

 Reaction of [3-N2-o-C2B10H11][BF4] with various kinds of nucleophiles gives a very broad spectrum of cage B(3)-substituted o-carborane derivatives, 3-X-o-C2B10H11 (X = OH, SCN, NH2, NO2, N3, CF3, PO(C6H5)2, etc.), and serves as a simple and efficient method for multiple functionalization of o-carborane.
Reaction of [3-N2-o-C2B10H11][BF4] with various kinds of nucleophiles gives a very broad spectrum of cage B(3)-substituted o-carborane derivatives, 3-X-o-C2B10H11 (X = OH, SCN, NH2, NO2, N3, CF3, PO(C6H5)2, etc.), and serves as a simple and efficient method for multiple functionalization of o-carborane.
Abstract
A simple and efficient method for selective cage B(3) multiple functionalization of o-carborane is described. Reaction of [3-N2-o-C2B10H11][BF4] with various kinds of nucleophiles gave a very broad spectrum of cage B(3)-substituted o-carborane derivatives, 3-X-o-C2B10H11 (X = OH, SCN, NH2, NO2, N3, CF3, PO(C6H5)2, etc). This reaction may serve as another efficient [18F]-radiolabeling method of carborane clusters for positron emission tomography applications.
Introduction
Carboranes, 3-dimensional relatives of benzenes, are a class of boron hydride clusters in which one or more BH vertices are replaced by CH units.1 Carboranes and organic molecules display different electronic, physical, chemical and geometrical properties, which highlights the feasibility or necessity to produce hybrid molecules incorporating both of these two types of fragments.2a,b Indeed, functional carboranes are now finding a broad range of applications encompassing organic synthesis, polymers, catalysis, metal–organic frameworks, electronic devices and more.2c–r As a result, considerable attention has been directed towards the functionalization of carborane molecules.3 In contrast to the relatively well-studied methods for cage carbon functionalization of carboranes,1,4 selective cage boron functionalization of carboranes still represents a challenging task and developing new methodologies for selective boron derivatization is eagerly desired.5,6
Diazonium compounds (R-N2+X–) constitute an important group of intermediates that have found wide applications in organic synthesis.7 Many prominent named reactions associated with aryl diazonium salts have been developed since their first discovery in 1858.8 In sharp contrast, diazonium derivatives of carboranes are little known.9 It has been documented that o-carboranyl diazonium salts are non-isolable, can only be prepared in situ and undergo substitution reactions with the reaction solvent, usually inorganic acids, in the presence of copper salts.10 Recently, we have reported the synthesis of a stable and isolable o-carboranyl diazonium salt, [3-N2-o-C2B10H11][BF4].11a It serves as an ideal precursor for the generation of 1,3-dehydro-o-carborane11 and boron-centered carboranyl radicals.12
On the other hand, it has been reported that B(9)-carboranyl iodonium salt can react with nucleophiles.13 Very recently, a similar approach for the functionalization of closo-borates via nucleophilic substitution reactions of the corresponding iodonium zwitterions has been developed.14 However, in these cases, only limited nucleophiles are tolerated and the chemoselectivity of the reaction is highly dependent on the nature of the nucleophiles or the reaction conditions.13c,14 As the most widely investigated among the carborane family, general and versatile methods for selected cage boron functionalization of o-carboranes still remain very limited.5 Previously, our group has reported that utilizing 3-diazonium-o-carborane tetrafluoroborate as the starting material, selective B(3)-arylation of o-carborane can be achieved via the aromatic ene reaction of 1,3-dehydro-o-carborane or a visible-light mediated B–C(sp2) coupling of a carboranyl boron-centered radical. However, the substrate scope is only limited to arenes.11a,12
Considering that dinitrogen is an excellent leaving group, carboranyl diazonium salt may easily undergo a substitution reaction in the presence of a nucleophile. Moreover, compared to aryl diazonium salts, carboranyl diazonium salt may exhibit higher reactivity due to the electron deficient nature of the boron atom and lack of conjugation between the carborane cage and the diazonium group. Herein, we report a proof-of-concept study demonstrating that carboranyl diazonium salt can serve as a powerful synthon for selective cage boron functionalization of o-carboranes (Scheme 1).
