

Abstract
Recent studies suggest that spontaneous and action potential-evoked neurotransmitter release processes are independently regulated. However, the mechanisms that uncouple the two forms of neurotransmission remain unclear. In cultured mouse and rat neurons, we show that the two C2 domain-containing protein copine-6 is localized to presynaptic terminals and binds to synaptobrevin2 as well as other SNARE proteins in a Ca2+-dependent manner. Ca2+-dependent interaction of copine-6 with synaptobrevin2 selectively suppresses spontaneous neurotransmission in a reaction that requires the tandem tryptophan residues at the C-terminal region of synaptobrevin2. Accordingly, copine-6 loss of function augmented presynaptic Ca2+ elevation-mediated neurotransmitter release. Intracellular Ca2+ chelation, on the other hand, occluded copine-6-mediated suppression of release. We also evaluated the molecular specificity of the copine-6-dependent regulation of spontaneous release and found that overexpression of copine-6 did not suppress spontaneous release in synaptobrevin2-deficient neurons. Together, these results suggest that copine-6 acts as a specific Ca2+-dependent suppressor of spontaneous neurotransmission.
SIGNIFICANCE STATEMENT Synaptic transmission occurs both in response to presynaptic action potentials and spontaneously, in the absence of stimulation. Currently, much more is understood about the mechanisms underlying action potential-evoked neurotransmission compared with spontaneous release. However, recent studies have shown selective modulation of spontaneous neurotransmission process by several neuromodulators, suggesting specific molecular regulation of spontaneous release. In this study, we identify copine-6 as a specific regulator of spontaneous neurotransmission. By both gain-of-function and loss-of-function experiments, we show that copine-6 functions as a Ca2+-dependent suppressor of spontaneous release. These results further elucidate the mechanisms underlying differential regulation of evoked and spontaneous neurotransmitter release.
Keywords: calcium regulation of release, spontaneous neurotransmission, synaptic vesicle fusion
Introduction
Spontaneous neurotransmitter release is a key feature of presynaptic terminals (Fatt and Katz, 1952). At synapses, most Ca2+-dependent action potential-evoked neurotransmitter release and a large fraction of spontaneous release events are mediated by the canonical synaptic SNARE (soluble N-ethylmaleimide-sensitive factor Attachment Protein Receptor) complex composed of synaptobrevin2 (syb2, also called VAMP2) on the synaptic vesicle and syntaxin1 and SNAP-25, both on the synaptic plasma membrane (Südhof and Rothman, 2009). Although syb2 is the most abundant vesicle-associated SNARE and is essential for fast synchronous neurotransmission (Schoch et al., 2001; Takamori et al., 2006), recent evidence suggests that a fraction of spontaneous neurotransmitter release events is mediated by alternative vesicular SNARE proteins, such as vti1a, VAMP7, and VAMP4 (Hua et al., 2011; Raingo et al., 2012; Ramirez et al., 2012; Bal et al., 2013; Kavalali, 2015). These alternative SNAREs may molecularly identify functionally distinct synaptic vesicle populations and render these vesicles insensitive to rapid Ca2+ transients but nevertheless enable them to fuse spontaneously or asynchronously in response to slow fluctuations in baseline Ca2+ levels (Crawford and Kavalali, 2015). Moreover, there is evidence that these alternative SNARE molecules may be enriched in synaptic vesicles that are retrieved spontaneously (Revelo et al., 2014). However, despite the evidence in support of a key role for these alternative SNAREs in spontaneous and asynchronous release processes, in mammalian central synapses a substantial fraction (70%–90%) of spontaneous release events are performed by the canonical vesicular SNARE syb2 (Schoch et al., 2001; Kaeser and Regehr, 2014; Schneggenburger and Rosenmund, 2015). Therefore, findings on the role of alternative SNARE molecules in neurotransmitter release do not fully explain why syb2-driven spontaneous release events are segregated from syb2-mediated evoked release events as suggested previously (Sara et al., 2005; Atasoy et al., 2008; Fredj and Burrone, 2009; Chung et al., 2010; Leitz and Kavalali, 2014).
In this study, we have identified copine-6, a two C2 domain-containing cytosolic protein, as a Ca2+-dependent binding partner for syb2 as well as other SNAREs. Copine-6 belongs to an evolutionarily conserved family of proteins (Copines) that bind negatively charged lipids and other proteins in a Ca2+-regulated manner (Tomsig and Creutz, 2002). There are 9 copine genes discovered in human and mammals. Copines share a highly conserved architecture containing a pair of tandem C2 domains (C2A and C2B) in the N terminus and a domain related to integrin A domain and a von Willebrand factor (vWA) domain interspersed by short linker sequences (see Fig. 1A) (Creutz et al., 1998). Similar to synaptotagmins and protein kinase C (PKC), the C2 domains of copines bind negatively charged phospholipids in the presence of Ca2+ with micromolar affinity (∼1–10 μm). The vWA domain of copines also binds to multiple proteins in vitro with unclear functional significance (Tomsig et al., 2003). Among copine protein family, copine-6 is also described as N-Copine as it is highly enriched in postnatal brain, especially in somatodendritic domains of hippocampus and olfactory bulb (Nakayama et al., 1998, 1999). It has been shown that copine-6 expression increased up to 25-fold during maturation in developing axonal projections (Yamatani et al., 2010), and its mRNA level was found to be upregulated upon kainate injection or high-frequency electrical stimulation that induces LTP in the hippocampus (Nakayama et al., 1998). Recent findings indicate that copine-6 may also play a key role in dendritic spine morphology through interactions with BDNF signaling (Burk et al., 2017). Here, we manipulated copine-6 expression in hippocampal neurons and found that overexpression of copine-6 resulted in a decrease in the frequency of spontaneous miniature postsynaptic currents, whereas knockdown of copine-6 increased their frequency. This regulation was presynaptic and specific to spontaneous events as evoked neurotransmission was largely unaffected by the same manipulations. Furthermore, we demonstrated that this regulation requires copine-6's interaction with the key SNARE component syb2. Our results support the notion that the copine-6 selectively regulates spontaneous neurotransmission within central synapses.
Figure 1.
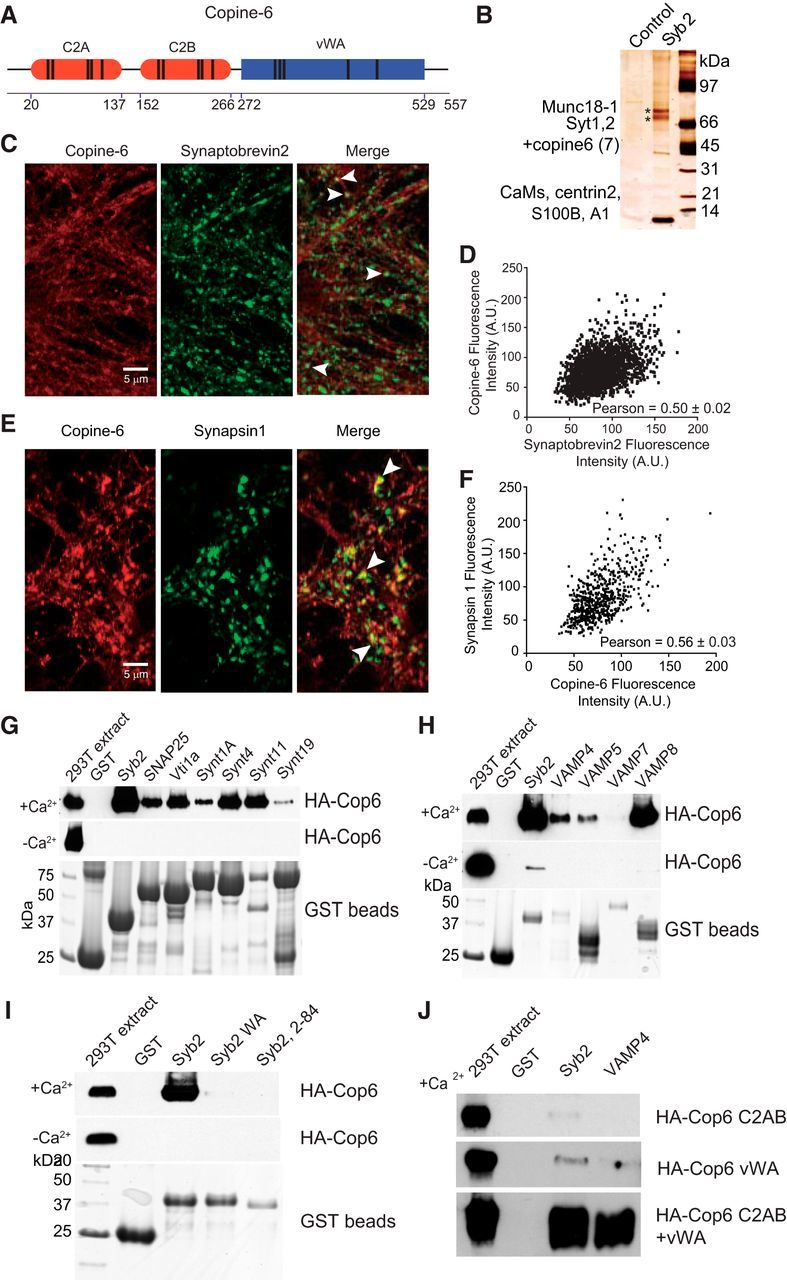
