

Abstract
Hydrogen sulfide (H2S) is a biologically-important small gaseous molecule that exhibits promising protective effects against a variety of physiological and pathological processes. To investigate the expanding roles of H2S in biology, researchers often use H2S donors to mimic enzymatic H2S synthesis or to provide increased H2S levels under specific circumstances. Aligned with the need for new broad and easily-modifiable platforms for H2S donation, we report here the preparation and H2S release kinetics from a series of isomeric caged-carbonyl sulfide (COS) compounds, including thiocarbamates, thiocarbonates, and dithiocarbonates, all of which release COS that is quickly converted to H2S by the ubiquitous enzyme carbonic anhydrase. Each donor is designed to release COS/H2S after the activation of a trigger by activation by hydrogen peroxide (H2O2). In addition to providing a broad palette of new, H2O2-responsive donor motifs, we also demonstrate the H2O2 dose-dependent COS/H2S release from each donor core, establish that release profiles can be modified by structural modifications, and compare COS/H2S release rates and efficiencies from isomeric core structures. Supporting our experimental investigations, we also provide computational insights into the potential energy surfaces for COS/H2S release from each platform. In addition, we also report initial investigations into dithiocarbamate cores, which release H2S directly upon H2O2-mediated activation. As a whole, the insights on COS/H2S release gained from these investigations provide a foundation for the expansion of the emerging area of responsive COS/H2S donors systems.
Graphical Abstract
INTRODUCTION
Hydrogen sulfide (H2S) is an important biological gaseous molecule that exhibits promising protective functions in mammals against various disease states. Due to its role in different physiological processes, H2S is now recognized as the third member of the gasotransmitter family, along with nitric oxide (NO) and carbon monoxide (CO).1–7 Endogenous H2S is generated enzymatically from cysteine and homocysteine by several enzymes, such as cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), cysteine aminotransferase (CAT), and 3-mercaptopyruvate sulfur transferase (3-MST), which can work either individually or in concert.8–12 Once produced, H2S causes relaxation of smooth muscle cells and can induce vasodilation by activating KATP channels.13–14 Endogenous H2S formation, as well as exogenous H2S administration, has been demonstrated to rescue cells, tissues, and organs from a variety of severe damages, thus raising the therapeutic potential of H2S-releasing materials.15–16 Although H2S is toxic at higher concentrations, lower levels often exert beneficial effects. For example, H2S exhibits anti-inflammatory activities in animal models13 and cardioprotective effects against oxidative stress by scavenging cellular reactive oxygen species (ROS).17–20
Due to the significant contributions of H2S in different (patho)physiological processes, researchers often use H2S releasing agents (H2S donors) to modulate H2S levels and enable new investigations.21–28 Common commercially-available H2S sources, such as sodium sulfide (Na2S) and sodium hydrogen sulfide (NaHS), generate H2S immediately upon addition to water and have been widely used in H2S studies. Although convenient, the instantaneous and uncontrollable H2S release from these salts does not mimic controlled enzymatic H2S synthesis and can often lead to contradictory results (i.e. anti-inflammation and pro-inflammation effects) depending on experimental conditions, handling, or commercial sources.29–30 Complementing these inorganic salts, garlic-derived polysulfide compounds, such as diallyl trisulfide (DATS, Figure 1), release H2S in the presence of reduced glutathione (GSH), and the resultant H2S from such compounds has been demonstrated to exhibit protective activities in animal models.31 Because such compounds generate persulfides en-route to H2S release, it remains unclear whether the observed protective effects are due to H2S alone or whether other sulfur-containing intermediates or products contribute to the observed activities.
Figure 1.
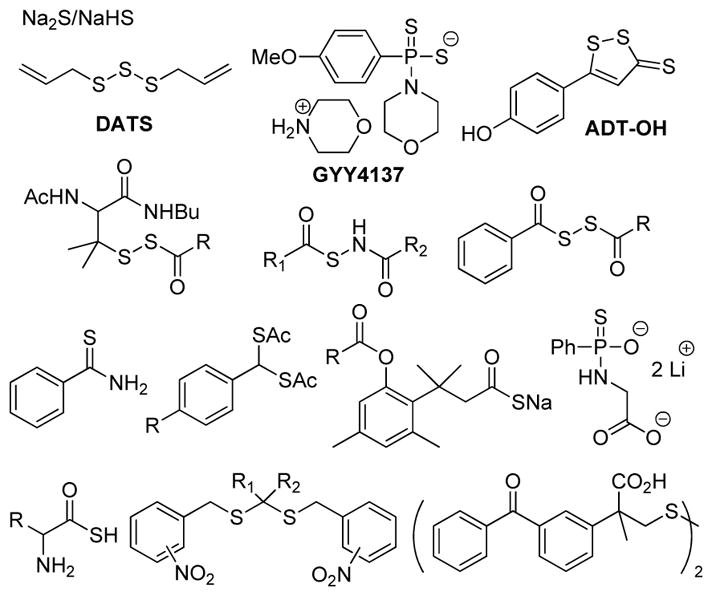
Selected small molecule H2S donors and motifs.
In addition to naturally-occurring H2S donors, researchers have also developed different classes of synthetic H2S releasing molecules with different properties and release mechanisms.21–23,25 For example, GYY4137, synthesized from Lawesson’s reagent, has been used widely as a hydrolysis-based H2S donor in systems ranging from simple cell culture experiments to animal models.32–37 Although GYY4137 exerts H2S-related outcomes in a wide array of systems, its H2S release is inefficient and unreacted GYY4137, or its H2S-depleted analog, may cause other side effects. Furthering the potential biomedical applications of synthetic donors, H2S-releasing ADT-OH has been coupled to a series of nonsteroidal anti-inflammation drugs (NSAIDs). Although the resultant H2S-hybrid NSAIDs exhibit greatly reduced NSAID-induced GI damage while maintaining NSAID activity in GI system, the detailed mechanism of H2S release from ADT-OH derived systems remains uncertain.38–40 Building on the need for mechanistically-understood systems, different controllable H2S donors have been designed and evaluated.21–23,25 Such donors are typically activated by specific triggers, such as cellular thiols,41–47 light,48–49 esterase,50 nitroreductase,51 or pH,52–53 to release H2S (Figure 1). In addition to their small-molecule counterparts, H2S-releasing biomaterials, including H2S releasing polymers, peptides, and microfibers, are also emerging as an important class of donor materials with modifiable macromolecular properties.54–59
Building from the need for new donor motifs that function through well-defined release mechanisms, we recently reported the first examples of H2S donors that function through the initial release of carbonyl sulfide (COS).60 Although COS has only recently emerged as a potential biologically-important molecule, it is the most prevalent sulfur-containing gas in the Earth’s atmosphere and has a long history in geological, atmospheric, and agricultural chemistry.61–62 In the global sulfur cycle, COS is generated from both abiotic sources, including volcanos, hot springs, and oceans, as well as biotic sources, including plants, soils, and biomass burning,63 and is eventually oxidized to sulfur dioxide (SO2) and sulfate (SO42−) in the atmosphere.61,64 In mammalian systems, however, COS functions as a competent substrate for the ubiquitous enzyme carbonic anhydrase (CA), which results in rapid conversion to H2S (kcat/KM for bovine carbonic anhydrase II: 2.2 × 104 M−1s−1).65–68 This property enables COS to serve as a H2S precursor and provides new avenues for H2S donor development based on COS-releasing motifs.68
Expanding from our initial report of using caged thiocarbamates as COS/H2S donor motifs, we recently reported a class of COS/H2S-releasing donors that were activated by reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), peroxynitrite (ONOO−), and superoxide (O2−), with H2O2 as the most potent trigger.69 H2S can scavenge H2O2 directly, albeit with modest second-order rate constants (0.73 M−1s−1 for HS−/H2O2 reaction at 37 °C, pH 7.4), and it is also well-established to react with other cellular oxidants.70 More recently, Kimura and co-workers demonstrated that NaSH significantly rescued Neuro2a cells from H2O2-induced oxidative stress, suggesting promising anti-oxidative effects of H2S.71 In our initial H2O2-triggered donor systems, COS was caged as an O-alkyl thiocarbamate, which upon activation of an H2O2-activated trigger, underwent a self-immolative decay to release COS/H2S in an H2O2 dose-dependent manner. Cellular investigations demonstrated that these donors, but not carbamate control compounds that release CO2/H2O, rescued cells from H2O2-induced oxidative stress, thus demonstrating that COS-releasing donors can access activities associated with H2S-releasing motifs. In addition to H2O2 activation, our group as well as other researchers have expanded the COS-releasing landscape to include donors activated by nucleophilic attack,72 light,73 click chemistry,53 and cellular esterases.74–75 Because such donors function by the intermediate release of COS en-route to H2S generation, it is possible that such donors could also reveal activities associated with COS directly, although the CA-mediated conversion of COS to H2S is fast. Despite the rapid expansion of this research area, the structural motifs comprising the caged COS core structures have remained somewhat limited. To expand our understanding of COS-based H2S donor systems to enable future expansion of this emerging area, we report here the preparation and COS/H2S-releasing capacities of six different isomeric cores, including O- and S-alkyl thiocarbamates and thiocarbonates, as well as dithiocarbonate derivatives (Figure 2). We complement our H2S releasing investigations with DFT investigations on the energetics on the self-immolative and COS-extrusion potential energy surfaces for each isomeric derivative. For the best releasing core structure, we demonstrate how electronic substitution can be used to tune the rate of COS/H2S release and highlight design requirements for efficient donor function. In addition, we also include an initial expansion of our platform to include dithiocarbamate motifs that are designed to release H2S and/or CS2, which has been recently reported to be protective in biosystems76–77 and may be an emerging small molecule of biological interest.
Figure 2.
