14; 16p11+/− plus saline, 0.70 ± 0.11; 16p11+/− plus CNO, 0.67 ± 0.09; WT plus CNO, 1.35 ± 0.06; n = 7 each group, F(1,24) (genotype) = 52.62, p < 0.0001, F(1,24) (treatment) = 1.22, p > 0.05, two-way ANOVA).
Next, the social approach behavior was examined. In Gq-DREADD-injected animals (Fig. 6C), 16p11+/− mice spent significantly less time interacting with a social stimulus and engaged in fewer social interactions compared to WT mice, and CNO injection significantly increased the amount of time that 16p11+/− mice spent interacting with the social stimulus and the number of social interactions (social time: WT plus saline, 166 ± 13.5 s; 16p11+/− plus saline, 108.3 ± 13.8 s; 16p11+/− plus CNO, 165.1 ± 11.5 s; WT plus CNO, 167.2 ± 13.0; n = 6–8 per group, F(1,24) (genotype) = 5.0, p < 0.05, F(1,24) (treatment) = 4.7, p < 0.05, two-way ANOVA; social number: WT plus saline, 28.5 ± 2.2; 16p11+/− plus saline, 20.3 ± 1.2; 16p11+/− plus CNO, 28.5 ± 1.6; WT plus CNO, 28.6 ± 1.8; n = 6–8 per group, F(1,24) (genotype) = 4.8, p < 0.05, F(1,24) (treatment) = 4.8, p < 0.05, two-way ANOVA). Conversely, in GFP-injected 16p11+/− mice (Fig. 6D), CNO injections failed to affect social interaction time (WT plus saline, 134.2 ± 12.04 s; 16p11+/− plus saline, 100.8 ± 8.8 s; 16p11+/− plus CNO, 94.4 ± 12.3 s; WT plus CNO, 136.8 ± 12.4; n = 7 per group, F(1,24) (genotype) = 10.44, p < 0.01, F(1,24) (treatment) = 0.03, p > 0.05, two-way ANOVA) and the number of social interactions (WT plus saline, 28.1 ± 1.2; 16p11+/− plus saline, 20.6 ± 1.8; 16p11+/− plus CNO, 21.1 ± 2.1; WT plus CNO, 27.9 ± 1.7; n = 7 per group, F(1,24) (genotype) = 17.6, p < 0.001, F(1,24) (treatment) = 0.007, p > 0.05, two-way ANOVA). Collectively, these results indicate that elevating PFC pyramidal neuronal activity in 16p11+/− mice is sufficient and necessary to ameliorate the cognitive and social deficits exhibited by these animals.
Discussion
Microdeletion of the 16p11.2 region is associated with several syndromes mainly characterized by intellectual and social deficits (Portmann et al., 2014; Tian et al., 2015; Yang et al., 2015a,b; Arbogast et al., 2016). Previous reports have implicated dysfunction of basal ganglia circuitry (Portmann et al., 2014; Maillard et al., 2015) and widespread brain structure irregularities (Horev et al., 2011) in the etiology of 16p11.2 deletion syndrome. In the present study, we show that 16p11.2 deletion mice exhibit deficient NMDAR function in the PFC. By using chemogenetic activation of PFC pyramidal neurons, we have demonstrated that NMDAR function is restored and cognitive and social deficits are ameliorated in 16p11+/− mice.
Dysfunctional glutamatergic activity in the PFC has not been previously reported in 16p11.2 deletion syndrome, although an increase in the AMPAR/NMDAR ratio was found in the nucleus accumbens (NAc) of 16p11+/− mice (Portmann et al., 2014). This result is analogous to the decreased NMDAR/AMPAR ratio observed in the PFC. Given the deficits in spatial memory shown previously (Yang et al., 2015b) and replicated here, disruptions in 16p11+/− hippocampal activity would also be conceivable; however, multiple studies have indicated normal synaptic function in the hippocampus of 16p11+/− mice (Tian et al., 2015; Arbogast et al., 2016). It is possible that NMDAR hypofunction in the PFC underlies the intellectual and social deficits associated with 16p11.2 deletion syndrome, as glutamatergic transmission in the PFC, and specifically NMDAR function, is a critical component for working memory and cognitive processing as well as for social behaviors (Goldman-Rakic, 1995; Lisman et al., 1998). Moreover, the disrupted signaling observed in the NAc and PFC may be related, as the NAc receives glutamatergic projections from the PFC (Britt et al., 2012) and NAc activity is regulated by PFC inputs (Del Arco and Mora, 2008).