Identification, localization, and binding properties of Copine-6. A, Diagram of the structure of Copine-6. Boundaries of C2A, C2B, and vWA domains are indicated below. Parallel bars represent positions of divalent cation binding sites within the domains. B, Proteins identified by pulldown reaction with GST-syb2 from rat brain extract in the presence of Ca2+. Proteins were eluted from GST-beads (control) or GST-syb2 by removing Ca2+, separated by SDS-PAGE and silver stained. The upper band labeled with asterisk contained Munc18-1, and the lower band contained comigrating synaptotagmin 1, synaptotagmin 2, and copine-6 (with minor amount of copine7 also present). Several low-molecular weight bands contained EF-hand motif proteins. All biochemical experiments were replicated 3 times with similar results. C, Representative confocal images of cultured hippocampal neurons immunostained with antibodies against endogenous copine-6 (left) and syb2 (middle). Right (merged image), White arrows indicate the colocalization of copine-6 and syb2 at hippocampal synaptic boutons. D, Intensity plot of endogenous copine-6 and syb2 from the merged image. The mean Pearson correlation coefficient for their colocalization is indicated (Pearson correlation coefficient = 0.50 ± 0.02, calculated using n = 15 images from 3 independent cultures). E, Representative confocal images of cultured hippocampal neurons immunostained with antibodies against endogenous copine-6 (left) and synapsin 1 (middle). Right (merged image), White arrows indicate the colocalization of copine-6 and synapsin 1 at hippocampal synaptic boutons. F, Intensity plot of endogenous copine-6 and synapsin 1 from the merged image. The mean Pearson correlation coefficient for their colocalization is indicated (Pearson correlation coefficient = 0.56 ± 0.03, calculated from n = 10 images from 2 independent cultures). G, Copine-6 binds to SNAREs in the presence of Ca2+. GST alone or GST fused to various SNARE proteins (without transmembrane region, except SNAP-25) were immobilized on glutathione Sepharose and incubated with extracts of HEK cells transfected with HA-tagged copine-6 in the presence (top) or absence of Ca2+ (middle). The eluted proteins (also in F,G) were analyzed by immunoblotting (top, middle), and GST beads were analyzed by Coomassie staining (bottom). H, Copine-6 binds to VAMPs in the presence of Ca2+. GST or GST fused to various VAMPs (without transmembrane regions) were immobilized on glutathione Sepharose and incubated with extracts of HEK cells transfected with HA-tagged copine-6 in the presence (top, bottom) or absence of Ca2+ (middle). I, The C-terminal linker region of syb2 is required for Ca2+-dependent binding to copine-6. GST or GST fused to various syb2 (without transmembrane regions) proteins were immobilized on glutathione Sepharose and incubated with extracts of HEK cells transfected with HA-tagged copine-6 in the presence (top, bottom) or absence of Ca2+ (middle). Syb2 WA mutant contains two alanine substitutions of vicinal W89A/W90A; syb2 2–84 lacks 10 juxtamembrane C-terminal residues. J, GST alone or GST fused to Syb2 and VAMP4 proteins were immobilized on glutathione Sepharose and incubated with extracts of HEK293T cells transfected with truncated HA-tagged copine-6 constructs (C2AB, vWA, or both) in the presence of Ca2+. After washing, proteins were eluted by removing Ca2+ and analyzed by immunoblotting with HA antibody.
Materials and Methods
Pulldowns and protein identification.
Stripped frozen rat brains from either sex (Pelfreeze) were homogenized in 150 mm NaCl, 25 mm HEPES, pH 7.4, 1 mm EDTA, protease inhibitor mixture Complete (Roche) and extracted by adding NP40 to 2%. Brain extract was cleared by centrifugation. Human HA-tagged full-length Copine-6, C2AB (2-271 aa), or vWA (271-557 aa) truncation mutants were produced in transfected HEK293 cells, and extracts were prepared as described above. Plasmids encoding GST fusion proteins with soluble fragments of various SNARE proteins were described previously or made using standard molecular biology procedures and verified by sequencing. Recombinant SNARE proteins were produced in BL21 Escherichia coli and purified on glutathione-Sepharose beads (GE Healthcare). For pulldown reactions, GST beads were incubated with rat brain extract or HEK293 cell extracts in the presence of calcium (2 mm CaCl2) or in the absence of calcium (0.1 mm EDTA) for 3 h at 4°C. Following extensive washing with binding buffer, beads were eluted with 150 mm NaCl, 25 mm HEPES, pH 7.4, 2 mm EGTA, 1% NP40. Eluates were concentrated on Amicon microconcentrators and loaded onto SDS-PAGE for immunoblotting analysis. Alternatively, gels were silver stained, and individual bands were excised and identified by trypsin digestion/LC mass spectrometry (University of Texas Southwestern proteomics core facility).
Antibody production.
C2AB (1-270aa) and vWA (268-557) domains of human Copine-6 were bacterially expressed and purified as GST fusion proteins on glutathione-Sepharose beads. Proteins were eluted by on-beads cleavage with thrombin. Purified Copine-6 fragments were used to produce rabbit polyclonal antibodies at Covance facility.
Immunocytochemistry in neuronal cultures.
High-density rat hippocampal cultures (2–3 weeks old) from either sex were fixed in 4% PFA for 15 min at room temperature. The primary polyclonal antibody to copine-6 was used at 1:1000 dilution and polyclonal antibody to syb2 (or synapsin 1) used at 1:5000 (or 1:400) dilution. The secondary antibodies, Alexa-594-conjugated goat anti-rabbit antibody and Alexa-488-conjugated goat anti-mouse antibody (Invitrogen), were used at 1:1000 dilution to detect copine-6 and syb2 (or synapsin 1), respectively. The immunocytochemistry experiment was processed as described previously (Ramirez et al., 2008). Stained coverslips were imaged on a Zeiss LSM510 confocal microscope using LSM 5 software. Synapsin-positive puncta were selected with the copine-6 signal (red channel) hidden. A total of 50–100 puncta of constant diameter were selected per image, and the Pearson correlations of the total red and green fluorescence intensities from 11 images within the synaptic puncta were calculated offline.
Lentivirus construction.
Lentiviral constructs were made in pFUGW transfer vector (for overexpression) or L307 (for shRNA knockdown) using standard molecular biology procedures and verified by sequencing. Full-length human copine-6, C2AB (2–271 aa) or vWA (271–557 aa) truncation constructs carried HA-tag at the N terminus or mOrange fluorescent protein separated from copine-6 sequence by the linker SPGSATGSAGTGAPGSATGSAGTGAPGSATGSAGTGAPGISGGGGGIQ. For knockdown experiments, oligonucleotides encoding rat Copine-6 shRNA sequences (5′-TCGAGGCAGCCTTTGCAATTCCACTTCAAGAGAGTGGAATTGCAAAGGCTGCTTA-3′ and 5′-CTAGTAAGCAGCCTTTGCAATTCCACTCTCTTGAAGTGGAATTGCAAAGGCTGCC-3′ for Copine-6 KD1; 5′-TCGAGCAGCCATTGTGCGAGCCTCTTCAAGAGAGAGGCTCGCACAATGGCTGTTA-3′ and 5′-CTAGTAACAGCCATTGTGCGAGCCTCTCTCTTGAAGAGGCTCGCACAATGGCTGC-3′ for Copine-6 KD2) were inserted into the XhoI/XbaI cloning site downstream of the human H1 promoter in the L307 lentiviral transfer vector (gift of Dr. T. C. Südhof, Stanford). The efficiency of the knockdown was established by measuring the endogenous levels of Copine-6 mRNA in infected rat hippocampal neurons using quantitative real-time PCR. Copine-6 knockdown in neuronal cultures was further verified by assaying endogenous Copine-6 levels using Western blotting. The syt-1 shRNA KD construct was a gift from Dr. T.C. Südhof (Stanford University) and has been previously validated (Xu et al., 2012, Li et al., 2017).
Production of recombinant lentiviruses.
Lentiviruses were produced in HEK293T cell by contransfection of pFUGW or L307 transfer vector and 3 packaging plasmids (pVsVg, pMdLg/pPRE and pRSV-Rev) using Fugene 6 reagent (Promega). One day after transfection, cells were exchanged into neuronal growth media and viruses were harvested after 48 h of accumulation. Viral media was clarified by low-speed centrifugation.