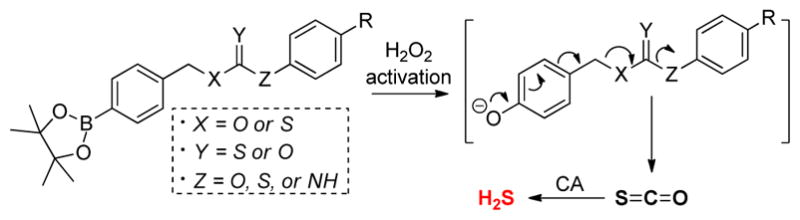
H2S release from H2O2-activated isomeric caged COS/H2S donors
RESULTS AND DISCUSSION
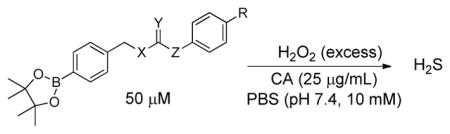
The general design of the H2O2-activated donor motifs requires H2O2-mediated boronate cleavage to generate a phenolic intermediate, which undergoes a subsequent self-immolative decomposition to release COS (Figure 2). In our initial report, we focused exclusively on the O-alkyl thiocarbamate core structure, although many other isomeric derivatives are accessible and should still release COS. To investigate the H2S releasing profile of such isomeric caged-COS molecules, we prepared isomeric O-alkyl and S-alkyl peroxy-sensitive thiocarbamates (OA-PeroxyTCM and SA-PeroxyTCM), thiocarbonates (OA-PeroxyTCN and SA-PeroxyTCN), and the S-alkyl dithiocarbonate (SA-PeroxyDTCN) with an arylboronate trigger. Upon activation, each of these donor platforms releases COS, which is quickly hydrolyzed to H2S by CA. In addition to these COS-releasing molecules, we also prepared peroxy-labile dithiocarbamate (PeroxyDTCM) compounds, which could potentially release CS2 or H2S directly by different pathways vide infra).
Donor Synthesis
One benefit of the caged COS donor platforms investigated is the simplicity and modularity of their preparation. The thiocarbamate-based OA-PeroxyTCMs were synthesized by treating the corresponding 4-(hydroxymethyl)phenylboronic acid pinacol ester with the desired aryl isothiocyanate in the presence of sodium hydride (Scheme 1a). Similarly, SA-PeroxyTCM-1 and the caged-CS2 PeroxyDTCMs were synthesized by treating 4-(thiomethyl)phenylboronic acid pinacol ester with the desired aryl isothiocyanate reagents (Scheme 1b). The thiocarbonates OA-PeroxyTCN-1 and SA-PeroxyTCN-1, were prepared by treating the desired pinacol ester benzyl alcohol or thiol starting materials with the corresponding aryl chloroformate or chlorothionoformate reagents (Scheme 1c). Similarly, SAPeroxyDTCN-1 was prepared by treating 4-(thiomethyl)phenylboronic acid pinacol ester with the aryl chloroformate in the presence of pyridine (Scheme 1c).
Scheme 1.
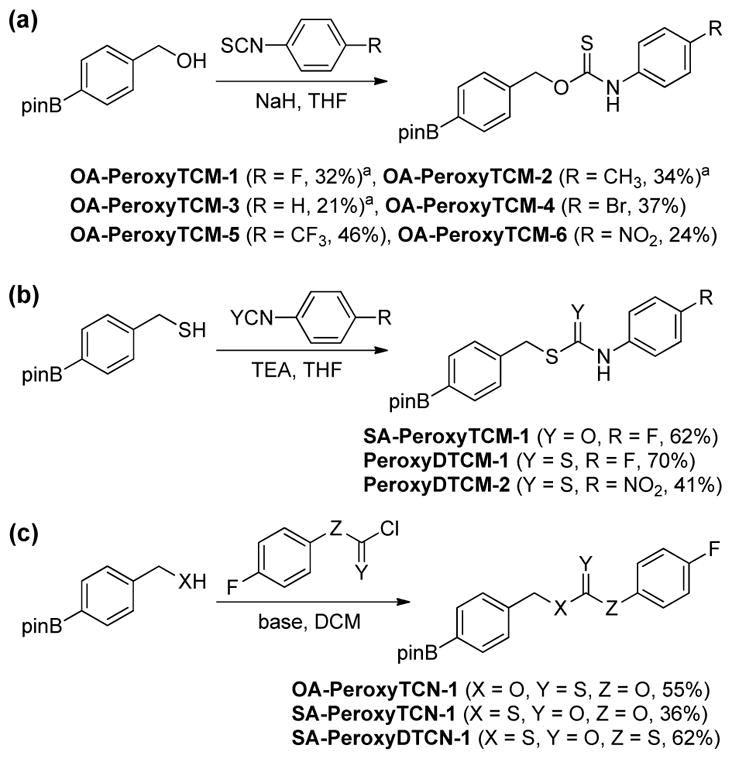
Synthesis of the isomeric thiocarbamate, thiocarbonate, and dithiocarbamate donors.
H2S Release from Caged Donors
Electronic Effects of H2S Release from O-alkyl Thiocarbamates
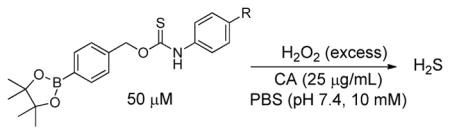
To evaluate the substituent effects on H2S release from OA-PeroxyTCMs, we chose to focus on the OA-PeroxyTCM family because of the well-defined release behavior, our previous investigations with O-alkyl thiocarbamate derivatives, and their high release efficiencies (vide infra). To measure rate data, we used an H2S-selective electrode to monitor H2S release from OA-PeroxyTCM-1 through 6 (50 μM) in the presence of excess H2O2 in PBS buffer (pH 7.4, 10 mM) containing cellularly-relevant concentrations of CA (25 μg/mL). We used excess H2O2 in this study to ensure the boronate cleavage was under pseudo-first order condition. The pseudo-first order rate constant (kobs) was obtained by plotting the H2S releasing response versus the time of measurement, and the second order rate constant (k2) was obtained by plotting the kobs versus H2O2 concentrations. For each donor in the series, we observed a dose-dependent increase in the rate of COS/H2S released with increased H2O2, but also a lower total peaking amount of H2S, which is consistent with an increase in H2S scavenging by H2O2 at higher concentrations (Figure S1). The OA-PeroxyTCM donors exhibited different k2 values for COS/H2S release ranging from 0.22 to 1.16 M−1s−1 (Table 1, Figure S3).
Table 1.
Comparison of H2S releasing efficiencies from differently substituted O-alkyl thiocarbamates.a
| ||||
|---|---|---|---|---|
|
| ||||
| Donor | Strucutre | k2(M−1s−1) | rel. rate | H2S (%)a |
| OA-PeroxyTCM-1 |
|
1.16 ± 0.05 | 1.27 | 60 |
| OA-PeroxyTCM-2 |
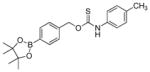
|
0.59 ± 0.03 | 0.65 | 90 |
| OA-PeroxyTCM-3 |
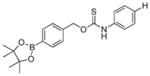
|
0.91 ± 0.01 | 1.00 | 80 |
| OA-PeroxyTCM-4 |
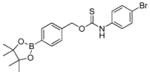
|
0.42 ± 0.02 | 0.46 | 76 |
| OA-PeroxyTCM-5 |
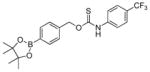
|
0.22 ± 0.02 | 0.24 | 64 |
| OA-PeroxyTCM-6 |
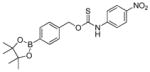
|
0.37 ± 0.02 | 0.41 | 46 |
H2S releasing efficiency was evaluated in the presence of 500 μM of H2O2. The H2S releasing yield (%) was calculated by using a NaSH calibration curve.
To better compare the effects of electronic substitution on COS/H2S release from the OA-PeroxyTCM donors, we normalized each rate to the parent OA-PeroxyTCM-3 (R = H) donor (Table 1). Our initial expectation was that inclusion of electron withdrawing groups (EWGs) would facilitate COS release, therefore accelerating H2S generation, whereas electron donating groups (EDGs) would lead to slower H2S release (Scheme 2a). Although OA-PeroxyTCM-2 (krel. = 0.65) and OA-PeroxyTCM-3 (krel. = 1.27) followed this trend, we observed slower H2S release from OA-PeroxyTCM-4 to 6 with krel. of 0.46, 0.24, and 0.41, respectively (Table 1). We attribute this slower H2S release to the acidification of the thiocarbamate NH proton by EWGs, thus potentially leading to isothiocyanate formation in aqueous buffer (Scheme 2b). Williams and co-workers have reported the similar degradation of thiocarbamates to isothiocyanates in aqueous solution, which supports our hypothesis.78 Although aryl isothiocyanates are known H2S donors, the H2S release from these compounds less efficient than from the OA-PeroxyTCM core and thus would be expected to reduce the overall rate of H2S formation.79–80 H2S Releasing efficiency was calculated by using a NaSH calibration curve. The low efficiency of H2S release EWG-containing donors, especially for OA-PeroxyTCM-6, highlighting a limitation of including highly electron withdrawing moieties on thiocarbamate-based donor motifs.
Scheme 2.
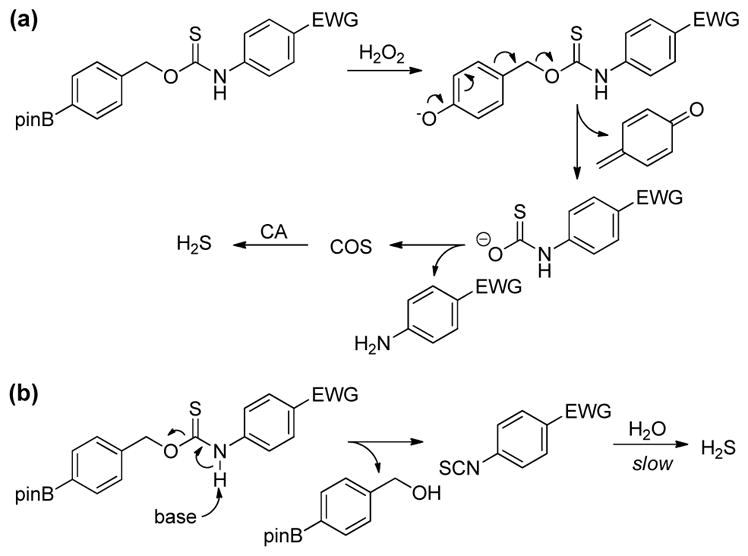
Competing H2S release pathways for O-alkyl thiocarbamates with electron withdrawing groups. (a) H2O2-mediated COS/H2S release and (b) base-mediated isothiocyanate formation followed by hydrolysis to generate H2S.