Scheme 1. Functionalization of arene and o-carborane via diazonium salt.

Results and discussion
3-Diazonium-o-carborane tetrafluoroborate ([3-N2-o-C2B10H11][BF4]; 1) was prepared in 77% isolated yield, by treatment of 3-amino-o-carborane with 1.5 equivalents of nitrosonium tetrafluoroborate.11a It is noted that the stability of 1 is dependent upon the counterions used and BF4– offers the highest thermal stability of the salt among the anions examined, such as PF6– and Cl–. A 1.0 g batch of carboranyl diazonium salt 1 stored at –5 °C showed no signs of decomposition over four months.
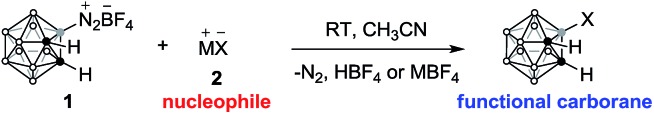
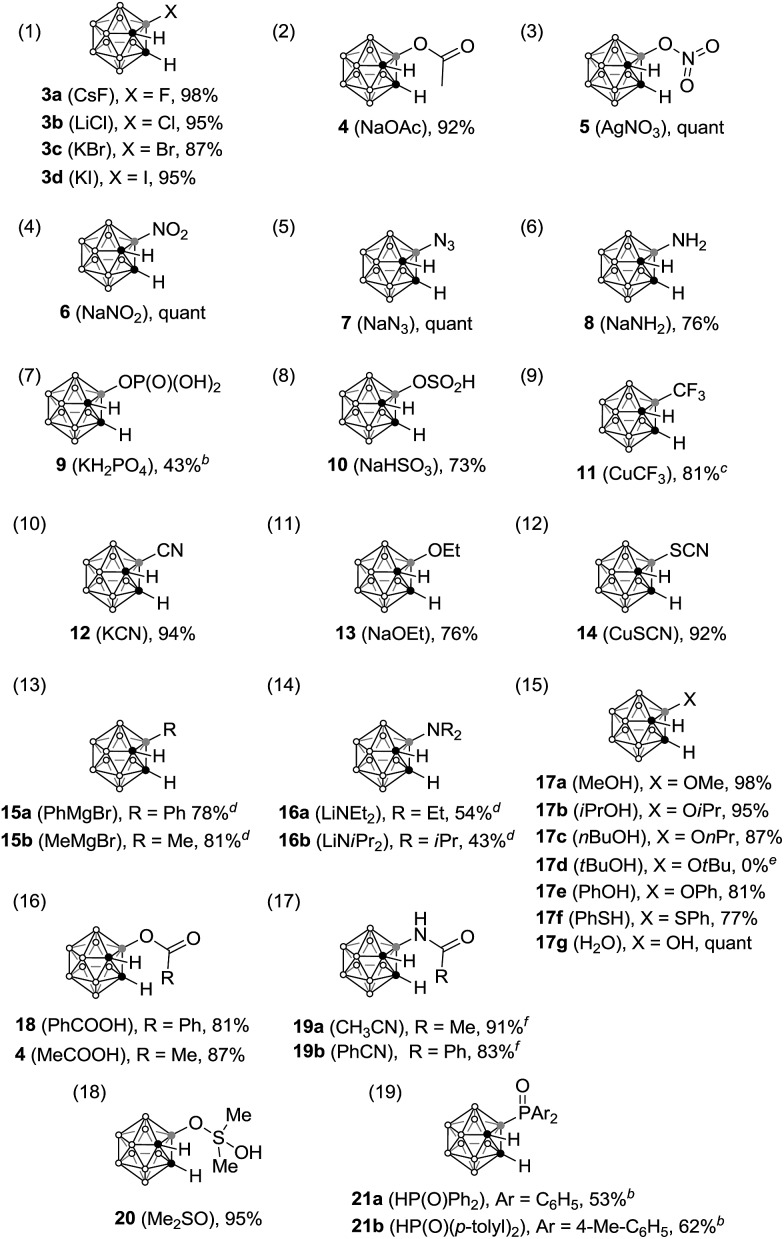
With this stable precursor in hand, we found that precursor 1 reacted rapidly with various nucleophiles (2) in acetonitrile, providing the corresponding B(3)-substituted o-carboranes in good to excellent yields (Table 1). Treatment of 1 with 1 equivalent of strong (charged) nucleophiles, such as halide ions, gave the corresponding halogenated carboranes in excellent yields in <5 min (Table 1, entry 1).