Dissociated hippocampal cultures and lentiviral infection.
Dissociated hippocampal cultures were prepared from Sprague Dawley rats of either sex using previously published protocols (Kavalali et al., 1999; Schoch et al., 2001). Neurons were infected at 4 DIV by adding 0.1–0.3 ml of viral media per coverslip in 24 well plates. All experiments were performed after 14 DIV to allow synapses to reach maturity. Neuronal cultures of syb2-deficient mice and their littermate controls were made from embryonic cortical tissue.
Electrophysiology.
Patch-clamp recordings were performed on hippocampal or cortical pyramidal neurons as described previously (Nosyreva and Kavalali, 2010). Pyramidal neurons were recorded using a MultiClamp 700B amplifier and Clampex 9.0 software (Molecular Devices), filtered at 2 kHz and sampled at 5 kHz at room temperature. The pipette solution contained the following (in mm): 115 CsMeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 tetraethylammonium chloride, 4 Mg-ATP, 0.3 Na2GTP, and 10 QX-314 (lidocaine N-ethyl bromide), pH 7.35, 300 mOsm. A modified Tyrode's solution was used as the extracellular solution, containing the following (in mm): 150 NaCl, 4 KCl, 2 MgCl2, 10 glucose, 10 HEPES, and 2 CaCl2, pH 7.4 (310 mOsm). To isolate mIPSCs, TTX (1 μm), the ionotropic glutamate receptor antagonist CNQX (10 μm; Sigma-Aldrich) and AP-5 (50 μm; Sigma-Aldrich) were added to the bath solution. For miniature EPSCs, TTX (1 μm), AP-5 (50 μm), and PTX (50 μm) were added. To test the Ca2+-dependent facilitation of copine-6, the membrane-permeable fast Ca2+ buffer BAPTA-AM was used. Hippocampal cultures were incubated with calcium-free modified Tyrode's solution containing 30 μm BAPTA-AM for 30 min at room temperature. Then BAPTA-AM was washed out with normal modified Tyrode's solution containing 2 mm Ca2+ and mIPSCs were recorded. In caffeine experiments, caffeine was added to the extracellular solution with a final concentration of 20 mm. For each experiment, mEPSCs were continually recorded before and after caffeine treatment on the same cell for ∼15 min. Evoked IPSCs were recorded in the presence of AP5 (50 μm) and CNQX (10 μm), and field stimulation was produced by parallel bipolar electrodes (FHC) immersed in the chamber with 33 mA pulses.
Fluorescence imaging.
A total of 16–21 DIV cultured pyramidal neurons expressing vGlut1-pHluorin were used for the imaging experiments. The modified Tyrode's solution from above containing 2 mm Ca2+ was used with TTX (1 μm), AP-5 (50 μm)[scap], and PTX (50 μm). Experiments were performed at room temperature using an Andor iXon+ back-illuminated EMCCD camera (model DU-897E-CSO-#BV) collected on a Nikon Eclipse TE2000-U microscope with a 100× Plan Fluor objective (Nikon). For illumination, we used a Lambda-DG4 (Sutter Instruments) with an FITC filter. Images were acquired at ∼10 Hz with an exposure time of 100 ms and binning of 4 × 4 to optimize the signal-to-noise ratio. Puncta were automatically selected based on local maximums generated by ammonium (50 mm) addition at the end (using a custom designed macro for ImageJ).
Fluorescence analysis.
Fluorescence intensity traces were analyzed using Microsoft Excel and a custom MATLAB script, based on our previous analysis with some modifications (Leitz and Kavalali, 2011, 2014). Photobleaching was corrected with an exponential decay and background was subtracted linearly, both photobleaching and background values were calculated based on fluorescence measurements of background in each imaging experiment and also for each ROI. For single-vesicle analysis, successful fusion events were those that meet the following restrictions: event fluorescence amplitude must be greater than twice the SD of the baseline (average of ∼2.5 s before the event) and smaller than the mean single-event amplitude + 2 SDs (to select single-vesicle fusion events, this value was determined experimentally in the laboratory using control cultures in 2 mm Ca2+).
Experimental design and statistical analysis.
Statistical analyses were performed using Excel (Microsoft) and Prism 7 (GraphPad Software). For parametric data with multiple groups, statistical significance was assessed by one-way ANOVA followed by Bonferroni post test. For nonparametric distributions, means were compared applying Mann–Whitney test. Cumulative distributions were compared using Kolmogorov–Smirnov test. Error bars indicate mean ± SEM.
Results
Presynaptic expression of Copine-6
We initially identified copine-6 (Fig. 1A,B) from rat brain extracts by pulldown reaction with glutathione S-transferase tagged synaptobrevin2 (GST-syb2, 2-94 aa) in the presence of Ca2+. As shown in Figure 1B, mass spectrometric analysis of protein fractions bound to GST-syb2 and eluted by removing Ca2+ revealed copine-6 as a major component in addition to munc18 (upper band) as well as synaptotagmins 1 and 2 and copine 7 (lower band) that comigrate with copine-6 on the gel (Tang et al., 2006; Dulubova et al., 2007; Xu et al., 2010, 2013). In addition, we identified several low molecular weight proteins (calmodulins, centrin2, S100B, and A1) that have been shown to interact with Syb2 by previous studies (Quetglas et al., 2002; Umbrecht-Jenck et al., 2010).
To validate the physiological relevance of this putative interaction, next, we investigated whether syb2 and copine-6 are colocalized in neurons. For this purpose, we used syb2 as a presynaptic marker, labeled neurons with anti-syb2 antibodies, then immunostained for native copine with anti-copine-6 antibodies. Representative images and an intensity plot are shown in Figure 1C,D. This analysis revealed that copine-6-positive puncta colocalized with syb2 staining (Fig. 1C, white arrows) with a mean Pearson correlation coefficient of 0.50 ± 0.02 (Fig. 1D). Furthermore, presynaptic localization of copine-6 is also supported by its colocalization with the synaptic vesicle marker synapsin 1 with a mean Pearson correlation coefficient of 0.56 ± 0.03 (Fig. 1E,F). This finding indicates that the native copine-6 colocalizes with synaptic vesicle proteins within presynaptic terminals. In addition, copine-6 also showed a broad subcellular distribution with prominent immunofluorescence staining in cell bodies and dendrites in syb2-negative area, which is consistent with the earlier report (Nakayama et al., 1999).
Copine-6 interacts with essential SNARE proteins
To examine the specificity of copine-6–syb2 interaction, we analyzed interactions of copine-6 with additional SNARE proteins involved in vesicle trafficking. We first immobilized soluble fragments of recombinant SNARE proteins (including syb2, SNAP25, Vti1a, Syntaxin1A, Syntaxin4, Syntaxin11, and Syntaxin19) on glutathione-Sepharose beads and incubated with extracts of HEK cells transfected with human influenza hemagglutinin (HA)-tagged copine-6 (HA-Cop6), and then analyzed the binding reactions by immunoblotting. We found that, although these proteins could bind to copine-6 in the presence of Ca2+, under Ca2+ free conditions minimal binding reaction could be observed (Fig. 1G). In the next set of experiments, we tested whether copine-6 could interact with other vesicular SNAREs under the same conditions as stated above (Fig. 1H). In addition to strong syb2—copine-6 interaction, these experiments revealed robust binding between VAMP8 and copine-6, relatively modest interactions of VAMP4 and VAMP5 with copine-6 and relatively weak coupling effects between VAMP7 and copine-6 in the presence of Ca2+. In the absence of Ca2+, however, copine-6 binding to vesicular SNAREs is greatly reduced. In some experiments (Fig. 1H), but not in others (Fig. 1G,H), we observed residual binding of syb2 and copine-6 in the absence of Ca2+, which suggests that syb2, albeit in a greatly reduced degree, can associate with copine-6 even under resting conditions in a Ca2+ independent manner.
Next, we focused on whether the specific regions of syb2 and copine-6 were required to support this robust interaction. For this purpose, we designed a point mutation construct, syb2 WA-containing alanine substitutions at the vicinal tryptophan residues W89A/W90A (Maximov et al., 2009) and a truncation construct, syb2 2-84 lacking 10 juxtamembrane residues (Fig. 1I). We found that both mutants almost completely lost the robust interaction with copine-6, suggesting that the C-terminal linker of syb2 and specifically the vicinal W89 and W90 residues in this region were required for copine-6 binding. Notably, the C-terminal region of VAMP8, which also shows strong Ca2+-dependent binding to copine-6 (Fig. 1H), is highly homologous to the syb2 C-terminal region. In parallel, we made two copine-6 recombinant proteins: HA-Cop6 C2AB lacking vWA domain and HA-Cop6 vWA missing the two C2 domains (Fig. 1J). The immunoblotting analysis revealed that both truncations severely impaired copine-6 binding to syb2. However, when the two parts of copine-6 were coincubated with syb2, they both interacted with syb2 similarly to full-length copine-6 (Fig. 1J), indicating that both domains bind in an independent manner to syb2.