H2S Release from S-Alkyl Thiocarbamates
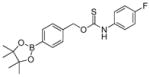
Having investigated the impacts of electronic substitution on the O-alkyl thiocarbamate scaffolds, we next compared the O- versus S-alkyl isomeric differences for thiocarbamate donors by evaluating the H2S releasing from SA-PeroxyTCM-1. We note that Chakrapani and co-workers recently reported esterase-cleavable S-alkyl thiocarbonate and thiocarbamate COS donor motifs, but their activities were not compared with O-alkyl donor analogues.75 Our expectation was that the S-alkyl isomer should have a greater ground state stability due to enhanced resonance stabilization in the amide moiety of the thiocarbamate by comparison to the thioamide moiety in the O-alkyl derivative. Although SA-PeroxyTCM-1 is more thermodynamically stable than the corresponding O-alkyl isomer, it is less stable in solution, likely due to the better leaving group ability of the benzyl thiol versus benzyl alcohol. For example, incubation of model compounds lacking the boronate trigger revealed complete recovery of the O-alkyl isomer from buffer after 24 hours, whereas only 40% of the S-alkyl isomer was recovered. In addition, over 80% of the S-alkyl isomer decomposed after 24-h incubation with ROS, such as H2O2, ONOO−, and O2−, suggesting that the S-alkyl thiocarbamate functional group is reactive toward ROS, which may lead to relatively lower COS/H2S releasing efficiency (Figure S6). As in the case of O-alkyl OA-PeroxyTCM-1, we observed dose-dependent H2O2-mediated COS/H2S release from SA-PeroxyTCM-1 (Figures 4 and S2a). The rate of COS/H2S release, however, was significantly slower (krel = 0.09) with respect to OA-PeroxyTCM-1 (krel = 1.00) with 42% of the expected H2S being captured (Table 2, Figure S3), indicating that H2S release from caged-thiocarbamate compounds can be tuned by isomeric changes to the caged COS core.
Figure 4.
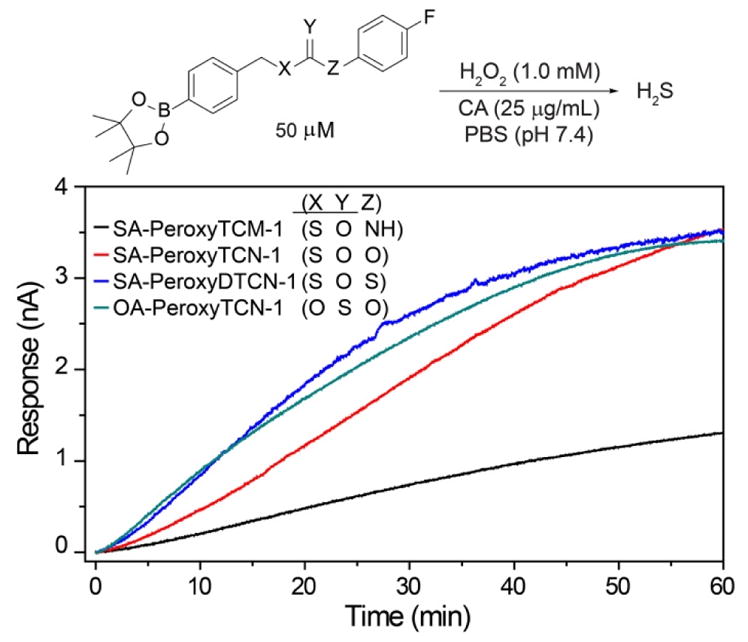
Representative H2S Release from caged-COS donors in the presence of H2O2.
Table 2.
H2S Release from caged-COS moleculesa
| ||||
|---|---|---|---|---|
|
| ||||
| Donor | Strucutre | k2(M−1s−1) | rel. rate | H2S (%)a |
| OA-PeroxyTCM-1 |
|
1.16 ± 0.05 | 1.0 | 60 |
| SA-PeroxyTCM-1 |
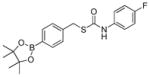
|
0.10 ± 0.01 | 0.09 | 42 |
| OA-PeroxyTCN-1 |
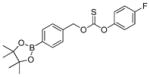
|
0.60 ± 0.04 | 0.52 | 36 |
| SA-PeroxyTCN-1 |
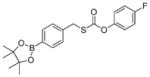
|
0.16 ± 0.01 | 0.14 | 36 |
| SA-PeroxyDTCN-1 |
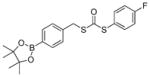
|
0.36 ± 0.02 | 0.31 | 46 |
H2S releasing efficiency was evaluated in the presence of 500 μM of H2O2. The H2S releasing yield (%) was calculated by using a NaSH calibration curve.
H2S Release from O- and S-Alkyl Thiocarbonates
In addition to thiocarbamates, thiocarbonate derivatives also provide access to phenolic payloads and also eliminate the potentially-acidic thiocarbamate proton. We envisioned that thiocarbonate compounds, including S- and O-alkyl isomers, with an arylboronate trigger would be new H2O2-responsive COS/H2S donors. Therefore, in addition to caging COS as thiocarbamates, thiocarbonates were also investigated as alternative COS/H2S donor motifs. We expected that COS/H2S release from these donors would still be triggered by the H2O2-mediated cleavage of the aryl boronate, and that the COS/H2S releasing kinetics would be different from that of thiocarbamate-based donors based on the inherent electronic differences between the thiocarbamate and thiocarbonate cores. To investigate our hypothesis, we measured the H2S release from the O-alkyl and S-alkyl isomers OA-PeroxyTCN-1 and SA-PeroxyTCN-1. As expected, both isomers exhibited an H2O2-dependent H2S release in the presence of CA, with the O-alkyl isomer exhibiting faster H2S-releasing kinetics (Figures 4, S2b, S2c, and S3) and also exhibiting excellent stability in buffer in the absence of H2O2 over a 24 hour period. Moreover, the O-alkyl thiocarbonate functional group exhibits better stability in the presence of ROS (i.e. H2O2, O2−, and ONOO−) compared to other isomeric counterparts, such as O-alkyl and S-alkyl thiocarbamates, making it a promising candidate for future COS/H2S donor design (Figure S6). In addition, we also prepared SA-PeroxyDTCN-1, in which the phenol payload was replaced with the corresponding thiophenol to determine whether dithiocarbonate cores were still functional as COS/H2S releasing moieties. We expected that the thiophenol would be a better leaving group than the parent phenol, thus leading to enhanced COS/H2S release rates. As expected, SA-PeroxyDTCN-1 exhibited faster COS/H2S release kinetics than the parent SA-PeroxyTCN-1 derivative (krel = 0.30 vs 0.14), and also established that dithiocarbonate cores are functional COS/H2S releasing donor motifs (Figures 4, S2d, S3 and Table 2).
H2S/CS2 Release from Dithiocarbamates
Building from the positive impacts that we envision COS donors will have on the biological roles of COS biology, we also envisioned that our general donor design approach could facilitate access to CS2-releasing motifs. Recent studies suggest that CS2 may have has specific protective activities in biological systems, and may be an emerging biologically-relevant sulfur-containing small molecule.76 Aligned with this idea, we prepared PeroxyDTCM-1 and 2, both of which cage CS2 into dithiocarbamate compounds. Much like H2O2-labile PeroxyTCMs and PeroxyTCNs, the PeroxyDTCMs should be activated by H2O2 to generate anionic dithiocarbamate intermediates, which could release CS2 instead of COS. (Scheme 3). It is also possible, however, that such dithiocarbamates could spontaneously extrude H2S en route to isothiocyanates generation by a pathway in which proton transfer from the nitrogen to the sulfur in the anionic dithiocarbamate intermediate results in the formation of aryl isothiocyanate and extrusion of H2S (Scheme 3).
Scheme 3.

Possible competing pathways for (a) H2S and (b) CS2 releasing pathways from caged dithiocarbamates.
To determine which of these pathways was operable, we treated PeroxyDTCM-1 with H2O2 and observed only a small amount of H2S release using the methylene blue assay (Figure 5). This result suggested that direct H2S extrusion may be possible, but was likely inefficient for PeroxyDTCM-1. To further investigate this platform, we also prepared PeroxyDTCM-2, which contains a nitro group in the para position, which should significantly acidify the NH proton, therefore facilitating isothiocyanate formation and H2S extrusion. As expected, we observed an enhanced H2S release from PeroxyDTCM-2 after addition of H2O2 (Figure 5). To rule out the possibility that released CS2 was reacting with H2O2 to produce H2S, we incubated authentic CS2 sample directly with H2O2 under the identical condition but failed to observe H2S release (Figure S4). Our attempts to measure CS2 released from the PeroxyDTCM donors using different CS2-responsive colorimetric methods81–83 as well as by GC have remained unsuccessful, suggesting that CS2 release is not facile from these compounds. This lack of CS2 release could be due, in part, to the greater stability of the dithiocarbamate anion formed after the initial self-immolative decay by comparison to the corresponding anionic thiocarbamate anions. Taken together, these dithiocarbamate platforms may provide a future scaffold for efficacious H2S or CS2 release after fine tuning of the intermediate stabilities.
Figure 5.