NMDAR function is highly regulated by GPCR signaling (Yang et al., 2014). Activation of protein kinases downstream of GPCR signaling have been shown to potentiate NMDAR currents in the PFC and other brain areas through mechanisms including receptor phosphorylation (Gurden et al., 2000; Chen et al., 2004; Rojas and Dingledine, 2013). Phosphorylation of NR1 subunit at S890 and S896 alters NMDAR clustering and surface expression (Tingley et al., 1997; Scott et al., 2001). The lack of changes in the level of synaptic NMDARs in 16p11+/− mice led us to focus on NR2B phosphorylation at S1303, a substrate of PKC and CaMKII (Chen and Roche, 2007). Previous studies have found that PKC can directly phosphorylate S1303 in the NR2B C terminus, leading to enhanced NMDAR currents (Liao et al., 2001). Interestingly, hM3Dq-DREADD significantly elevated the diminished level of S1303-phosphorylated NR2B in 16p11+/− mice, which correlated well with the DREADD-induced restoration of NMDAR function in these animals. It suggests that NMDAR hypofunction in 16p11+/− mice is likely caused by the dysregulation of NMDAR phosphorylation, which can be reversed by chemogenetic stimulation of the GPCR signaling.
In conclusion, our results suggest that dysregulated NMDAR function in the PFC is strongly implicated in the manifestation of cognitive and social impairments in 16p11.2 deletion syndrome. Chemogenetically activating PFC pyramidal neurons in 16p11+/− mice is sufficient to ameliorate behavioral deficits, potentially revealing a novel intervention strategy for the treatment of 16p11.2 deletion syndrome.
Footnotes
This work was supported by the Nancy Lurie Marks Family Foundation and the National Institutes of Health (Grants R41-MH112237, R01-MH108842, and R01-DA037618 to Z.Y.). We thank Xiaoqing Chen and Kaijie Ma for excellent technical support.
The authors declare no competing financial interests.
References
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, McNamara JO, Roth BL (2009) Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63:27–39. 10.1016/j.neuron.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodio DM, Frith CD (2006) Meeting of minds: the medial frontal cortex and social cognition. Nat Rev Neurosci 7:268–277. 10.1038/nrn1884 [DOI] [PubMed] [Google Scholar]
- Arbogast T, Ouagazzal A-M, Chevalier C, Kopanitsa M, Afinowi N, Migliavacca E, Cowling BS, Birling M-C, Champy M-F, Reymond A, Herault Y (2016) Reciprocal effects on neurocognitive and metabolic phenotypes in mouse models of 16p11.2 deletion and duplication syndromes. PLoS Genet 12:e1005709. 10.1371/journal.pgen.1005709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL (2007) Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 104:5163–5168. 10.1073/pnas.0700293104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, Lundin J, Lapunzina P, Pérez Jurado LA, Delle Chiaie B, Loeys B, Menten B, Oostra A, Verhelst H, Amor DJ, Bruno DL, van Essen AJ, Hordijk R, Sikkema-Raddatz B, Verbruggen KT, et al. (2009) Extending the phenotype of current rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet 52:77–87. 10.1016/j.ejmg.2009.03.006 [DOI] [PubMed] [Google Scholar]
- Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A (2012) Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76:790–803. 10.1016/j.neuron.2012.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Roche KW (2007) Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53:362–368. 10.1016/j.neuropharm.2007.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Greengard P, Yan Z (2004) Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci U S A 101:2596–2600. 10.1073/pnas.0308618100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian SL, Brune CW, Sudi J, Kumar RA, Liu S, Karamohamed S, Badner JA, Matsui S, Conroy J, McQuaid D, Gergel J, Hatchwell E, Gilliam TC, Gershon ES, Nowak NJ, Dobyns WB, Cook EH Jr (2008) Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry 63:1111–1117. 10.1016/j.biopsych.2008.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo D, Lebon S, Chen Q, Martin-Brevet S, Snyder LG, Hippolyte L, Hanson E, Maillard AM, Faucett WA, Macé A, Pain A, Bernier R, Chawner SJ, David A, Andrieux J, Aylward E, Baujat G, Caldeira I, Conus P, Ferrari C, et al. (2016) Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 73:20–30. 10.1001/jamapsychiatry.2015.2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco A, Mora F (2008) Prefrontal cortex-nucleus accumbens interaction: in vivo modulation by dopamine and glutamate in the prefrontal cortex. Pharmacol Biochem Behav 90:226–235. 10.1016/j.pbb.2008.04.011 [DOI] [PubMed] [Google Scholar]
- Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, Qin L, Ma K, Dietz DM, Kajiwara Y, Buxbaum JD, Yan Z (2015) Autism-like deficits in shank3-deficient mice are rescued by targeting actin regulators. Cell Rep 11:1400–1413. 10.1016/j.celrep.2015.04.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, Joseph-George AM, Mackay S, Whitten K, Noble B, Vardy C, Crosbie V, Luscombe S, Tucker E, Turner L, Marshall CR, Scherer SW (2010) Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet 47:195–203. 10.1136/jmg.2009.069369 [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. (1995) Cellular basis of working memory. Neuron 14:477–485. 10.1016/0896-6273(95)90304-6 [DOI] [PubMed] [Google Scholar]
- Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, Ellis RJ, Richie CT, Harvey BK, Dannals RF, Pomper MG, Bonci A, Michaelides M (2017) Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357:503–507. 10.1126/science.aan2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurden H, Takita M, Jay TM (2000) Essential role of D1 but not D2 receptors in the NMDA receptor-dependent long-term potentiation at hippocampal–prefrontal cortex synapses in vivo. J Neurosci 20:RC106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson E, Nasir RH, Fong A, Lian A, Hundley R, Shen Y, Wu BL, Holm IA, Miller DT, 16p11.2 Study Group Clinicians (2010) Cognitive and behavioral characterization of 16p11.2 deletion syndrome. J Dev Behav Pediatr 31:649–657. 10.1097/DBP.0b013e3181ea50ed [DOI] [PubMed] [Google Scholar]
- Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, Snow AV, Wallace AS, Campe KL, Zhang Y, Chen Q, D'Angelo D, Moreno-De-Luca A, Orr PT, Boomer KB, Evans DW, Kanne S, Berry L, Miller FK, Olson J, et al. (2015) The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry 77:785–793. 10.1016/j.biopsych.2014.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M, Mills AA (2011) Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc Natl Acad Sci U S A 108:17076–17081. 10.1073/pnas.1114042108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH Jr, Dobyns WB, Christian SL (2008) Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 17:628–638. [DOI] [PubMed] [Google Scholar]
- Liao GY, Wagner DA, Hsu MH, Leonard JP (2001) Evidence for direct protein kinase-C mediated modulation of N-methyl-D-aspartate receptor current. Mol Pharmacol 59:960–964. 10.1124/mol.59.5.960 [DOI] [PubMed] [Google Scholar]
- Lisman JE, Fellous JM, Wang XJ (1998) A role for NMDA-receptor channels in working memory. Nat Neurosci 1:273–275. 10.1038/1086 [DOI] [PubMed] [Google Scholar]
- Locklear MN, Bhamidipaty S, Kritzer MF (2015) Local N-methyl-d-aspartate receptor antagonism in the prefrontal cortex attenuates spatial cognitive deficits induced by gonadectomy in adult male rats. Neuroscience 288:73–85. 10.1016/j.neuroscience.2014.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF (1999) G-protein-coupled receptors act via protein kinase C and src to regulate NMDA receptors. Nat Neurosci 2:331–338. 10.1038/7243 [DOI] [PubMed] [Google Scholar]
- Maffei A, Turrigiano GG (2008) Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci 28:4377–4384. 10.1523/JNEUROSCI.5298-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard AM, Ruef A, Pizzagalli F, Migliavacca E, Hippolyte L, Adaszewski S, Dukart J, Ferrari C, Conus P, Männik K, Zazhytska M, Siffredi V, Maeder P, Kutalik Z, Kherif F, Hadjikhani N, Beckmann JS, Reymond A, Draganski B, Jacquemont S, et al. (2015) The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol Psychiatry 20:140–147. 10.1038/mp.2014.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, Krause V, Kumar RA, Grozeva D, Malhotra D, Walsh T, Zackai EH, Kaplan P, Ganesh J, Krantz ID, Spinner NB, et al. (2009) Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet 41:1223–1227. 10.1038/ng.474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portmann T, Yang M, Mao R, Panagiotakos G, Ellegood J, Dolen G, Bader PL, Grueter BA, Goold C, Fisher E, Clifford K, Rengarajan P, Kalikhman D, Loureiro D, Saw NL, Zhengqui Z, Miller MA, Lerch JP, Henkelman M, Shamloo M, et al. (2014) Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep 7:1077–1092. 10.1016/j.celrep.2014.