Copine-6 is not critical for maintenance of synchronous-evoked neurotransmitter release
To investigate the functional impact of copine-6 interaction with SNARE proteins on synaptic transmission, we tested whether overexpression or loss of function of copine-6 could alter evoked neurotransmitter release in hippocampal synapses. When we overexpressed fluorescently tagged (mOrange) copine-6 in the neurons via lentiviral infection, synaptic responses to paired pulse stimulation (at 50 ms, 100 ms, and 1 s intervals) were unaltered. The ratio of IPSCs evoked in response to paired pulse stimulation (P2/P1) did not show any difference between noninfected controls (Ctrl) and copine-6-overexpressing (Cop6) neurons, suggesting lack of alterations in release probability in synapses overexpressing copine-6 (Fig. 2C,D). This was not due to low infectivity of copine-6-overexpressing lentivirus because we observed a 5.8 ± 1.3-fold increase in protein levels compared with controls. In copine-6-overexpressing neurons, cumulative charge histograms of individual evoked IPSCs recorded at 1 Hz stimulation were also identical to the controls, indicating that copine-6 expression did not alter the kinetics of evoked IPSCs (Fig. 2E,F). Next, we repeated the same experimental protocols with neurons deficient in copine-6. The copine-6 expression dramatically declined after infection with two lentiviral short hairpin RNA (shRNA) constructs (KD1 and KD2) where copine-6 mRNA levels showed 80%–90% reduction (Fig. 2A). A representative immunoblot of neuronal protein samples harvested from cells expressing shRNA against copine-6 is shown in Figure 2B. Similarly, downregulation of copine-6 level did not alter paired pulse ratio of evoked IPSCs and their release kinetics (Fig. 2D–F) compared with the L307 vehicle-infected cells. In addition, under all the conditions described above, we did not detect statistically significant differences among the absolute amplitudes of evoked IPSCs (data not shown). Together, these observations suggest that copine-6 is not essential for maintenance of evoked synchronous neurotransmitter release.
Figure 2.
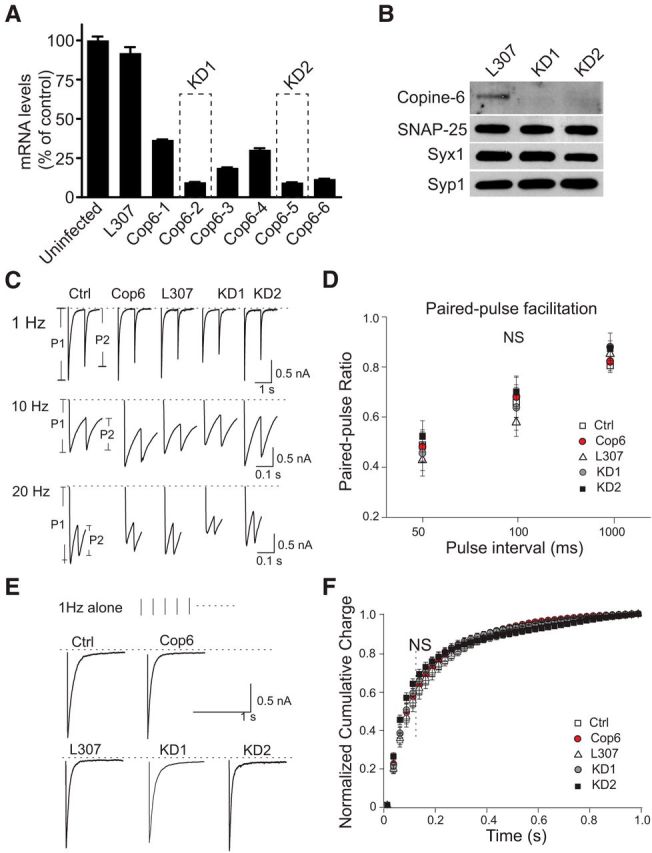
Copine-6 is not critical for maintaining synchronous-evoked neurotransmission. A, Bar graph represents copine-6 mRNA levels in rat primary hippocampal cultures under control condition (uninfected) or after infection with an empty (L307) or shRNA-producing lentivirus. Data are from representative experiment performed in quadruplicates. The best knockdown efficiency was observed for two lentiviruses termed KD1 and KD2, which were used throughout the study. B, Western blot analysis of rat hippocampal cultures infected with L307 virus (control) or copine-6 knockdown viruses (KD1 and KD2). Neurons from 4 coverslips were pooled together for each condition. Syx1, Syntaxin 1; Syp1, Synaptophysin 1. Both KD constructs reduced copine-6 expression substantially compared with the neurons infected with L307 virus. C, Representative IPSC traces of postsynaptic response triggered by a paired-pulse stimulation from noninfected control (Ctrl), mOrange-copine-6 (Cop6) overexpressing, empty L307 vector virus (L307) infected, and copine-6 knockdown (KD1 and KD2) hippocampal neurons in the presence of physiological Ca2+. Stimulus intervals: 1000 (1 Hz), 100 (10 Hz), 50 ms (20 Hz). D, Statistical analysis of paired-pulse ratio (P2/P1) shows no significant differences under the various conditions stated above. 1 Hz, Ctrl 0.8 ± 0.03, n = 16; and Cop6 0.8 ± 0.04, n = 14, p > 0.7, compared with Ctrl; L307, 0.8 ± 0.04, n = 12; KD1, 0.9 ± 0.06, n = 10, p1 > 0.6, KD2, 0.9 ± 0.03, n = 15, p2 > 0.6, compared with L307. 10 Hz: Ctrl, 0.7 ± 0.06, n = 23; Cop6, 0.7 ± 0.08, n = 15, p > 0.8, compared with Ctrl; L307, 0.6 ± 0.06, n = 19; KD1, 0.6 ± 0.09, n = 13, p1 > 0.5; KD2, 0.7 ± 0.06, n = 15, p2 > 0.1, compared with L307. 20 Hz: Ctrl, 0.5 ± 0.06, n = 19; and Cop6, 0.5 ± 0.06, n = 19, p > 0.9 compared with Ctrl; L307, 0.4 ± 0.06, n = 17; KD1, 0.5 ± 0.07, n = 19, p1 > 0.7; KD2, 0.5 ± 0.06, n = 15, p2 > 0.2, compared with L307. E, Representative IPSC traces during 1 Hz stimulation from control neurons and neurons infected with lentiviruses expressing Cop6, L307 control, KD1, and KD2. F, Average cumulative charge histograms from the first IPSC trace under 1 Hz stimulation depicting the synchronization of synaptic responses from cultured hippocampal neurons described above, and no statistical significance was observed among different conditions: Ctrl, n = 12; Cop6, n = 12; L307, n = 11; KD1, n = 10; KD2, n = 10.
Copine-6 selectively regulates spontaneous release in a Ca2+-dependent manner
In the next set of experiments, we investigated whether copine-6 could regulate spontaneous neurotransmitter release. mEPSCs and mIPSCs were recorded from neurons overexpressing full-length mOrange-tagged copine-6 (Cop6) or after copine-6 loss of function using the two shRNA constructs described above (KD1 and KD2). Representative mEPSC traces for each condition are shown in Figure 3A. Compared with control neurons, a significant reduction of mEPSC frequency was observed in neurons that overexpress copine-6, without a change in average mEPSC amplitudes (Fig. 3A–C). In a subsequent set of experiments, we assessed the impact of copine-6 expression on mIPSCs and found a similar effect where copine-6 overexpression decreased mIPSC frequency (Fig. 3D–F). These results suggest that copine-6 may play a role in the regulation of spontaneous release. These findings are further supported by experiments where we detected an increase in the frequency of both excitatory (Fig. 3A–C) and inhibitory (Fig. 3D–F) events after knockdown of copine-6 levels while the average amplitudes of these events were not altered. To confirm that copine-6 regulation of spontaneous release was dependent on its action at the presynaptic terminal, we used fluorescence imaging using pH-sensitive GFP (pHluorin) probes. We expressed pHluorin tagged to the vesicle lumen region of the vesicular glutamate transporter-1 (vGlut1-pHluorin) in both control and copine-6 KD neurons (Fig. 3G) (Voglmaier et al., 2006). By analyzing single-vesicle spontaneous fusion events (Leitz and Kavalali, 2014), we found a corresponding increase in the frequency of spontaneous events in the copine-6 loss-of-function neurons compared with control neurons (Fig. 3H). This result indicates that copine-6 loss of function indeed increases the rate of spontaneous fusion from presynaptic terminals.
Figure 3.