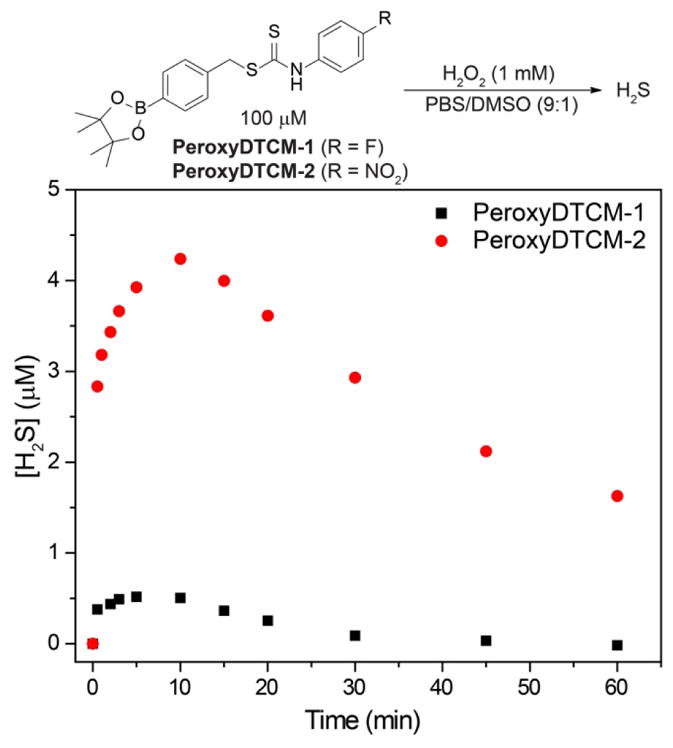
H2S release from PeroxyDTCMs in the presence of H2O2.
Computational Investigations
To provide further insights into the energetics of COS/H2S release from the different isomeric donor motifs described above, we conducted conformational searches for the ground state, intermediate, and transition state geometries using Gaussian 09 at the B3LYP/6–311++G(d,p) level of theory applying the IEF-PCM water solvation model. Each potential energy surface (PES) begins from the phenol product generated after boronate cleavage by H2O2 to provide equivalent starting structures for each energy surface. Graphical summaries of the potential energy surfaces for each donor motif are shown in Figure 6. Attempt to locate the transition state for quinone methide extrusion from the neutral phenol for any of the structures were unsuccessful, which is consistent with prior reports that the self-immolative decomposition proceeds from the anionic phenolate.84–85
Figure 6.

Calculated potential energy surfaces for thiocarbamate, thiocarbonate, and dithiocarbonate donors at the B3LYP/6-311++G(d,p) level of theory with PCM solvation (H2O). (a) O-Alkyl and S-alkyl thiocarbamates OA-PeroxyTCM and SA-PeroxyTCM. (b) O-Alkyl and S-alkyl thiocarbonates OA-PeroxyTCN and SA-PeroxyTCN (c) Dithiocarbonate SA-PeroxyDTCN-1. (d) Hypothetical S-alkyl dithiocarbonate SA-PeroxyDTCN-2, which should release CS2 upon cleavage. All energies are reported in kcal/mol and referenced to the lowest energy phenolate generated after initial donor deprotonation.
For OA-PeroxyTCM, initial proton transfer from the phenol generates the anionic phenolate intermediate and H3O+, which then proceeds through TS1 to generate the monothiocarbamate intermediate and the quinone methide with a barrier of 5.3 kcal/mol. This local barrier is lower than that for SA-PeroxyTCM (8.2 kcal/mol), which is consistent with the different inherent stabilities of the thiocarbamate isomers. During our searches for COS extrusion pathways from the monothiocarbamate anionic intermediate, we were surprised to observe a significant barrier of 42.3 and 28.4 kcal/mol for the O-alkyl and S-alkyl derivatives, respectively. These barriers are in contrast with our lack of observable intermediates during the course of these reaction. Because prior work by Ghadiri and co-workers has highlighted the importance of different additives and/or Lewis acids in COS extrusion from thiocarbamates en route to peptide bond formation,86 we also investigated whether lower energy assisted pathways might be present. To simplify such investigations, we focused on water-assisted COS extrusion, in which proton transfer from H2O occurs concomitantly with COS extrusion (TS2′), and also on general acid catalyzed COS extrusion, in which proton transfer from H2PO4− accompanies COS extrusion (TS2″). Both assisted pathways eliminate the direct formation of the high-energy deprotonated amine intermediate that results from direct COS extrusion, which would be unlikely in an aqueous environment. Notably, we were able to locate transition states corresponding to concomitant proton transfer and COS release for both assisted reactions, which are 8.5 and 15.6 kcal/mole lower in energy than the un-assistant pathway for the H2O and H2PO4− assisted routes, respectively. The ease of locating these assisted COS extrusion pathways, as well as their lower energy barriers, suggesting the viability of other solvent or other additive assisted routes of COS release. For both the O-alkyl and S-alkyl isomers, COS extrusion from the monothiocarbamate anion intermediate is exotheric. To complete the final PES for each isomer, we assumed that the produced quinone methide would be scavenged by H2O to form the p-hydroxy benzyl alcohol, which is consistent with previous experimental observations in similar systems. In total, the overall reaction is only exothermic for the O-alkyl thiocarbamate but endothermic, and thus entropically-driven, for the S-alkyl thiocarbamate donor.
We carried out similar calculations for isomeric O-alkyl and S-alkyl thiocarbonate donor derivatives and observed similar ground state energy differences between the two isomers (Figure 6b). The S-alkyl isomer, SA-PeroxyTCN, was 15.7 kcal/mol lower in energy than the O-alkyl isomer, OA-PeroxyTCN, which we attribute to better orbital overlap between the ester moiety with the aromatic ring. Much like the thiocarbamate counterparts, the O-alkyl and S-alkyl isomers both proceed through self-immolative transition states TS1 (2.6 kcal/mol) and TS1′ (7.2 kcal/mol) respectively, en route to the common thiocarbonate anion intermediate. Direct COS extrusion from the monothiocarbonate anion intermediate proceeds through a significantly lower transition state TS2 (17.2 and 1.5 kcal/mol for the S-alkyl and O-alkyl isomers, respectively) than for the thiocarbamate analogues, which we attribute to the greater leaving group ability of the phenolate by comparison to the analogous deprotonated amine product for the corresponding thiocarbamate donors. Attempts to locate transition states for H2O or H2PO4− assisted COS extrusion were unsuccessful, likely due to the lower basicity of the developing phenolate by contrast to the adeprotonated amine. Subsequent COS extrusion, proton transfer to generate the neutral phenol, and p-hydroxy benzyl alcohol formation complete the overall PES. Similar to the overall thermodynamics of the PES for the thiocarbamate isomers, only the O-alkyl thiocarbonate donor is exothermic, with the more thermodynamically stable S-alkyl thiocarbonate being endothermic, and entropically controlled. One additional insight afforded from the thiocarbonate energy surfaces is that the ground state destabilization of the O-alkyl thiocarbonate generates a system in which the initial self-immolative step to release the quinone methide is rate-limiting, rather than COS extrusion from the anionic thiocarbonate intermediate.
In contrast to the two thiocarbonate isomers, the two dithiocarbonate isomers proceed through different intermediates that should result in COS and CS2 extrusion from the OA-PeroxyDTCN-1 and SA-PeroxyDTCN-2 isomers, respectively. OA-PeroxyDTCN-1, proceeds through a modest 5.6 kcal/mol activation barrier (TS1) to generate the anionic dithiocarbonate intermediate, which proceeds through TS2 to generate thiophenol and extrude COS en route to an overall endothermic reaction (Figure 6c). Although we were unable to prepare the SA-PeroxyDTCN-2, our DFT investigations show that such donors could potentially function as CS2-releasing molecules. SA-PeroxyDTCN-2 proceeds through a modest activation barrier of 6.0 kcal/mol en route to the dithiocarbonate intermediate, which requires a significantly higher (17.9 kcal/mol) activation barrier to result in CS2 release (Figure 6d). Attempts to locate transition states for H2O or H2PO4− assisted pathways for CS2 extrusion pathways were unsuccessful. After CS2 extrusion, proton transfer to generate the phenol product, and generation of the p-hydroxy benzyl alcohol, the overall reaction is endothermic by 3.8 kcal/mol. As a final point of comparison, the OA-PeroxyDTCN-1 is isomeric to SA-PeroxyDTCN-2, allowing for the energetics of the two reactions to be compared directly. Interestingly, referencing to the more thermodynamically-stable SA-PeroxyDTCN-1 isomer reveals that the anionic dithiocarbamate intermediate formed from SA-PeroxyDTCN-1 is 15.3 kcal/mol higher than the dithiocarbamate intermediate formed in the SA-PeroxyDTCN-2 PES. Additionally, TS2 is 19.9 kcal/mol higher in energy for SA-PeroxyDTCN-2 than for SA-PeroxyDTCN-1, suggesting that CS2 release is less kinetically favorable than COS release from the corresponding isomers (see Supporting Information for a direct energy comparison).