Table 1. Reaction of nucleophiles with precursor 1 a .
|
|
aReaction conditions: precursor 1 (0.1 mmol) was treated with nucleophile 2 (0.1 mmol for inorganic salt and phosphine oxide; 1.0 mmol for alcohol, acid and ketone; 0.4 mmol for Grignard reagent and lithium amide; nitriles were utilized as solvent) in CH3CN solution for 5 min; yields of isolated products are given.
bDeboronation occurred during purification.
cCuCF3 was prepared in situ from TMSCF3, CuSCN and Cs2CO3 in acetonitrile.16
d–78 °C, THF, 15 min.
eThe only product was 3-F-C2B10H11.
f50 °C, 6 h.
A large variety of nucleophiles, including inorganic salts, water, alcohols, acids, organometallic reagents, ketones, nitriles and phosphine oxides are compatible with this reaction, resulting in the formation of B–C, B–N, B–P, B–O, B–S and B–X (X = F, Cl, Br, I) bonds. More importantly, various functional groups that were previously unable to be introduced into the carborane unit can now be installed in a very simple and efficient manner. For instance, common functional groups can be easily installed on the o-carborane cage boron position using simple inorganic salts in 5 min, affording the corresponding B(3)-functionalized o-carboranes 3–14 (Table 1, entries 1–12). Reaction of precursor 1 with Grignard reagents or lithium amides also gave the B(3)-substituted o-carboranes in moderate to good yields (Table 1, entries 13 and 14).
Weak nucleophiles also work well in this reaction. For instance, in the presence of 10 equivalents of alcohols or water, B(3)-oxygenated carboranes 17 were produced in 81–98% yield (Table 1, entry 15). However, no desired product was observed for tert-butyl alcohol, probably due to the steric hindrance imposed by the tert-butyl group. Instead, 3-F-o-carborane 3a, generated via decomposition of precursor 1, was the only isolated product. Compared to other neutral nucleophiles, the reaction of nitriles is slower even at elevated temperature (Table 1, entry 17).15 The reactivity of precursor 1 towards nucleophiles containing P O and S O double bonds was also examined. For example, the reaction of dimethyl sulfoxide furnished compound 20 after hydrolysis (Table 1, entry 18). Although 31P and 11B NMR spectra indicated high conversions, reactions with phosphine oxide nucleophiles resulted in lower yields due to the deboronation of the product during the purification process (Table 1, entry 19).17 Notably, this metal-free approach provides a rare example of B-carboranyl phosphines.18 The rich chemistry of the carboranyl diazonium salt towards various nucleophiles suggests that it can serve as a very promising synthon for selective cage boron functionalization of o-carboranes. It is noteworthy that the reaction also works well when performed on a 0.5 mmol scale.17
All new compounds were fully characterized by 1H, 13C, and 11B NMR spectroscopy as well as HRMS spectrometry.
The molecular structures of compounds 4 and 6 were further confirmed by single-crystal X-ray analyses.17
Interestingly, precursor 1 did not react with anhydrous ether (Scheme 2, eqn (1)); however, it reacted rapidly with wet ethereal solvents. For instance, upon treatment with wet diethyl ether, compound 13, resulting from the C–O bond cleavage of ether, was isolated in 95% yield (Scheme 2, eqn (2)). When treated with anhydrous THF, polymerization occurred, leading to gel formation, which suggests the intermediacy of cationic species (Scheme 2, eqn (3)).17 When tert-butyl methyl ether was examined under the same reaction conditions, compound 17a, bearing a methoxy substituent at B(3) position, was formed quantitatively (Scheme 2, eqn (4)), which may shed some light on the reaction mechanism (vide infra).