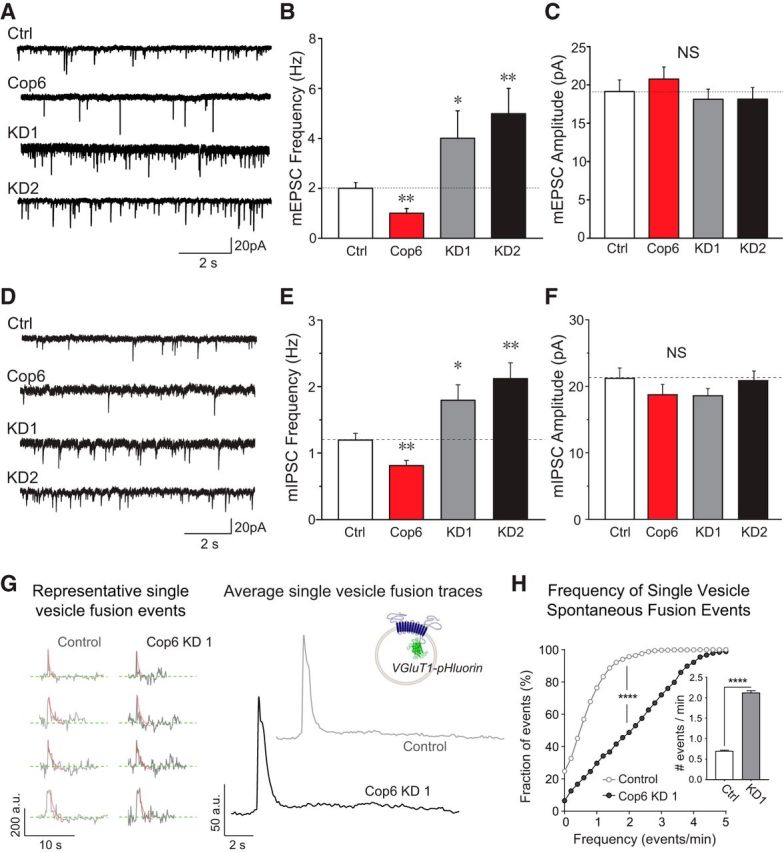
Copine-6 suppresses presynaptic spontaneous vesicle fusion. A, Example traces of mEPSC recording from control hippocampal neurons (Ctrl), neurons overexpressing copine-6 and copine-6 knockdown shRNA constructs (KD1 and KD2) in the presence of 2 mm Ca2+. B, C, Quantification of mEPSC frequency (B) and amplitude (C) from control, copine-6 overexpression, and copine-6 KD neurons. Overexpressing copine-6 suppressed mEPSC frequency and knockdown of copine-6 increased mEPSC frequency (Ctrl, 2.0 ± 0.2 Hz, n = 16; HA-Cop6, 1.0 ± 0.2 Hz, n = 15, p = 0.002; KD1, 4.0 ± 1.1 Hz, n = 11, p = 0.04; KD2, 4.9 ± 1.1 Hz, n = 9, p = 0.001). The average amplitudes of each condition were not significantly different (19.1 ± 1.5 pA for Ctrl, 20.8 ± 1.6 pA for HA-Cop6, 18.1 ± 1.3 pA for KD1, and 18.1 ± 1.5 pA for KD2). D, Example traces of mIPSCs recorded in noninfected control (Ctrl), copine-6 (Cop6) overexpression, and copine-6 knockdown (KD1 and KD2) hippocampal neurons in the presence of 2 mm Ca2+. E, F, Quantification of average mIPSC frequency (E) and amplitude (F) of the cells as mentioned in A. Copine-6 overexpression resulted in suppression on spontaneous release rate with no change of amplitudes. Ctrl,1.2 ± 0.1 Hz, 21.2 ± 1.6 pA, n = 14; Cop6, 0.8 ± 0.1 Hz, 18.7 ± 1.6 pA, n = 14; L307, 1.1 ± 0.2 Hz, 18.2 ± 1.2 pA, n = 11; KD1, 1.8 ± 0.2 Hz, 18.6 ± 1.1 pA, n = 15, KD2, 2.1 ± 0.2 Hz, 20.9 ± 1.4 pA, n = 15. G, Left, Representative single-vesicle fusion spontaneous fusion events measured with vGlut1-pHluorin in control (light gray) and copine-6 KD (black) neurons. Right, Average fluorescence traces from single-vesicle spontaneous fusion events in control neurons (light gray) and copine-6 KD neurons (black). Inset, Schematic of vGlut1-pHluorin fusion protein in a synaptic vesicle. H, Cumulative histogram of frequency of single-vesicle spontaneous fusion events for control and copine-6 KD neurons. Cumulative distributions were compared using Kolmogorov–Smirnov test. ****p < 0.0001. Inset, Bar graph represents the average number of events per minute for both conditions: control, 0.70 ± 0.02 events/min, copine-6 KD, 2.12 ± 0.04 events/min. Means were compared using Mann–Whitney test. ****p < 0.0001. Data from 5 independent cultures. Control, 1237 boutons from 12 coverslips; copine-6 KD, 976 boutons from 7 coverslips. *p < 0.05. **p < 0.01.
Next, we investigated the structural determinants of copine-6's impact on spontaneous neurotransmitter release and tested the function of truncated copine-6 mutants: C2AB (missing the vWA domain) and vWA (missing the C2A and B domains). We found that overexpressing each mutant alone did not mimic the effect of full-length copine-6 but produced an increase in mIPSC frequency, akin to the copine-6 knockdown experiments, suggesting a potential dominant negative effect of these mutants (Fig. 4A,B). In addition, coexpression of the two truncation mutants resulted in a decrease in spontaneous release as well (Fig. 4A,B), which is similar to the result of the binding experiments shown in Figure 1J, suggesting that both C2AB and vWA domains are independently required for copine-6 to perform its function.
Figure 4.

Copine-6 regulates spontaneous neurotransmission in a Ca2+-dependent manner. A, Example traces of mIPSC recording from WT neurons (Ctrl) and neurons overexpressing copine-6 truncation mutants (C2AB, vWA, or coexpressing both). B, Quantification of mIPSC frequency of the four conditions shown in A. Expressing copine-6 mutants (C2AB or vWA) alone increased the mIPSC frequency, whereas coincubating them suppressed mIPSCs events. Ctrl, 1.2 ± 0.1 Hz, n = 19; C2AB, 2.0 ± 0.3 Hz, n = 15, p = 0.002; vWA, 1.9 ± 0.2 Hz, n = 14, p = 0.002; C2AB+vWA, 0.8 ± 0.1 Hz, n = 15, p = 0.02. C, Example traces of mIPSCs recorded from wild-type and copine-6-overexpressing neurons without or with 30 μm BAPTA-AM treatment. D, E, Quantification of average mIPSC frequency (D) and amplitude (E) of the cells as mentioned in D. Ctrl, 1.2 ± 0.1 Hz, 13.6 ± 0.7 pA, n = 16 without and 0.4 ± 0.03 Hz, 13.8 ± 0.7 pA, n = 15 with BAPTA-AM treatment; Cop 6, 0.8 ± 0.1 Hz, 13 ± 1.1 pA, n = 18 without and 0.5 ± 0.1 Hz, 14.9 ± 1.0 pA, n = 17 with BAPTA-AM treatment. G, Example traces of mEPSCs of noninfected control and copine-6 knockdown neurons before and after 20 mm caffeine treatment. H, Quantification of average mEPSC frequency of the cells as mentioned in G. Ctrl, 1.5 ± 0.2 Hz before and 3.7 ± 0.7 Hz after caffeine treatment, n = 11; KD1, 4.0 ± 1.1 Hz before and 7.9 ± 1.3 Hz after caffeine treatment, n = 11; KD2, 5.0 ± 1 Hz before and 9.4 ± 1.8 Hz after caffeine treatment, n = 9. Error bars indicate ± SEM. *p < 0.05. **p < 0.01.
The results presented above (Fig. 1G–J) indicate that copine-6 binds to its targets in a Ca2+-dependent manner. This finding suggests that copine-6's suppression of spontaneous neurotransmitter release may require Ca2+. To test this premise, we incubated the noninfected control and copine-6-overexpressing neurons with the membrane-permeable fast Ca2+ chelator BAPTA-AM for 30 min. After removal of extracellular BAPTA-AM, we recorded mIPSCs and compared their properties with those recorded from neurons that were not subjected to BAPTA-AM preincubation. Figure 4C depicts the sample traces of each condition. The average data in Figure 4D show that, without BAPTA-AM, copine-6 overexpression, as before, attenuated spontaneous fusion at rest compared with the control conditions. In contrast, BAPTA-AM application not only dramatically inhibited the baseline level of spontaneous release events in control and copine-6-overexpressing cells, but also abolished the relative suppression of spontaneous events in copine-6-overexpressing cells. The amplitudes of mIPSCs were not altered in any of these conditions (Fig. 4E).