To further interrogate the observed H2S release from the dithiocarbamate PeroxyDTCM, we employed similar computational methods. The initial self-immolative decay from the phenolate intermediate proceeds with a modest activation barrier of 5.7 kcal/mol en route to formation of the dithiocarbamate anion intermediate (Figure 7). Direct CS2 extrusion from this intermediate, however, is enthalpically unfavorable with a high-energy TS2 of 41.9 kcal/mol. In parallel to our investigations on the COS-releasing thiocarbamate donors, we also located transition states for H2O and H2PO4− assisted CS2 release pathways, which maintain activation enthalpies of 34.0 and 27.8 kcal/mol, respectively. The final product of all of these routes, which include CS2, PhNH2, and p-hydroxy benzyl alcohol, is endothermic by 6.2 kcal/mol. We were also able to locate a PES for direct HS− release, which proceeds through initial proton transfer from the NH to RS− in the anionic dithiocarbamate intermediate, followed by direct HS− extrusion and isothiocyanate formation with an activation barrier (TS3) of 25.4 kcal/mol. The resultant isothiocyanate should also release COS through hydrolysis, leading to a slightly exothermic product formation of H2S, COS, PhNH2, and p-hydroxy benzyl alcohol. Notably, this transition state for direct HS− extrusion is lower in energy than any of the assisted CS2 extrusion pathways, suggesting that sulfide extrusion, which in solution is almost certainly assisted through hydrogen bonding, is a lower energy pathway than CS2 release. We also investigated whether a similar direct HS− extrusion pathway was energetically accessible from the thiocarbamate-based donors, but found that the transition state for HS− to be 44.9 kcal/mol, which is even higher than the unassisted COS-release pathway. As a whole, these computational insights support the observed direct H2S release from the caged dithiocarbamate donor motifs, but also suggest that such platforms may be modifiable to access CS2 releasing systems as well.
Figure 7.

Calculated potential energy surfaces for CS2 and H2S extrusion from the dithiocarbamate donor PeroxyDTCM at the B3LYP/6-311++G(d,p) level of theory with PCM solvation (H2O). All energies are reported in kcal/mol and referenced to the phenolate generated after initial donor deprotonation.
CONCLUSIONS
By preparing and directly comparing thiocarbamate, thiocarbonate, and dithiocarbonate donor platforms, we demonstrate access to a wide array of structural motifs that function as controllable COS-based H2S donors. In addition to providing access to new H2O2-responsive H2S donors, the combined experimental and computational investigations provide a cohesive platform for understanding how donor core modifications impact efficiency, stability, and rates. Finally, our initial investigations into dithiocarbamate motifs suggest that these platforms provide access to new classes of triggerable H2S releasing motifs, which could also possibly be tuned to release CS2. In conclusion, the insights on COS/H2S release gained from these investigations provide a foundation for the expansion of the emerging area of responsive COS/H2S donors systems.
EXPERIMENTAL SECTION
Materials and Methods
Reagents were purchased from Sigma-Aldrich, Tokyo Chemical Industry (TCI), Fisher Scientific, and VWR and used directly as received. Carbonic anhydrase (CA) from bovine erythrocytes was purchased from Sigma-Aldrich (C2624). Silica gel (SiliaFlash F60, Silicycle, 230–400 mesh) was used for column chromatography. Deuterated solvents were purchased from Cambridge Isotope Laboratories (Tewksbury, Massachusetts, USA). 1H, 19F and 13C{1H} NMR spectra were recorded on Varian INOVA 500 MHz, Bruker 500 MHz, or Bruker 600 MHz NMR instruments at the indicated frequencies. Chemical shifts are reported in ppm relative to residual protic solvent resonances. H2S Detection was monitored by using an H2S electrode (ISO-H2S-2, World Precision Instruments, Inc. Sarasota, Florida, USA) or UV-Vis spectrometer (Cary 60, Agilent Technologies, Santa Clara, California, USA) by following methylene blue assay in PBS buffer. Stability test was performed on HPLC (1260 Infinity II, Agilent Technologies, Santa Clara, California, USA). OA-PeroxyTCM-1, OA-PeroxyTCM-2, OA-PeroxyTCM-3, and OA-TCM-1 were synthesized by following our previous study.69
Synthesis
General procedure for the synthesis of O-alkylthiocarbamate compounds
4-(Hydroxymethyl)phenylboronic acid pinacol ester (1.0 equiv.) was combined with substituted phenyl isothiocyanate (1.0 equiv.) in anhydrous THF (15 mL) at 0 °C, followed by the addition of NaH (60% in paraffin liquid, 1.2 equiv.). The resultant mixture was stirred at 0 °C for 20 min, after which the ice bath was removed, and the reaction mixture was stirred at r.t. until the completion of the reaction indicated by TLC. The reaction was quenched by adding brine (30 mL), and the aqueous solution was extracted with ethyl acetate (3 ×15 mL). The organic layers were combined, dried over MgSO4, and evaporated under vacuum. The crude product was purified by column chromatography. We note that thiocarbamates show two sets of NMR resonances at room temperature due to slow rotation around the thiocarbamate functional group.
OA-PeroxyTCM-4 was prepared from 4-(hydroxymethyl)phenylboronic acid pinacol ester and 4-bromophenyl isothiocyanate using the general synthetic procedure described above (125 mg, 37% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.35 (s, 1H), 7.70 (s, 3 H), 7.51 (m, 4H), 7.29 (br, 1H), 5.59 (br, 2H), 1.30 (s, 12H). 13C{1H} NMR (150 MHz, DMSO-d6) δ (ppm): 187.8, 187.4, 139.6, 139.2, 138.4, 137.3, 135.0, 132.1, 131.8, 128.7, 127.8, 125.1, 124.3, 117.5, 84.2, 72.6, 70.6, 40.5, 25.1. IR (cm−1): 3208, 3097, 3031, 2975, 1615, 1590, 1532, 1487, 1390, 1360, 1325, 1273, 1168, 1141, 1091, 1007. HRMS m/z [M - H]− calcd. for [C20H22BBrNO3S]− 446.0597; found 446.0599.
OA-PeroxyTCM-5 was prepared from 4-(hydroxymethyl)phenylboronic acid pinacol ester and 4-trifluoromethylphenyl isothiocyanate using the general synthetic procedure described above (100 mg, 46% yield). 1H NMR (500 MHz, Acetone-d6) δ (ppm): 10.45 (s, 1H), 7.95 (br, 2H), 7.80 (d, J = 10.0 Hz, 2H), 7.72 (d, J = 10.0 Hz, 2H), 7.51 (d, J = 10.0 Hz, 2H), 5.66 (s, 2H), 1.36 (s, 12H). 13C{1H} NMR (125 MHz, Acetone-d6) δ (ppm): 188.0, 138.8, 134.8, 127.4, 127.1, 125.9, 125.3, 123.5, 121.9, 83.7, 59.7, 24.3. 19F NMR (470 MHz, Acetone-d6) δ (ppm): −62.6. IR (cm−1): 3208, 3097, 3031, 2975, 1615, 1590, 1532, 1487, 1390, 1360, 1325, 1273, 1184, 1168, 1141, 1091, 1017, 816. HRMS m/z [M - H]− calcd. for [C21H22BF3NO3S]− 436.1366; found 436.1369.
OA-PeroxyTCM-6 was prepared from 4-(hydroxymethyl)phenylboronic acid pinacol ester and 4-nitrophenyl isothiocyanate using the general synthetic procedure described above (82 mg, 24% yield). 1H NMR (500 MHz, CDCl3) δ (ppm): 8.50 (s, 1H), 8.18 (d, J = 10.0 Hz, 2H), 7.85 (d, J = 10.0 Hz, 2H), 7.56 (br, 2H), 7.42 (d, J = 10.0 Hz, 2H), 5.62 (s, 2H), 1.35 (s, 12H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 187.7, 144.1, 142.6, 137.3, 135.2, 127.7, 125.0, 120.6, 118.7, 84.0, 74.0, 24.9. IR (cm−1): 3265, 2973, 2926, 1609, 1598, 1553, 1508, 1493, 1396, 1359, 1332, 1319, 1268, 1169, 1144, 1068, 1015, 857, 842, 656. HRMS m/z [M - H]− calcd. for [C20H22BN2O5S]− 413.1342; found 413.1345.
Synthesis of S-alkylthiocarbamate compounds
The corresponding benzyl thiol species (1.0 equiv.) was combined with 4-fluorophenyl isocyanate (1.0 equiv.) in anhydrous THF (15 mL) at 0 °C, followed by the addition of triethylamine (1.2 equiv.). The resultant solution was stirred at 0 °C for 20 min, after which the ice bath was removed, and the reaction mixture was stirred at r.t. until the completion of the reaction indicated by TLC. The reaction was quenched by adding brine (30 mL), and the aqueous solution was extracted with ethyl acetate (3 ×15 mL). The organic layers were combined, dried over MgSO4, and evaporated under vacuum. The crude product was purified by column chromatography.
SA-PeroxyTCM-1 was prepared from 4-(thiomethyl)phenylboronic acid pinacol ester (2) using the general synthetic procedure described above (87 mg, 62% yield). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.76 (d, J = 10.0 Hz, 2H), 7.36 (m, 4H), 7.21 (s, 1H), 6.99 (t, J = 5.0 Hz, 2H), 4.21 (s, 2H), 1.34 (s, 12H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 165.5, 160.6, 158.6, 141.0, 135.2, 133.5, 128.3, 121.9, 115.9, 115.7, 83.9, 34.6, 29.7, 24.9. 19F NMR (470 MHz, CDCl3) δ (ppm): −117.8. IR (cm−1): 3293, 2982, 2920, 1691, 1611, 1530, 1508, 1398, 1382, 1303, 1270, 1136, 1084, 854, 833. HRMS m/z [M - H]− calcd. for [C20H22BFNO3S]− 386.1397; found 386.1398.
SA-TCM-1 was prepared from benzyl thiol using the general synthetic procedure described above (120 mg, 49% yield). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.39 (m, 4H), 7.34 (t, J = 10.0 Hz, 2H). 7.28 (m, 1H), 7.07 (br, 1H), 7.04 (t, J = 10.0 Hz, 2H), 4.25 (s, 2H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 165.6, 160.6, 158.6, 137.8, 133.5, 128.9, 128.7, 127.4, 121.9, 115.9, 34.5. 19F NMR (470 MHz, CDCl3) δ (ppm): −117.6. IR (cm−1): 3254, 3143, 3060, 2924, 1644, 1613, 1538, 1504, 1453, 1407, 1301, 1211, 1153, 1096, 698. HRMS m/z [M + H]+ calcd. for [C14H13FNOS]+ 262.0702; found 262.0700.