Scheme 2. Reaction of precursor 1 with ethers.

The nucleophilic reaction of the carboranyl diazonium salt was expected to proceed through an SN1 type of mechanism (Scheme 3).9,19 Although precursor 1 is stable in solution, it can undergo nucleophile-induced heterolytic B–N bond cleavage, producing a boronium intermediate A.14 Similar to the reaction of the dinitrogen derivatives of closo-borates, the rate-determining step is the B–N bond cleavage.9 The resultant reactive boronium intermediate can be trapped by various nucleophiles. For instance, when charged nucleophiles such as inorganic salts were employed as nucleophiles, the corresponding substituted compounds 3–14 were formed in very high yields within 5 min. If the nucleophiles are strong bases, the addition products 15–17 might also be produced via the intermediacy of 1,3-dehydro-o-carborane intermediates.11 Addition of neutral nucleophiles to the boronium intermediate A, alcohols for example, generates an oxonium ion B, which is further deprotonated by the BF4– anion to afford 17. For weakly nucleophilic ethers, such as tert-butyl methyl ether, no reaction occurs under anhydrous conditions. However, in the presence of a catalytic amount of water, the oxygenated products, such as 17a, were produced within 5 min. This reaction probably proceeds through a sequence of C–H bond cleavage/isobutylene elimination in intermediate C, which is generated by the nucleophilic addition of the ether to the naked boron vertex of intermediate A.20 The role of the catalytic amount of water is to facilitate the isobutylene elimination that was detected by GC-MS analyses. The formation of HBF4 was also confirmed by 11B and 19F NMR spectra.17
Scheme 3. Proposed reaction pathways.

The present strategy provides a straightforward and practical access to cage boron functionalized o-carboranes. It has been documented that 18F-labelled (t1/2 = 109.8 min) carboranes are promising radiotracers in Positron Emission Tomography (PET). Previously, 18[F]-fluorination of o-carborane was achieved by nucleophilic substitution of a B(9)-carboranyl iodonium bromide.21 However, the overall synthesis time of 20 min limits its possible application, probably due to the low reactivity of the carboranyl iodonium bromide. As a proof of concept, we opted to improve the efficiency of the fluorination process by using precursor 1 as the starting material. Under similar reaction conditions to those reported in the literature,17 the fluorinated product 3a was formed quantitatively within 1 min and it can be easily purified (eqn (5)).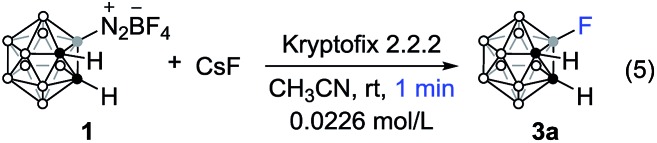
Conclusions
A practical method for selective cage boron functionalization of o-carborane has been developed. By utilizing B-carboranyl diazonium salt as a synthon, a large class of o-carborane derivatives bearing previously inaccessible functional groups can now be efficiently prepared, which may find applications in materials sciences.
This work demonstrates that B-carboranyl diazonium salt can serve not only as a source of boron-centered radicals12 or 1,3-dehydro-o-carborane,11 but also as a source of boronium cations in the presence of nucleophiles.9,20 These intermediates serve different purposes and are complementary to each other, building up a useful toolbox for cage boron functionalization of o-carboranes.
Compared to aryl diazonium salts, the exceptionally high reactivity of B-carboranyl diazonium salt may be due to the lack of conjugation between the carborane cage and the diazonium group. Such a method may find useful applications in the efficient and fast synthesis of 18F-labelled o-carborane derivatives for medical applications.21
Supplementary Material
Acknowledgments
The work described in this paper was supported by grants from the Research Grants Council of the Hong Kong Special Administration Region (Project No. CUHK7/CRF/12G and 14304115) and NSFC/RGC Joint Research Scheme (Project No. N_CUHK442/14).