In the next set of experiments, we aimed to test whether endogenous copine-6 impaired Ca2+-dependent augmentation of spontaneous release. For this purpose, we treated the neurons with 20 mm caffeine to induce Ca2+ release from intracellular stores and detected a robust increase in mEPSC frequency, as previously observed in hippocampal synapses (Sharma and Vijayaraghavan, 2003). Under the same conditions, knockdown of copine-6 substantially augmented the increase in mEPSC frequency seen after treatment with caffeine (Fig. 4F,G). Together, these results indicate that endogenous copine-6 not only suppresses spontaneous neurotransmitter release under resting conditions but also curtails intracellular Ca2+-mediated augmentation of spontaneous neurotransmission. These results also strongly support the premise that Ca2+ is required for copine-6 to perform its function.
Syb2 is involved in the regulation of copine-6 on synaptic transmission at rest
As indicated above, copine-6 targets several proteins involved in vesicle fusion including syb2 (Schoch et al., 2001). In the next set of experiments, we asked whether the ability of copine-6 to bind syb2 could underlie its impact on spontaneous release. To address this question, we tested the ability of copine-6 to suppress synaptic transmission in syb2-deficient neurons. We infected high-density cortical neuronal cultures from syb2 knock-out (KO) mouse embryos with copine-6 constructs. Syb2 KO did not impact copine-6 levels because noninfected wild-type and syb2 KO neurons showed similar protein levels of copine-6 (average optical density of 1.0 ± 0.3 and 0.98 ± 0.4, respectively). Electrophysiology experiments showed that, in the absence of syb2 copine-6 overexpression did not result in a decrease in spontaneous release, compared with noninfected (Ctrl) or GFP-infected (GFP) syb2-deficient neurons (Fig. 5A–C). This result suggests that syb2 is critically required for the regulation of spontaneous release by copine-6. To validate this premise, we performed a series of rescue experiments in syb2-deficient cultures. Because we have previously shown that the C-terminal linker of syb2 was needed for syb2 to bind copine-6 (Fig. 1I) and the syb2 WA mutant impaired this interaction, we expressed wild-type syb2 and syb2 WA mutant in syb2-deficient neurons to examine their ability to rescue copine-6-mediated suppression of spontaneous release. When we coexpressed copine-6 or GFP together with syb2 or syb2 WA, we found that expressing wild-type syb2 restored spontaneous release, whereas the mutant syb2 WA gave rise to a robust increase in spontaneous event frequency compared with the control (Ctrl = 0.2 ± 0.02 Hz; syb2 + GFP = 1.28 ± 0.21 Hz; syb2 WA + GFP = 2.53 ± 0.49 Hz), in agreement with a previous report using the syb2 WA mutant (Maximov et al., 2009). Furthermore, spontaneous release rescued by wild-type syb2 was suppressed by coexpression of copine-6, whereas spontaneous events rescued by the syb2WA mutant were unaffected by copine-6 overexpression (syb2 + GFP = 1.28 ± 0.21 Hz vs syb2 + Cop6 = 0.60 ± 0.09 Hz, p = 0.004; syb2 WA + GFP = 2.53 ± 0.49 Hz vs syb2WA + Cop6 = 2.54 ± 0.41 Hz, p = 0.99). The amplitudes of mIPSCs were unaltered in most conditions except for the syb2WA mutant-expressing conditions consistent with a potential role for syb2 in postsynaptic receptor trafficking (Fig. 5C) (Jurado et al., 2013; Arendt et al., 2015). Together, these data indicate that copine-6 exerts its effect on spontaneous neurotransmission by interacting with the tryptophan residues within the juxtamembrane region of syb2.
Figure 5.
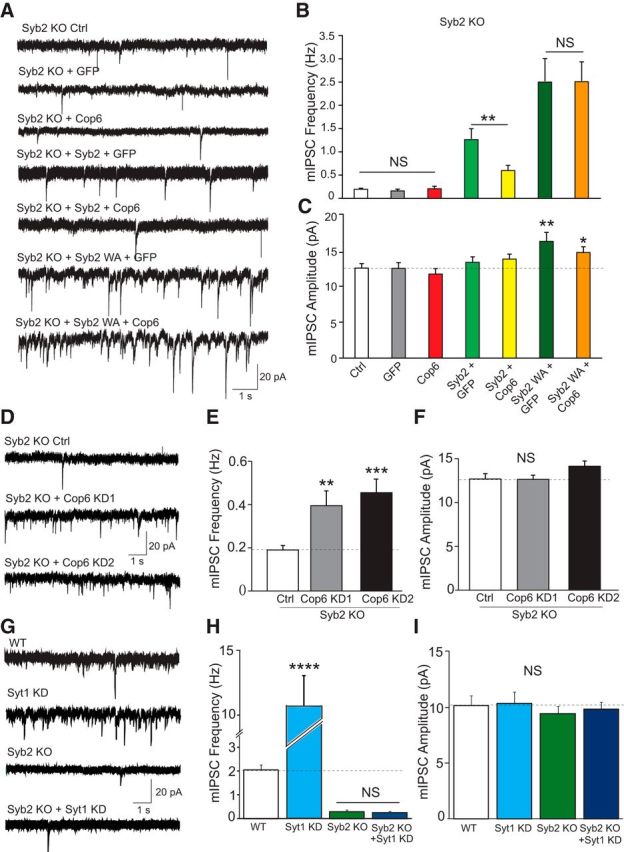
Syb2 is required for copine-6-mediated regulation of spontaneous neurotransmitter release. A, Recordings were performed (in 2 mm Ca2+) in syb2-deficient neurons under various conditions: noninfected cells (Ctrl), syb2-deficient neurons expressing GFP alone (GFP), full-length mOrange-copine-6 (Cop6), wild-type syb2 and GFP (syb2 + GFP), wild-type Sy2 and mOrange-copine-6 (syb2 + Cop6), mutant syb2 WA and GFP (syb2 WA + GFP), mutant syb2 WA and mOrange-copine-6 (syb2 WA + Cop6), or copine-6 knockdown constructs (KO + KD1, KO+KD2). Representative traces of mIPSC recordings in syb2-deficient neurons under different conditions as stated above: Ctrl, GFP, Cop6, syb2 + GFP, syb2 + Cop6, syb2 WA + GFP, and syb2 WA + Cop6. B, C, Quantification of average mIPSC frequency (B) and amplitude (C) from neurons metioned in A. Overexpression of copine-6 in syb2-deficient neurons did not change mIPSC rate and amplitude Ctrl, 0.2 ± 0.02 Hz and 12.5 ± 0.6 pA, n = 24; GFP, 0.2 ± 0.04 Hz and 12.5 ± 0.8 pA, n = 12; Cop6, 0.2 ± 0.05 Hz and 11.7 ± 0.7 pA, n = 12. Copine-6-overexpressing suppressed mIPSCs when wild-type syb2 expression rescued spontaneous neurotransmission but showed no impact on mutant syb2 WA expression. syb2 + GFP, 1.3 ± 0.2 Hz and 13.3 ± 0.8 pA, n = 18; syb2 + Cop6, 0.6 ± 0.1 Hz and 13.8 ± 0.7 pA, n = 21; syb2 WA + GFP, 2.5 ± 0.5 Hz and 16.2 ± 1.2 pA, n = 23; syb2 WA + Cop6, 2.5 ± 0.4 Hz and 14.7 ± 0.8 pA, n = 23. D, Representative traces of mIPSC recordings from syb2 KO control cells and syb2 KO cells with copine-6 knockdown: Ctrl, KO+KD1, and KO+KD2. E, F, Quantification of average mIPSC frequency (E) and amplitude (F) from neurons described in D. Knockdown of endogenous copine-6 in syb2-deficient neurons caused a significant elevation of spontaneous release frequency with no change of amplitude compared with the control cells. Ctrl, 0.2 ± 0.02 Hz and 12.7 ± 0.6 pA, n = 34; KO + KD1, 0.4 ± 0.07 Hz and 12.6 ± 0.5 pA, n = 21, p1 = 0.002; KO + KD2, 0.46 ± 0.06 Hz and 14.1 ± 0.5 pA, n = 21, p2 = 0.00004. The total analyzed neurons were from five independent cultures. G, Representative traces of mIPSC recordings from WT and syt1 KD neurons as well as syb2 KO neurons and syb2 KO neurons with syt1 KD. H, I, Quantification of average mIPSC frequency (H) and amplitude (I) from neurons described in G. Syt1 KD neurons exhibited significantly higher mIPSC frequency, whereas syb2 KO neurons had significantly lower mIPSC frequency regardless of syt1 loss of function compared with WT. ****p < 0.0001. Data from 3 independent cultures. WT, n = 8, Syt1KD: n = 9, syb2KO, n = 7, syb2KO+Syt1KD, n = 11. *p < 0.05. **p < 0.01. ***p < 0.001.