Synthesis of SA-PeroxyTCN-1
4-(Thiomethyl)-phenylboronic acid pinacol ester (160 mg, 0.64 mmol, 1.0 equiv.) was combined with 4-fluorophenyl chloroformate (113 mg, 0.64 mmol, 1.0 equiv.) in anhydrous DCM (15 mL) at 0 °C, followed by the addition of NEt3 (65 mg, 0.64 mmol 1.0 equiv.). The resultant solution was stirred at 0 °C for 20 min, after which the ice bath was removed, and the reaction mixture was stirred at r.t. for 16 h. The reaction was quenched by adding brine (30 mL), and the aqueous solution was extracted with ethyl acetate (3 ×15 mL). The organic layers were combined, dried over MgSO4, and evaporated under vacuum. The crude product was purified by column chromatography (90 mg, 36% yield). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.82 (d, J = 5.0 Hz, 2H), 7.41 (d, J = 5.0 Hz, 2H), 7.13 (m, 2H), 7.08 (t, J = 5.0 Hz, 2H), 4.20 (s, 2H), 1.37 (s, 12H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 169.9, 161.4, 159.4, 147.0, 139.7, 135.2, 128.3, 122.8, 116.3, 116.1, 83.9, 35.8, 24.9. 19F NMR (470 MHz, CDCl3) δ (ppm): −116.2. IR (cm−1): 2979, 2922, 1724, 1610, 1498, 1403, 1355, 1322, 1175, 1143, 1105, 1010. HRMS m/z [M + H]+ calcd. for [C20H23BFO4S]+ 389.1394; found 389.1387.
4-Fluorophenyl thiochloroformate (3)
4-Fluorophenyl mercaptan (384 mg, 3.0 mmol, 1.0 equiv.) was dissolved in DCM (20 mL), followed by the addition of triphosgene (356 mg, 1.2 mmol, 0.4 equiv.) at 0 °C. To this solution was then added pyridine (237 mg, 3.0 mmol, 1.0 equiv.) dropwise. The resultant solution was stirred at r.t. for 3 h. The reaction was quenched by adding HCl (10 mL, 1 M) and the aqueous solution was extracted with DCM (3 × 15 mL). The organic layers were combined, dried over MgSO4, and evaporated under vacuum. The crude product was purified by column chromatography (3, 404 mg, 71%). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.55 (t, J = 5.0 Hz, 2H), 7.18 (t, J = 5.0 Hz, 2H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 165.3, 163.2, 136.4, 122.6, 117.0. 19F NMR (470 MHz, CDCl3) δ (ppm): −107.9. IR (cm−1): 1759, 1589, 1489, 1400, 1231, 1158, 1093, 1013, 800. HRMS m/z M+ calcd. for [C7H4ClFOS]+• 189.9655; found 189.9652.
Synthesis of SA-PeroxyDTCN-1
4-(Thiomethyl)phenylboronic acid pinacol ester (2) (154 mg, 0.60 mmol, 1.0 equiv.) was combined with 4-fluorophenyl thiochloroformate (3, 114 mg, 0.60 mmol, 1.0 equiv.) in anhydrous DCM (15 mL) at 0 °C, followed by the addition of NEt3 (61 mg, 0.60 mmol 1.0 equiv.). The resultant solution was stirred at 0 °C for 20 min, after which the ice bath was removed, and the reaction mixture was stirred at r.t. for 16 h. The reaction was quenched by adding brine (30 mL), and the aqueous solution was extracted with ethyl acetate (3 ×15 mL). The organic layers were combined, dried over MgSO4, and evaporated under vacuum. The crude product was purified by column chromatography (150 mg, 62% yield). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.79 (d, J = 5.0 Hz, 2H), 7.53 (m, 2H), 7.32 (d, J = 5.0 Hz, 2H), 7.14 (t, J = 10.0 Hz, 2H), 4.22 (s, 2H), 1.37 (s, 12H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 188.9, 165.0, 163.0, 139.7, 137.7, 135.2, 128.3, 122.3, 116.8, 116.6, 83.9, 35.3, 24.9. 19F NMR (470 MHz, CDCl3) δ (ppm): −109.5. IR (cm−1): 2980, 2920, 1730, 1644, 1610, 1587, 1488, 1396, 1354, 1320, 1270, 1225, 1143, 1087, 962. HRMS m/z [M + H]+ calcd. for [C20H23BFO3S2]+ 405.1166; found 405.1165.
Synthesis of O-alkylthiocarbonate
The benzyl alcohol species (1.0 equiv.) was combined with 4-fluorophenyl chlorothionoformate (1.0 equiv.) in anhydrous DCM (15 mL) at 0 °C, followed by the addition of pyridine (1.0 equiv.). The resultant solution was stirred at 0 °C for 20 min, after which the ice bath was removed, and the reaction mixture was stirred at r.t. until the completion of the reaction indicated by TLC. The reaction was quenched by adding brine (30 mL), and the aqueous solution was extracted with ethyl acetate (3 × 15 mL). The organic layers were combined, dried over MgSO4, and evaporated under vacuum. The crude product was purified by column chromatography.
OA-PeroxyTCN-1 was prepared from 4-(hydroxymethyl)phenylboronic acid pinacol ester using the general synthetic procedure described above (106 mg, 55% yield). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.89 (d, J = 10.0 Hz, 2H), 7.48 (d, J = 5.0 Hz, 2H), 7.12 (d, J = 5.0 Hz, 4H), 5.60 (s, 2H), 1.39 (s, 12H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 195.0, 161.7, 159.7, 149.4, 149.3, 137.0, 135.1, 127.5, 123.5, 123.4, 116.4, 116.2, 84.0, 75.5, 24.9. 19F NMR (470 MHz, CDCl3) δ (ppm): −115.6. IR (cm−1): 2978, 2921, 1615, 1500, 1400, 1359, 1292, 1183, 1140, 1008. HRMS m/z [M + Na]+ calcd. for [C20H22BFNaO4S]+ 411.1214; found 411.1216.
OA-TCN-1 was prepared from benzyl alcohol using the general synthetic procedure described above (60 mg, 85% yield). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.47 (m, 4H), 7.15 (m, 4H), 5.60 (s, 2H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 195.0, 161.7, 159.7, 149.4, 149.3, 134.1, 128.9, 128.8, 128.6, 123.5, 116.4, 116.2, 75.8. 19F NMR (470 MHz, CDCl3) δ (ppm): −115.6. IR (cm−1): 3034, 2926, 1500, 1454, 1379, 1274, 1174, 1144, 1088, 1009, 836. HRMS m/z M+• calcd. for C14H11FO2S+• 262.0464; found 262.0472.
Synthesis of dithiocarbamate compounds
The corresponding benzyl thiol species (1.0 equiv.) was combined with aryl isothiocyanate (1.0 equiv.) in anhydrous THF (15 mL) at 0 °C, followed by the addition of triethylamine (1.2 equiv.). The resultant solution was stirred at 0 °C for 20 min, after which the ice bath was removed, and the reaction mixture was stirred at r.t. until the completion of the reaction indicated by TLC. The reaction was quenched by adding brine (30 mL), after which the resultant aqueous solution was extracted with ethyl acetate (3 × 15 mL). The organic layers were combined, dried over MgSO4, and evaporated under vacuum. The crude product was purified by column chromatography.
PeroxyDTCM-1 was prepared from 4-fluorophenyl isothiocyanate using the general synthetic procedure described above (173 mg, 70% yield). 1H NMR (500 MHz, CDCl3) δ (ppm): 8.87 (s, 1H), 7.77 (d, J = 10.0 Hz, 2H), 7.38 (m, 4H), 7.09 (t, J = 5.0 Hz, 2H), 4.58 (s, 2H), 1.35 (s, 12H). 13C{1H} NMR (125 MHz, DMSO-d6) δ (ppm): 196.2,140.9, 136.5, 135.1, 129.0, 127.8, 126.3, 115.8, 84.1, 39.0, 25.1. 19F NMR (470 MHz, CDCl3) δ (ppm): −112.0. IR (cm−1): 3307, 2975, 2922, 1605, 1506, 1451, 1352, 1319, 1268, 1204, 1142, 1086, 994. HRMS m/z [M - H]− calcd. for [C20H22BFNO2S2]− 402.1169; found 402.1166.
PeroxyDTCM-2 was prepared from 4-nitrophenyl isothiocyanate using the general synthetic procedure described above (88 mg, 41% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 12.17 (s, 1H), 8.27 (d, J = 10.0 Hz, 2H), 8.08 (d, J = 10.0 Hz, 2H), 7.64 (d, J = 10.0 Hz, 2H), 7.43 (d, J = 10.0 Hz, 2H), 4.61 (s, 2H), 1.29 (s, 12H). 13C{1H} NMR (125 MHz, DMSO-d6) δ (ppm): 197.2, 145.8, 144.3, 140.5, 135.1, 129.1, 125.0, 123.4, 84.1, 39.1, 25.1. IR (cm−1): 2982, 1610, 1594, 1514, 1355, 1328, 1312, 1270, 1193, 1087, 804 HRMS m/z [M - H]− calcd. for [C20H22BN2O4S2]− 429.1114; found 429.1116.
H2S Release Measurements
H2S Release measurement from PeroxyTCMs and PeroxyTCNs
A donor stock solution (50 μL, 20 mM in DMSO) was added to 20 mL of PBS (pH 7.4, 10 mM) containing CA (25 μg/mL) in a scintillation vial. After stirring at room temperature for 5 min, the H2O2 stock solution (1.0 M in H2O) was added to reach the desired final H2O2 concentration, and the H2S release was monitored using an H2S microsensor. The k2 value of each donor was obtained by plotting kobs vs H2O2 concentrations. Relative rates were obtained by dividing k2 of each donor by that of PeroxyTCM-3.