Footnotes
†Electronic supplementary information (ESI) available: Experimental details, complete characterization data, and X-ray data in CIF format for 4 and 6. CCDC 1473011 and 1473012. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01566b
References
- (a) Grimes R. N., Carboranes, Academic Press, 2nd edn, New York, 2011. [Google Scholar]; (b) Hosmane N. S., Boron Science: New Technologies and Applications, CRC Press, Boca Raton, 2012. [Google Scholar]; (c) Zhang J., Xie Z. Acc. Chem. Res. 2014;47:1623–1633. doi: 10.1021/ar500091h. [DOI] [PubMed] [Google Scholar]
- For selected reviews and examples on applications of o-carboranes, see: ; (a) Grimes R. N. Dalton Trans. 2015;44:5939–5956. doi: 10.1039/c5dt00231a. [DOI] [PubMed] [Google Scholar]; (b) Xie Z. Acc. Chem. Res. 2003;36:1–9. doi: 10.1021/ar010146i. [DOI] [PubMed] [Google Scholar]; (c) Mukherjee S., Thilagar P. Chem. Commun. 2016;52:1070–1093. doi: 10.1039/c5cc08213g. [DOI] [PubMed] [Google Scholar]; (d) Li X., Yan H., Zhao Q. Chem.–Eur. J. 2016;22:1888–1898. doi: 10.1002/chem.201503456. [DOI] [PubMed] [Google Scholar]; (e) Housecroft C. E. J. Organomet. Chem. 2015;798:218–228. [Google Scholar]; (f) Hawthorne M. F., Pushechnikov A. Pure Appl. Chem. 2012;84:2279–2288. [Google Scholar]; (g) Scholz M., Hey-Hawkins E. Chem. Rev. 2011;111:7035–7062. doi: 10.1021/cr200038x. [DOI] [PubMed] [Google Scholar]; (h) Reed C. A. Acc. Chem. Res. 2010;43:121–128. doi: 10.1021/ar900159e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Armstrong A. F., Valliant J. F. Dalton Trans. 2007:4240–4251. doi: 10.1039/b709843j. [DOI] [PubMed] [Google Scholar]; (j) Valliant J. F., Guenther K. J., King A. S., Morel P., Schaffer P., Sogbein O. O., Stephenson K. A. Coord. Chem. Rev. 2002;232:173–230. [Google Scholar]; (k) Xie Z. Coord. Chem. Rev. 2002;231:23–46. [Google Scholar]; (l) Schwartz J. J., Mendoza M. A., Wattanatorn N., Zhao Y., Nguyen V. T., Spokoyny A. M., Mirkin C. A., Baše T., Weiss P. S. J. Am. Chem. Soc. 2016;138:5957–5967. doi: 10.1021/jacs.6b02026. [DOI] [PubMed] [Google Scholar]; (m) Kirlikovali K. O., Axtell J. C., Gonzalez A., Phung A. C., Khan S. I., Spokoyny A. M. Chem. Sci. 2016 doi: 10.1039/c6sc01146b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Eleazer B. J., Smith M. D., Peryshkov D. V. Organometallics. 2016;35:106–112. [Google Scholar]; (o) Wong Y. O., Smith M. D., Peryshkov D. V. Chem.–Eur. J. 2016;22:6764–6767. doi: 10.1002/chem.201601194. [DOI] [PubMed] [Google Scholar]; (p) Estrada J., Lugo C. A., McArthur S. G., Lavallo V. Chem. Commun. 2016;52:1824–1826. doi: 10.1039/c5cc08377j. [DOI] [PubMed] [Google Scholar]; (q) McArthur S. G., Geng L., Lavallo V. Inorg. Chem. Front. 2015;2:1101–1104. [Google Scholar]; (r) Estrada J., Woen D. H., Tham F. S., Miyake G. M., Lavallo V. Inorg. Chem. 2015;54:5142–5144. doi: 10.1021/acs.inorgchem.5b00576. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Kracke G. R., VanGordon M. R., Sevryugina Y. V., Kueffer P. J., Kabytaev K., Jalisatgi S. S., Hawthorne M. F. ChemMedChem. 2015;10:62–67. doi: 10.1002/cmdc.201402369. [DOI] [PubMed] [Google Scholar]; (b) Zhao D., Zhang J., Xie Z. J. Am. Chem. Soc. 2015;137:13938–13942. doi: 10.1021/jacs.5b09074. [DOI] [PubMed] [Google Scholar]; (c) Zhao D., Zhang J., Xie Z. J. Am. Chem. Soc. 2015;137:9423–9428. doi: 10.1021/jacs.5b05426. [DOI] [PubMed] [Google Scholar]; (d) Zhao D., Zhang J., Xie Z. Angew. Chem., Int. Ed. 2014;53:12902–12906. doi: 10.1002/anie.201409141. [DOI] [PubMed] [Google Scholar]; (e) Wee K. R., Cho Y. J., Song J. K., Kang S. O. Angew. Chem., Int. Ed. 2013;52:9682–9685. doi: 10.1002/anie.201304321. [DOI] [PubMed] [Google Scholar]; (f) Brusselle D., Bauduin P., Girard L., Zaulet A., Viñas C., Teixidor F., Ly I., Diat O. Angew. Chem., Int. Ed. 2013;52:12114–12118. doi: 10.1002/anie.201307357. [DOI] [PubMed] [Google Scholar]; (g) Wee K. R., Cho Y. J., Jeong S., Kwon S., Lee J. D., Suh I. H., Kang S. O. J. Am. Chem. Soc. 2012;134:17982–17990. doi: 10.1021/ja3066623. [DOI] [PubMed] [Google Scholar]; (h) Cioran A. M., Musteti A. D., Teixidor F., Krpetić ž., Prior I. A., He Q., Kiely C. J., Brust M., Viñas C. J. Am. Chem. Soc. 2012;134:212–221. doi: 10.1021/ja203367h. [DOI] [PubMed] [Google Scholar]; (i) Wee K. R., Han W. S., Cho D. W., Kwon S., Pac C., Kang S. O. Angew. Chem., Int. Ed. 2012;51:2677–2680. doi: 10.1002/anie.201109069. [DOI] [PubMed] [Google Scholar]; (j) Koshino M., Tanaka T., Solin N., Suenaga K., Isobe H., Nakamura E. Science. 2007;316:853. doi: 10.1126/science.1138690. [DOI] [PubMed] [Google Scholar]
- (a) Zhao D., Xie Z. Coord. Chem. Rev. 2016;314:14–33. [Google Scholar]; (b) Qiu Z., Xie Z. Dalton Trans. 2014;43:4925–4934. doi: 10.1039/c3dt52711e. [DOI] [PubMed] [Google Scholar]
- For reviews on the functionalization of cage borons, see: ; (a) Olid D., Nuñez R., Viñas C., Teixidor F. Chem. Soc. Rev. 2013;42:3318–3336. doi: 10.1039/c2cs35441a. [DOI] [PubMed] [Google Scholar]; (b) Qiu Z. Tetrahedron Lett. 2015;56:963–971. [Google Scholar]; (c) Xie Z. Sci. China: Chem. 2014;57:1061–1063. [Google Scholar]
- For recent examples on catalytic B-H functionalization of C2B10H12, CB11H12–, C2B9H112– and boranes, see: ; (a) Quan Y., Xie Z. Angew. Chem., Int. Ed. 2016;55:1295–1298. doi: 10.1002/anie.201507697. [DOI] [PubMed] [Google Scholar]; (b) Lyu H., Quan Y., Xie Z. Angew. Chem., Int. Ed. 2015;54:10623–10626. doi: 10.1002/anie.201504481. [DOI] [PubMed] [Google Scholar]; (c) Cao K., Huang Y., Yang J., Wu J. Chem. Commun. 