In the next set of experiments, we asked whether syb2 is exclusively required for copine-6's function. To address this question, we knocked down endogenous copine-6 in syb2-deficient neuron by lentiviral-mediated expression of KD1 and KD2 shRNA constructs and recorded mIPSCs. In the absence of syb2 expression, suppression of copine-6 mRNA level gave rise to an elevation of spontaneous release rate compared with the control neurons (Fig. 5D–F), consistent with our previous observations in wild-type neurons (Fig. 3). However, the enhanced mIPSC rate was still much lower than the spontaneous release rate seen in syb2-deficient neurons rescued with syb2 (0.4 ± 0.1 Hz and 0.5 ± 0.1 Hz vs 1.28 ± 0.21 Hz), indicating that endogenous copine-6 may not only suppress syb2-mediated spontaneous events but also inhibit events mediated by alternative SNAREs, such as vti1a or VAMP4 (Raingo et al., 2012; Ramirez et al., 2012) and possibly synaptobrevin1 (Liu et al., 2011; Zimmermann et al., 2014). As a comparison, we investigated the syb2 dependence of synaptotagmin-1's regulation of spontaneous release. Synaptotagmin-1 loss-of-function neurons exhibit a well-documented increase in spontaneous release rates (Fig. 5G,H) (Xu et al., 2009). By expressing syt-1 shRNA KD in syb2 KO neurons, we show that this increase in spontaneous release is syb2-dependent (Fig. 5G–I). These results suggest that syt-1 inhibits spontaneous release that is mediated by syb2, whereas copine-6 inhibits spontaneous release that may be mediated by both canonical and noncanonical SNARE complexes.
Discussion
In this study, we used an unbiased approach to identify Ca2+-dependent binding partners of the vesicular SNARE protein syb2 and uncovered copine-6 as a key syb2-interacting protein and a specific regulator of spontaneous neurotransmitter release. Our results show that copine-6 is partly localized to hippocampal presynaptic terminals where it colocalizes with syb2 and the synaptic vesicle protein synapsin 1. In functional experiments, overexpression of full-length copine-6 decreases the frequency of spontaneous neurotransmitter release events and shRNA-mediated knockdown of endogenous copine-6 increases the frequency of spontaneous release without altering evoked neurotransmission. Furthermore, our experiments revealed that syb2, especially its C-terminal region, was required for copine-6-mediated regulation of spontaneous release as targeted disruption of syb2–copine-6 interaction blocked copine-6's impact on release events. Importantly, the suppression of spontaneous release by copine-6 required intracellular Ca2+ as it was abolished by treatment of neurons with the membrane-permeable Ca2+ chelator BAPTA-AM. In contrast, knockdown of copine-6 augmented the increase seen in spontaneous release events after treatment with caffeine, which triggers Ca2+ release from intracellular stores. We also evaluated the molecular specificity of the copine-6-dependent regulation of spontaneous release by expressing syb2 mutants on the syb2 KO background. In this setting, we found that overexpression of copine-6 did not suppress spontaneous release in syb2-deficient neurons. Moreover, the decrease in spontaneous release rate seen after copine-6 overexpression depended on the C-terminal WW region of syb2 as point mutations at the vicinal tryptophan residues W89A/W90A abolished the effect. Interestingly, copine-6 loss of function on the syb2 KO background caused an increase in the normally low spontaneous release rate seen in these neurons. This effect is presumably due to desuppression of spontaneous release mediated by alternative vesicular SNAREs, such as vti1a, which also binds to copine-6 (Ramirez et al., 2012), or possibly synaptobrevin1, which has recently been shown to mediate some spontaneous release in the absence of syb2 (Liu et al., 2011; Zimmermann et al., 2014).
Copines are an evolutionarily conserved family of proteins that bind negatively charged lipids and other proteins in a manner that is highly regulated by Ca2+ (Tomsig and Creutz, 2002). Copines were initially discovered in Paramecium where they constitute the bulk of Ca2+ binding proteins in the cell similar to the annexin, a highly abundant Ca2+-binding protein in other organisms (Creutz et al., 1998). Similar to synaptotagmins, copine-6 contains tandem C2 domains (C2A and C2B). Accordingly, in our biochemistry and electrophysiological experiments, robust binding of copine-6 to its target proteins, in particular SNAREs, and its impact on spontaneous release events were significantly disrupted when Ca2+ was eliminated. Our results are consistent with earlier studies on copine-6 function. For instance, among the 9 known copine genes in humans and other mammalian copines, 6 and 7 are highly enriched in postnatal brain (Nakayama et al., 1998), especially in the somatodendritic regions of the hippocampus and the olfactory bulb (Nakayama et al., 1999). Copine-6 levels increase 20- to 25-fold during maturation of axonal projections (Yamatani et al., 2010), suggesting a potential presynaptic function. Our results do not exclude a potential postsynaptic role for copine-6 in receptor trafficking and plasticity as proposed by earlier work (Nakayama et al., 1998; Reinhard et al., 2016; Burk et al., 2017). Indeed, recent studies have identified a critical role for syb2 in postsynaptic receptor trafficking, especially during long-term potentiation and homeostatic synaptic plasticity (Jurado et al., 2013; Arendt et al., 2015). In agreement with this premise, in our experiments, we also detected a statistically significant increase in postsynaptic response amplitudes after expression of syb2 mutants that cannot interact with copine-6 (Fig. 5).
Previous studies have shown that the heterogeneous distribution of synaptic vesicle-associated SNARE proteins underlies the functional diversity among synaptic vesicles (Hua et al., 2011; Raingo et al., 2012; Ramirez et al., 2012; Bal et al., 2013). These studies have suggested that vesicular SNARE protein VAMP4 specifically marks a pool of synaptic vesicles that give rise to evoked asynchronous neurotransmitter release (Raingo et al., 2012), whereas alternative SNAREs vti1a and VAMP7 contribute to spontaneous neurotransmission (Hua et al., 2011; Ramirez et al., 2012; Bal et al., 2013). In a recent study, our group demonstrated that secreted glycoprotein Reelin can act presynaptically, resulting in robust rapid enhancement of spontaneous neurotransmitter release without affecting properties of evoked neurotransmission (Bal et al., 2013). These results suggest a novel presynaptic regulatory pathway that uses the heterogeneity of synaptic vesicle-associated SNAREs and selectively regulate spontaneous neurotransmission, indicating that syb2-independent neurotransmitter release is physiologically relevant. Although these observations provided molecular evidence of how distinct synaptic vesicle-associated SNAREs contribute to the diversity of neurotransmitter release properties, they did not address how syb2-mediated spontaneous release events may be differentially regulated from their evoked counterparts. Therefore, the findings we present here provide insight into this question by demonstrating that copine-6 primarily targets syb2-dependent SNARE machinery and in turn selectively regulates spontaneous release. These results indicate that presynaptic terminals possess a network of synaptic proteins that include synaptotagmin1, complexins, and doc2, along with copine-6, that mediate differential Ca2+-dependent regulation of spontaneous release events from their evoked counterparts, thus enabling autonomous modulation of spontaneous neurotransmission (Xu et al., 2009; Groffen et al., 2010; Pang et al., 2011; Dai et al., 2015). However, unlike synaptotagmins and complexins, but similar to doc2, copine-6 selectively targets spontaneous release events albeit in the opposite direction. Elevation in presynaptic Ca2+ levels is a well-characterized facilitator of spontaneous release (Schneggenburger and Rosenmund, 2015). Copine-6 provides a counterbalance to this facilitation by inhibiting syb2-mediated spontaneous fusion in a Ca2+-dependent manner. This negative feedback regulation by copine-6 may constitute a safeguard against excessive increases in Ca2+-dependent spontaneous release and ensure stability of neuronal networks.
Footnotes
This work was supported by National Institutes of Health Grant MH066198 to E.T.K. We thank Brent Trauterman for excellent technical assistance; and other members of the E.T.K. laboratory for helpful comments.
The authors declare no competing financial interests.