H2S Release measurement from PeroxyDTCMs
A donor stock solution (200 μL, 10 mM in DMSO) was added to 20 mL of PBS (pH 7.4, 10 mM)/DMSO (9:1) in a scintillation vial. After stirring at room temperature for 5 min, the H2O2 stock solution (20 μL, 1.0 M in H2O) was added, and the H2S release was monitored using methylene blue (MB) protocol. Briefly, 0.5 mL of reaction aliquots were taken to 1-cm UV cuvettes containing 0.5 mL of MB cocktail (0.1 mL zinc acetate (1% w/v), 0.2 mL FeCl3 (30 mM in 1.2 M HCl), and 0.2 mL N,N-dimethyl-p-phenylene diamine (20 mM in 7.2 M HCl)) periodically. The absorbance at 670 nm was then measured after 30 min and was converted to H2S concentration by using the H2S calibration curve.
Stability Investigations
An OA-TCM-1, SA-TCM-1, or OA-TCN-1 stock solution (5 μL, 10 mM in DMSO) was added to 500 μL of PBS (pH 7.4, 10 mM)/CH3CN (1:1) in a 1.5 mL GC vial, followed by the addition of the ROS stock solution (5 μL, 30 mM in H2O). The reaction was analyzed by HPLC, using the following conditions: HPLC gradient: 50% – 0% solvent A (5% MeOH in H2O) in solvent B (MeCN) over 4 min then 100% solvent B over 4 min and 0% – 50% solvent A in solvent B over 4 min. Flow rate: 1 mL/min, 2 μL injection. Column: Agilent Poroshell 120 EC-C18, 4.6 × 100 mm, 2.7 μm (particle size)
Computational Methods
Calculations were performed using the Gaussian 09 software package.87 Conformational searches were conducted through the geometry optimizations of all reasonable input dihedral angles using the 6–311++G(d,p) basis set at the B3LYP level of theory applying the IEFPCM solvation model for water. Frequency calculations were performed on each located stationary point to ensure that it was a local minimum or saddlepoint. Relative energies were calculated using the zero-point-energy-corrected electronic energies obtained in the frequency calculations. A superfine integration grid in Gaussian was used for all computations. Graphical representations of optimized structures were prepared using VESTA.88
Supplementary Material
Figure 3.
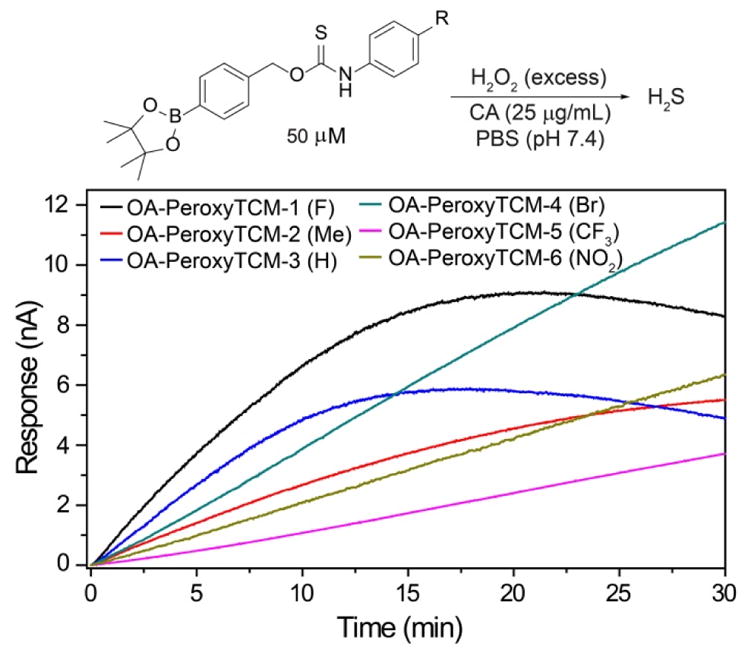
Representative H2S release from OA-PeroxyTCM-1 to 6 in the presence of H2O2.
Acknowledgments
Research reported in this publication was supported in part by the Sloan Foundation, Dreyfus Foundation, and the NIH (R01GM113030). NMR (CHE-1427987) and computational (OCI-0960354) infrastructure at the UO is supported by the NSF.
Footnotes
Notes: The authors declare no competing financial interests.
Supporting Information. H2S release curves, CS2 evaluation, stability studies, NMR spectra, and tabulated coordinates for calculated structures. This material is available free of charge via the Internet at http://pubs.acs.org.”
References
- 1.Wang R. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 2.Li L, Rose P, Moore PK. Annu Rev Pharmacol Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 3.Fukuto JM, Carrington SJ, Tantillo DJ, Harrison JG, Ignarro LJ, Freeman BA, Chen A, Wink DA. Chem Res Toxicol. 2012;25:769–793. doi: 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ono K, Akaike T, Sawa T, Kumagai Y, Wink DA, Tantillo DJ, Hobbs AJ, Nagy P, Xian M, Lin J, Fukuto JM. Free Radic Biol Med. 2014;77:82–94. doi: 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szabo C. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 6.Kolluru GK, Shen X, Bir SC, Kevil CG. Nitric Oxide. 2013;35:5–20. doi: 10.1016/j.niox.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson KR. Antioxid Redox Signal. 2012;17:32–44. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kabil O, Banerjee R. Antioxid Redox Signal. 2014;20:770–782. doi: 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paul BD, Snyder SH. Nat Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 10.Bruce King S. Free Radic Biol Med. 2013;55:1–7. doi: 10.1016/j.freeradbiomed.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szabo C. Antioxid Redox Signal. 2012;17:68–80. doi: 10.1089/ars.2011.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu LF, Lu M, Hon Wong PT, Bian JS. Antioxid Redox Signal. 2011;15:405–419. doi: 10.1089/ars.2010.3517. [DOI] [PubMed] [Google Scholar]
- 13.Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Gastroenterology. 2009;137:569–578. doi: 10.1053/j.gastro.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 14.Zhao W, Zhang J, Lu Y, Wang R. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace JL, Wang R. Nat Rev Drug Discov. 2015;14:329–345. doi: 10.1038/nrd4433. [DOI] [PubMed] [Google Scholar]
- 16.Polhemus DJ, Lefer DJ. Circ Res. 2014;114:730–737. doi: 10.1161/CIRCRESAHA.114.300505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang L, Geng B, Yu F, Zhao J, Jiang H, Du J, Tang C. Amino Acids. 2008;34:573–585. doi: 10.1007/s00726-007-0011-8. [DOI] [PubMed] [Google Scholar]
- 18.Geng B, Chang L, Pan C, Qi Y, Zhao J, Pang Y, Du J, Tang C. Biochem Biophys Res Commun. 2004;318:756–763. doi: 10.1016/j.bbrc.2004.04.094. [DOI] [PubMed] [Google Scholar]
- 19.Calvert JW, Coetzee WA, Lefer DJ. Antioxid Redox Signal. 2010;12:1203–1217. doi: 10.1089/ars.2009.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Predmore BL, Lefer DJ. Expert Rev Clin Pharmacol. 2011;4:83–96. doi: 10.1586/ecp.10.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hartle MD, Pluth MD. Chem Soc Rev. 2016;45:6108–6117. doi: 10.1039/c6cs00212a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Biggs TD, Xian M. Chem Commun. 2014;50:11788–11805. doi: 10.1039/c4cc00968a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y, Pacheco A, Xian M. Handb Exp Pharmacol. 2015;230:365–388. doi: 10.1007/978-3-319-18144-8_18. [DOI] [PubMed] [Google Scholar]
- 24.Kashfi K, Olson KR. Biochem Pharmacol. 2013;85:689–703. doi: 10.1016/j.bcp.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pluth MD, Bailey TS, Hammers MD, Hartle MD, Henthorn HA, Steiger AK. Synlett. 2015;26:2633–2643. [Google Scholar]
- 26.Zheng Y, Yu B, De La Cruz LK, Roy Choudhury M, Anifowose A, Wang B. Med Res Rev. 2017 doi: 10.1002/med.21433. [DOI] [PubMed] [Google Scholar]
- 27.Szabo C, Papapetropoulos A. Pharmacol Rev. 2017;69:497–564. doi: 10.1124/pr.117.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang CT, Chen L, Xu S, Day JJ, Li X, Xian M. Front Pharmacol. 2017;8:664. doi: 10.3389/fphar.2017.00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamizhselvi R, Moore PK, Bhatia M. Pancreas. 2008;36:e24–e31. doi: 10.1097/MPA.0b013e31816857bb. [DOI] [PubMed] [Google Scholar]
- 30.Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- 31.Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, Darley-Usmar VM, Doeller JE, Kraus DW. Proc Natl Acad Sci U S A. 2007;104:17977–17982. doi: 10.1073/pnas.0705710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li L, Whiteman M, Guan YY, Neo KL, Cheng Y, Lee SW, Zhao Y, Baskar R, Tan CH, Moore PK. Circulation. 2008;117:2351–2360. doi: 10.1161/CIRCULATIONAHA.107.753467. [DOI] [PubMed] [Google Scholar]
- 33.Vela-Anero A, Hermida-Gomez T, Gato-Calvo L, Vaamonde-Garcia C, Diaz-Prado S, Meijide-Failde R, Blanco FJ, Burguera EF. Nitric Oxide. 2017;70:42–50. doi: 10.1016/j.niox.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 34.Sun X, Wang W, Dai J, Jin S, Huang J, Guo C, Wang C, Pang L, Wang Y. Sci Rep. 2017;7:3541. doi: 10.1038/s41598-017-03941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Untereiner AA, Olah G, Modis K, Hellmich MR, Szabo C. Biochem Pharmacol. 2017;136:86–98. doi: 10.1016/j.bcp.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weber GJ, Pushpakumar SB, Sen U. Am J Physiol Heart Circ Physiol. 2017;312:H874–H885. doi: 10.1152/ajpheart.00637.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patil A, Singh S, Opere C, Dash A. AAPS PharmSciTech. 2017;18:2291–2302. doi: 10.1208/s12249-017-0712-6. [DOI] [PubMed] [Google Scholar]