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Dingledine R (2013) Ionotropic glutamate receptors: regulation by G-protein-coupled receptors. Mol Pharmacol 83:746–752. 10.1124/mol.112.083352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Coppinger J, Bejjani BA, Girirajan S, Eichler EE, Shaffer LG, Ballif BC (2010) Speech delays and behavioral problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications. J Neurodev Disord 2:26–38. 10.1007/s11689-009-9037-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL. (2016) DREADDs for neuroscientists. Neuron 89:683–694. 10.1016/j.neuron.2016.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA (2000) Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci 3:1027–1034. 10.1038/79848 [DOI] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD (2001) An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci 21:3063–3072. 10.1523/JNEUROSCI.21-09-03063.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Rakoczy S, Brown-Borg H (2010) Assessment of spatial memory in mice. Life Sci 87:521–536. 10.1016/j.lfs.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, Clark G, Lee J, Proud M, Stocco A, Rodriguez DL, Kozel BA, Sparagana S, Roeder ER, McGrew SG, Kurczynski TW, et al. (2010) Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet 47:332–341. 10.1136/jmg.2009.073015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Stoppel LJ, Heynen AJ, Lindemann L, Jaeschke G, Mills AA, Bear MF (2015) Contribution of mGluR5 to hippocampal pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat Neurosci 18:182–184. 10.1038/nn.3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL (1997) Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem 272:5157–5166. 10.1074/jbc.272.8.5157 [DOI] [PubMed] [Google Scholar]
- Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, Falchi M, Chen F, Andrieux J, Lobbens S, Delobel B, Stutzmann F, El-Sayed Moustafa JS, Chèvre JC, Lecoeur C, Vatin V, Bouquillon S, Buxton JL, Boute O, Holder-Espinasse M, et al. (2010) A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature 463:671–675. 10.1038/nature08727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Zhong P, Qin L, Tan T, Yan Z (2018) Chemicogenetic restoration of the prefrontal cortex to amygdala pathway ameliorates stress-induced deficits. Cereb Cortex 28:1980–1990. 10.1093/cercor/bhx104:1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, et al. (2008) Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 358:667–675. 10.1056/NEJMoa075974 [DOI] [PubMed] [Google Scholar]
- Yang K, Jackson MF, MacDonald JF (2014) Recent progress in understanding subtype specific regulation of NMDA receptors by G protein coupled receptors (GPCRs). J Mol Sci 15:3003–3024. 10.3390/ijms15023003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Mahrt EJ, Lewis F, Foley G, Portmann T, Dolmetsch RE, Portfors CV, Crawley JN (2015a) 16p11.2 deletion syndrome mice display sensory and ultrasonic vocalization deficits during social interactions. Autism Res 8:507–521. 10.1002/aur.1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Lewis FC, Sarvi MS, Foley GM, Crawley JN (2015b) 16p11.2 deletion mice display cognitive deficits in touchscreen learning and novelty recognition tasks. Learn Mem 22:622–632. 10.1101/lm.039602.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Kafri T, van Praag H, Yan Z (2010) Regulation of AMPA receptor channels and synaptic plasticity by cofilin phosphatase slingshot in cortical neurons. J Physiol 588:2361–2371. 10.1113/jphysiol.2009.186353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z (2012) Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73:962–977. 10.1016/j.neuron.2011.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Yan Z (2016) Distinct physiological effects of dopamine D4 receptors on prefrontal cortical pyramidal neurons and fast-spiking interneurons. Cereb Cortex 26:180–191. 10.1093/cercor/bhu190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. (1989) Short-term synaptic plasticity. Ann Rev Neurosci 12:13–31. 10.1146/annurev.ne.12.030189.000305 [DOI] [PubMed] [Google Scholar]
- Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, Macé A, Ferrari C, Kutalik Z, Andrieux J, Aylward E, Barker M, Bernier R, Bouquillon S, Conus P, Delobel B, Faucett WA, Goin-Kochel RP, Grant E, Harewood L, et al. (2012) A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet 49:660–668. 10.1136/jmedgenet-2012-101203 [DOI] [PMC free article] [PubMed] [Google Scholar]