References
- Arendt KL, Zhang Y, Jurado S, Malenka RC, Südhof TC, Chen L (2015) Retinoic acid and LTP recruit postsynaptic AMPA receptors using distinct SNARE-dependent mechanisms. Neuron 86:442–456. 10.1016/j.neuron.2015.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET (2008) Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci 28:10151–10166. 10.1523/JNEUROSCI.2432-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal M, Leitz J, Reese AL, Ramirez DM, Durakoglugil M, Herz J, Monteggia LM, Kavalali ET (2013) Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron 80:934–946. 10.1016/j.neuron.2013.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk K, Ramachandran B, Ahmed S, Hurtado-Zavala JI, Awasthi A, Benito E, Faram R, Ahmad H, Swaminathan A, McIlhinney J, Fischer A, Perestenko P, Dean C (2017) Regulation of dendritic spine morphology in hippocampal neurons by copine-6. Cereb Cortex 2017:1–18. 10.1093/cercor/bhx009 [DOI] [PubMed] [Google Scholar]
- Chung C, Barylko B, Leitz J, Liu X, Kavalali ET (2010) Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J Neurosci 30:1363–1376. 10.1523/JNEUROSCI.3427-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Kavalali ET (2015) Molecular underpinnings of synaptic vesicle pool heterogeneity. Traffic 16:338–364. 10.1111/tra.12262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creutz CE, Tomsig JL, Snyder SL, Gautier MC, Skouri F, Beisson J, Cohen J (1998) The copines, a novel class of C2 domain-containing, calcium-dependent, phospholipid-binding proteins conserved from paramecium to humans. J Biol Chem 273:1393–1402. 10.1074/jbc.273.3.1393 [DOI] [PubMed] [Google Scholar]
- Dai J, Chen P, Tian H, Sun J (2015) Spontaneous vesicle release is not tightly coupled to voltage-gated calcium channel-mediated Ca2+ influx and is triggered by a Ca2+ sensor other than synaptotagmin-2 at the juvenile mice calyx of held synapses. J Neurosci 35:9632–9637. 10.1523/JNEUROSCI.0457-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Khvotchev M, Liu S, Huryeva I, Südhof TC, Rizo J (2007) Munc18-1 binds directly to the neuronal SNARE complex. Proc Natl Acad Sci U S A 104:2697–2702. 10.1073/pnas.0611318104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatt P, Katz B (1952) Spontaneous subthreshold activity at motor nerve endings. J Physiol 117:109–128. 10.1113/jphysiol.1952.sp004735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredj NB, Burrone J (2009) A resting pool of vesicles is responsible for spontaneous vesicle fusion at the synapse. Nat Neurosci 12:751–758. 10.1038/nn.2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M (2010) Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 327:1614–1618. 10.1126/science.1183765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z, Leal-Ortiz S, Foss SM, Waites CL, Garner CC, Voglmaier SM, Edwards RH (2011) v-SNARE composition distinguishes synaptic vesicle pools. Neuron 71:474–487. 10.1016/j.neuron.2011.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJ, Südhof TC, Malenka RC (2013) LTP requires a unique postsynaptic SNARE fusion machinery. Neuron 77:542–558. 10.1016/j.neuron.2012.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG (2014) Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol 76:333–363. 10.1146/annurev-physiol-021113-170338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET. (2015) The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16:5–16. 10.1038/nrn3875 [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Klingauf J, Tsien RW (1999) Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci U S A 96:12893–12900. 10.1073/pnas.96.22.12893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitz J, Kavalali ET (2011) Ca(2)(+) influx slows single synaptic vesicle endocytosis. J Neurosci 31:16318–16326. 10.1523/JNEUROSCI.3358-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitz J, Kavalali ET (2014) Fast retrieval and autonomous regulation of single spontaneously recycling synaptic vesicles. Elife 3:e03658. 10.7554/eLife.03658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Chanaday NL, Xu W, Kavalali ET (2017) Synaptotagmin-1- and synaptotagmin-7-dependent fusion mechanisms target synaptic vesicles to kinetically distinct endocytic pathways. Neuron 93:616–631.e3. 10.1016/j.neuron.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sugiura Y, Lin W (2011) The role of Synaptobrevin1/VAMP1 in Ca2+-triggered neurotransmitter release at the mouse neuromuscular junction. J Physiol 589:1603–1618. 10.1113/jphysiol.2010.201939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC (2009) Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323:516–521. 10.1126/science.1166505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Yaoi T, Kuwajima G (1999) Localization and subcellular distribution of N-copine in mouse brain. J Neurochem 72:373–379. 10.1046/j.1471-4159.1999.0720373.x [DOI] [PubMed] [Google Scholar]
- Nakayama T, Yaoi T, Yasui M, Kuwajima G (1998) N-copine: a novel two C2-domain-containing protein with neuronal activity-regulated expression. FEBS Lett 428:80–84. 10.1016/S0014-5793(98)00497-9 [DOI] [PubMed] [Google Scholar]
- Nosyreva E, Kavalali ET (2010) Activity-dependent augmentation of spontaneous neurotransmission during endoplasmic reticulum stress. J Neurosci 30:7358–7368. 10.1523/JNEUROSCI.5358-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC (2011) Doc2 supports spontaneous synaptic transmission by a Ca2+-independent mechanism. Neuron 70:244–251. 10.1016/j.neuron.2011.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quetglas S, Iborra C, Sasakawa N, De Haro L, Kumakura K, Sato K, Leveque C, Seagar M (2002) Calmodulin and lipid binding to synaptobrevin regulates calcium-dependent exocytosis. EMBO J 21:3970–3979. 10.1093/emboj/cdf404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingo J, Khvotchev M, Liu P, Darios F, Li YC, Ramirez DM, Adachi M, Lemieux P, Toth K, Davletov B, Kavalali ET (2012) VAMP4 directs synaptic vesicles to a pool that selectively maintains asynchronous neurotransmission. Nat Neurosci 15:738–745. 10.1038/nn.3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Andersson S, Russell DW (2008) Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol 507:1676–1693. 10.1002/cne.21605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Khvotchev M, Trauterman B, Kavalali ET (2012) Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron 73:121–134. 10.1016/j.neuron.2011.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard JR, Kriz A, Galic M, Angliker N, Rajalu M, Vogt KE, Ruegg MA (2016) The calcium sensor copine-6 regulates spine structural plasticity and learning and memory. Nat Comm 7:11613. 10.1038/ncomms11613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelo NH, Kamin D, Truckenbrodt S, Wong AB, Reuter-Jessen K, Reisinger E, Moser T, Rizzoli SO (2014) A new probe for super-resolution imaging of membranes elucidates trafficking pathways. J Cell Biol 205:591–606. 10.1083/jcb.201402066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara Y, Virmani T, Deák F, Liu X., Kavalalí ET (2005) An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 45:563–573. 10.1016/j.neuron.2004.12.056 [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Rosenmund C (2015) Molecular mechanisms governing Ca2+ regulation of evoked and spontaneous release. Nat Neurosci 18:935–941. 10.1038/nn.4044 [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET (2001) SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294:1117–1122. 10.1126/science.1064335 [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S (2003) Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 38:929–939. 10.1016/S0896-6273(03)00322-2 [DOI] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323:474–477. 10.1126/science.1161748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, et al. (2006) Molecular anatomy of a trafficking organelle. Cell 127:831–846. 10.1016/j.cell.2006.10.030 [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC (2006) A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126:1175–1187. 10.1016/j.cell.2006.08.030 [DOI] [PubMed] [Google Scholar]
- Tomsig JL, Creutz CE (2002) Copines: a ubiquitous family of Ca(2+)-dependent phospholipid-binding proteins. Cell Mol Life Sci 59:1467–1477. 10.1007/s00018-002-8522-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomsig JL, Snyder SL, Creutz CE (2003) Identification of targets for calcium signaling through the copine family of proteins. characterization of a coiled-coil copine-binding motif. J Biol Chem 278:10048–10054. 10.1074/jbc.M212632200 [DOI] [PubMed] [Google Scholar]
- Umbrecht-Jenck E, Demais V, Calco V, Bailly Y, Bader MF, Chasserot-Golaz S (2010) S100A10-mediated translocation of annexin-A2 to SNARE proteins in adrenergic chromaffin cells undergoing exocytosis. Traffic 11:958–971. 10.1111/j.1600-0854.2010.01065.x [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH (2006) Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron 51:71–84. 10.1016/j.neuron.2006.05.027 [DOI] [PubMed] [Google Scholar]
- Xu J, Brewer KD, Perez-Castillejos R, Rizo J (2013) Subtle interplay between synaptotagmin and complexin binding to the SNARE complex. J Mol Biol 425:3461–3475. 10.1016/j.jmb.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC (2009) Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci 12:759–766. 10.1038/nn.2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Südhof TC (2012) Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron 73:990–1001. 10.1016/j.neuron.2011.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Su L, Rizo J (2010) Binding of Munc18-1 to synaptobrevin and to the SNARE four-helix bundle. Biochemistry 49:1568–1576. 10.1021/bi9021878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamatani H, Kawasaki T, Mita S, Inagaki N, Hirata T (2010) Proteomics analysis of the temporal changes in axonal proteins during maturation. Dev Neurobiol 70:523–537. 10.1002/dneu.20794 [DOI] [PubMed] [Google Scholar]
- Zimmermann J, Trimbuch T, Rosenmund C (2014) Synaptobrevin 1 mediates vesicle priming and evoked release in a subpopulation of hippocampal neurons. J Neurophysiol 112:1559–1565. 10.1152/jn.00340.2014 [DOI] [PubMed] [Google Scholar]