2015;51:7257–7260. doi: 10.1039/c5cc01331c. [DOI] [PubMed] [Google Scholar]; (d) Quan Y., Xie Z. J. Am. Chem. Soc. 2015;137:3502–3505. doi: 10.1021/jacs.5b01169. [DOI] [PubMed] [Google Scholar]; (e) Qiu Z., Quan Y., Xie Z. J. Am. Chem. Soc. 2013;135:12192–12195. doi: 10.1021/ja405808t. [DOI] [PubMed] [Google Scholar]; (f) Johnson H. C., McMullin C. L., Pike S. D., Macgregor S. A., Weller A. S. Angew. Chem., Int. Ed. 2013;52:9776–9780. doi: 10.1002/anie.201304382. [DOI] [PubMed] [Google Scholar]; (g) Liu D., Dang L., Sun Y., Chan H.-S., Lin Z., Xie Z. J. Am. Chem. Soc. 2008;130:16103–16110. doi: 10.1021/ja8067098. [DOI] [PubMed] [Google Scholar]; (h) Molinos E., Brayshaw S. K., Kociok-Köhn G., Weller A. S. Organometallics. 2007;26:2370–2382. [Google Scholar]; (i) Rojo I., Teixidor F., Kivekäs R., Sillanpää R., Viñas C. J. Am. Chem. Soc. 2003;125:14720–14721. doi: 10.1021/ja037279e. [DOI] [PubMed] [Google Scholar]
- For selected reviews on aryl diazonium compounds, see: ; (a) Mo F., Dong G., Zhang Y., Wang J. Org. Biomol. Chem. 2013;11:1582–1593. doi: 10.1039/c3ob27366k. [DOI] [PubMed] [Google Scholar]; (b) Hari D. P., Konig B. Angew. Chem., Int. Ed. 2013;52:4734–4743. doi: 10.1002/anie.201210276. [DOI] [PubMed] [Google Scholar]; (c) Mahouche-Chergui S., Gam-Derouich S., Mangeney C., Chehimi M. M. Chem. Soc. Rev. 2011;40:4143–4166. doi: 10.1039/c0cs00179a. [DOI] [PubMed] [Google Scholar]; (d) Galli C. Chem. Rev. 1988;88:765–792. [Google Scholar]; (e) Heinrich M. R. Chem.–Eur. J. 2009;15:821–833. [Google Scholar]
- For selected examples, see: ; (a) Griess J. P. Ann. Chem. 1858;106:123–125. [Google Scholar]; (b) Sandmeyer T. Ber. Dtsch. Chem. Ges. 1884;17:1633–1635. [Google Scholar]; (c) Pschorr R. Ber. Dtsch. Chem. Ges. 1896;29:496–501. [Google Scholar]; (d) Gomberg M., Bachmann W. E. J. Am. Chem. Soc. 1924;46:2339–2343. [Google Scholar]; (e) Balz G., Schiemann G. Ber. Dtsch. Chem. Ges. B. 1927;60:1186–1190. [Google Scholar]; (f) Meerwein H., Buchner E., van Emsterk K. J. Prakt. Chem. 1939;152:237–266. [Google Scholar]; (g) Doyle M. P., Siegfried B., Dellaria Jr J. F. J. Org. Chem. 1977;42:2426–2431. [Google Scholar]; (h) Kikukawa K., Matsuda T. Chem. Lett. 1977:159–162. [Google Scholar]
- (a) Ringstrand B., Kaszyński P., Young V. G., Janoušek Z. Inorg. Chem. 2010;49:1166–1179. doi: 10.1021/ic9021323. [DOI] [PubMed] [Google Scholar]; (b) Ringstrand B., Kaszyński P., Young Jr V. G. Inorg. Chem. 2011;50:2654–2660. doi: 10.1021/ic102557s. [DOI] [PubMed] [Google Scholar]
- (a) Zakharkin L. I., Kalinin V. N. Izv. Akad. Nauk SSSR, Ser. Khim. 1967:2585–2586. [Google Scholar]; (b) Sevryugina Y., Julius R., Hawthorne M. F. Inorg. Chem. 2010;49:10627–10634. doi: 10.1021/ic101620h. [DOI] [PubMed] [Google Scholar]; (c) Kasar R. A., Knudsen G. M., Kahl S. B. Inorg. Chem. 1999;38:2936–2940. doi: 10.1021/ic990037o. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zakharkin L. I., Kalinin V. N., Gedymin V. V. J. Organomet. Chem. 1969;16:371–379. [Google Scholar]
- (a) Zhao D., Zhang J., Xie Z. Angew. Chem., Int. Ed. 2014;53:8488–8491. doi: 10.1002/anie.201405023. [DOI] [PubMed] [Google Scholar]; (b) Zhao D., Zhang J., Xie Z. Chem.–Eur. J. 2015;21:10334–10337. doi: 10.1002/chem.201501911. [DOI] [PubMed] [Google Scholar]
- Zhao D., Xie Z. Angew. Chem., Int. Ed. 2016;55:3166–3170. doi: 10.1002/anie.201511251. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Grushin V. V. Acc. Chem. Res. 1992;25:529–536. [Google Scholar]; (b) Grushin V. V., Demkina I. I., Tolstaya T. P. Inorg. Chem. 1991;30:4860–4863. [Google Scholar]; (c) Grushin V. V., Shcherbina T. M. J. Organomet. Chem. 1985;292:105–117. [Google Scholar]
- Kaszyński P., Ringstrand B. Angew. Chem., Int. Ed. 2015;54:6576–6581. doi: 10.1002/anie.201411858. [DOI] [PubMed] [Google Scholar]
- Ringstrand B., Kaszynski P., Acc. Chem. Res., 2013, 46 , 214 –225 , , and references therein . [DOI] [PubMed] [Google Scholar]
- Danoun G., Bayarmagnai B., Grunberg M. F., Gooßen L. J. Angew. Chem., Int. Ed. 2013;52:7972–7975. doi: 10.1002/anie.201304276. [DOI] [PubMed] [Google Scholar]
- For experimental details, safety precautions, complete characterization data, and X-ray data, see the ESI.
- For selected examples on B–P bond formation of o-carboranes, see: ; (a) Kabytaev K. Z., Safronov A. V., Sevryugina Y. V., Barnes C. L., Jalisatgi S. S., Hawthorne M. F. Inorg. Chem. 2015;54:4143–4150. doi: 10.1021/acs.inorgchem.5b00414. [DOI] [PubMed] [Google Scholar]; (b) Spokoyny A. M., Lewis C. D., Teverovskiy G., Buchwald S. L. Organometallics. 2012;31:8478–8481. doi: 10.1021/om301116x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bregadze V. I., Kosenko I. D., Lobanova I. A., Starikova Z. A., Godovikov I. A., Sivaev I. B. Organometallics. 2010;29:5366–5372. [Google Scholar]; (d) Bernard R., Cornu D., Luneau D., Naoufal D., Scharff J.-P., Miele P. J. Organomet. Chem. 2005;690:2745–2749. [Google Scholar]; (e) Viñas C., Núñez R., Teixidor F., Sillanpää R., Kivekäs R. Organometallics. 1999;18:4712–4717. [Google Scholar]; (f) Jasper S. A., Jones R. B., Mattern J., Huffman J. C., Todd L. J. Inorg. Chem. 1994;33:5620–5624. [Google Scholar]; (g) Teixidor F., Casabo J., Romerosa A. M., Viñas C., Rius J., Miravitlles C. J. Am. Chem. Soc. 1991;113:9895–9896. [Google Scholar]
- Jankowiak A., Baliński A., Harvey J. E., Mason K., Januszko A., Kaszyński P., Young V. G., Persoons A. J. Mater. Chem. C. 2013;1:1144–1159. [Google Scholar]
- Zharov I., Havlas Z., Orendt A. M., Barich D. H., Grant D. M., Fete M. G., Michl J. J. Am. Chem. Soc. 2006;128:6089–6100. doi: 10.1021/ja054256m. [DOI] [PubMed] [Google Scholar]
- Gona K. B., Gómez-Vallejo V., Padro D., Llop J. Chem. Commun. 2013;49:11491–11493. doi: 10.1039/c3cc46695g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.