- 38.Caliendo G, Cirino G, Santagada V, Wallace JL. J Med Chem. 2010;53:6275–6286. doi: 10.1021/jm901638j. [DOI] [PubMed] [Google Scholar]
- 39.Sparatore A, Santus G, Giustarini D, Rossi R, Del Soldato P. Expert Rev Clin Pharmacol. 2011;4:109–121. doi: 10.1586/ecp.10.122. [DOI] [PubMed] [Google Scholar]
- 40.Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P, Moore PK. Free Radic Biol Med. 2007;42:706–719. doi: 10.1016/j.freeradbiomed.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Y, Wang H, Xian M. J Am Chem Soc. 2011;133:15–17. doi: 10.1021/ja1085723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Y, Bhushan S, Yang C, Otsuka H, Stein JD, Pacheco A, Peng B, Devarie-Baez NO, Aguilar HC, Lefer DJ, Xian M. ACS Chem Biol. 2013;8:1283–1290. doi: 10.1021/cb400090d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Y, Kang J, Park CM, Bagdon PE, Peng B, Xian M. Org Lett. 2014;16:4536–4539. doi: 10.1021/ol502088m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y, Yang C, Organ C, Li Z, Bhushan S, Otsuka H, Pacheco A, Kang J, Aguilar HC, Lefer DJ, Xian M. J Med Chem. 2015;58:7501–7511. doi: 10.1021/acs.jmedchem.5b01033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foster JC, Powell CR, Radzinski SC, Matson JB. Org Lett. 2014;16:1558–1561. doi: 10.1021/ol500385a. [DOI] [PubMed] [Google Scholar]
- 46.Martelli A, Testai L, Citi V, Marino A, Pugliesi I, Barresi E, Nesi G, Rapposelli S, Taliani S, Da Settimo F, Breschi MC, Calderone V. ACS Med Chem Lett. 2013;4:904–908. doi: 10.1021/ml400239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roger T, Raynaud F, Bouillaud F, Ransy C, Simonet S, Crespo C, Bourguignon MP, Villeneuve N, Vilaine JP, Artaud I, Galardon E. Chembiochem. 2013;14:2268–2271. doi: 10.1002/cbic.201300552. [DOI] [PubMed] [Google Scholar]
- 48.Devarie-Baez NO, Bagdon PE, Peng B, Zhao Y, Park CM, Xian M. Org Lett. 2013;15:2786–2789. doi: 10.1021/ol401118k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fukushima N, Ieda N, Sasakura K, Nagano T, Hanaoka K, Suzuki T, Miyata N, Nakagawa H. Chem Commun. 2014;50:587–589. doi: 10.1039/c3cc47421f. [DOI] [PubMed] [Google Scholar]
- 50.Zheng Y, Yu B, Ji K, Pan Z, Chittavong V, Wang B. Angew Chem Int Ed. 2016;55:4514–4518. doi: 10.1002/anie.201511244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shukla P, Khodade VS, SharathChandra M, Chauhan P, Mishra S, Siddaramappa S, Pradeep BE, Singh A, Chakrapani H. Chem Sci. 2017;8:4967–4972. doi: 10.1039/c7sc00873b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kang J, Li Z, Organ CL, Park CM, Yang CT, Pacheco A, Wang D, Lefer DJ, Xian M. J Am Chem Soc. 2016;138:6336–6339. doi: 10.1021/jacs.6b01373. [DOI] [PubMed] [Google Scholar]
- 53.Steiger AK, Yang Y, Royzen M, Pluth MD. Chem Commun. 2017;53:1378–1380. doi: 10.1039/c6cc09547j. [DOI] [PubMed] [Google Scholar]
- 54.Feng S, Zhao Y, Xian M, Wang Q. Acta Biomater. 2015;27:205–213. doi: 10.1016/j.actbio.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu J, Li Y, He C, Kang J, Ye J, Xiao Z, Zhu J, Chen A, Feng S, Li X, Xiao J, Xian M, Wang Q. ACS Appl Mater Interfaces. 2016;8:27474–27481. doi: 10.1021/acsami.6b06466. [DOI] [PubMed] [Google Scholar]
- 56.Qian Y, Matson JB. Adv Drug Deliv Rev. 2017;110–111:137–156. doi: 10.1016/j.addr.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 57.Carter JM, Qian Y, Foster JC, Matson JB. Chem Commun. 2015;51:13131–13134. doi: 10.1039/c5cc04883d. [DOI] [PubMed] [Google Scholar]
- 58.Hasegawa U, van der Vlies AJ. Bioconjug Chem. 2014;25:1290–1300. doi: 10.1021/bc500150s. [DOI] [PubMed] [Google Scholar]
- 59.Foster JC, Radzinski SC, Zou X, Finkielstein CV, Matson JB. Mol Pharm. 2017;14:1300–1306. doi: 10.1021/acs.molpharmaceut.6b01117. [DOI] [PubMed] [Google Scholar]
- 60.Steiger AK, Pardue S, Kevil CG, Pluth MD. J Am Chem Soc. 2016;138:7256–7259. doi: 10.1021/jacs.6b03780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Svoronos PDN, Bruno TJ. Ind Eng Chem Res. 2002;41:5321–5336. [Google Scholar]
- 62.Hanst PL, Spiller LL, Watts DM, Spence JW, Miller MF. J Air Pollut Control Assoc. 1975;25:1220–1226. [Google Scholar]
- 63.Watts SF. Atmos Environ. 2000;34:761–779. [Google Scholar]
- 64.Taubman SJ, Kasting JF. Geophys Res Lett. 1995;22:803–805. [Google Scholar]
- 65.Chengelis CP, Neal RA. Toxicol Appl Pharm. 1980;55:198–202. doi: 10.1016/0041-008x(80)90236-7. [DOI] [PubMed] [Google Scholar]
- 66.Kesselmeier J, Teusch N, Kuhn U. J Geophys Res-Atmos. 1999;104:11577–11584. [Google Scholar]
- 67.Schenk S, Kesselmeier J, Anders E. Chem-Eur J. 2004;10:3091–3105. doi: 10.1002/chem.200305754. [DOI] [PubMed] [Google Scholar]
- 68.Steiger AK, Zhao Y, Pluth MD. Antioxid Redox Signal. 2017 doi: 10.1089/ars.2017.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao Y, Pluth MD. Angew Chem Int Ed. 2016;55:14638–14642. doi: 10.1002/anie.201608052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carballal S, Trujillo M, Cuevasanta E, Bartesaghi S, Moller MN, Folkes LK, Garcia-Bereguiain MA, Gutierrez-Merino C, Wardman P, Denicola A, Radi R, Alvarez B. Free Radical Biology and Medicine. 2011;50:196–205. doi: 10.1016/j.freeradbiomed.2010.10.705. [DOI] [PubMed] [Google Scholar]
- 71.Kimura Y, Goto Y, Kimura H. Antioxid Redox Signal. 2010;12:1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- 72.Powell CR, Foster JC, Okyere B, Theus MH, Matson JB. J Am Chem Soc. 2016:13477–13480. doi: 10.1021/jacs.6b07204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao Y, Bolton SG, Pluth MD. Org Lett. 2017;19:2278–2281. doi: 10.1021/acs.orglett.7b00808. [DOI] [PubMed] [Google Scholar]
- 74.Steiger AK, Marcatti M, Szabo C, Szczesny B, Pluth MD. ACS Chem Biol. 2017;12:2117–2123. doi: 10.1021/acschembio.7b00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chauhan P, Bora P, Ravikumar G, Jos S, Chakrapani H. Org Lett. 2017;19:62–65. doi: 10.1021/acs.orglett.6b03336. [DOI] [PubMed] [Google Scholar]
- 76.DeMartino AW, Zigler DF, Fukuto JM, Ford PC. Chem Soc Rev. 2017;46:21–39. doi: 10.1039/c6cs00585c. [DOI] [PubMed] [Google Scholar]
- 77.DeMartino AW, Souza ML, Ford PC. Chem Sci. 2017;8:7186–7196. doi: 10.1039/c7sc02727c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hill SV, Thea S, Williams A. J Chem Soc Perk T. 1983;2:437–446. [Google Scholar]
- 79.Testai L, Marino A, Piano I, Brancaleone V, Tomita K, Di Cesare Mannelli L, Martelli A, Citi V, Breschi MC, Levi R, Gargini C, Bucci M, Cirino G, Ghelardini C, Calderone V. Pharmacol Res. 2016;113:290–299. doi: 10.1016/j.phrs.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 80.Martelli A, Testai L, Citi V, Marino A, Bellagambi FG, Ghimenti S, Breschi MC, Calderone V. Vascul Pharmacol. 2014;60:32–41. doi: 10.1016/j.vph.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 81.Ensafi AA, Mansour HR, Majlesi R. Anal Sci. 2003;19:1679–1681. doi: 10.2116/analsci.19.1679. [DOI] [PubMed] [Google Scholar]
- 82.Cullen TE. Anal Chem. 1964;36:221–224. [Google Scholar]
- 83.Schwack W, Nyanzi S. Fresen J Anal Chem. 1993;345:705–711. [Google Scholar]
- 84.Schmid KM, Jensen L, Phillips ST. J Org Chem. 2012;77:4363–4374. doi: 10.1021/jo300400q. [DOI] [PubMed] [Google Scholar]
- 85.Blencowe CA, Russell AT, Greco F, Hayes W, Thornthwaite DW. Polymer Chem. 2011;2:773–790. [Google Scholar]
- 86.Leman L, Orgel L, Ghadiri MR. Science. 2004;306:283–286. doi: 10.1126/science.1102722. [DOI] [PubMed] [Google Scholar]
- 87.Frisch MJT, GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Wallingford, CT: 2009. [Google Scholar]
- 88.Momma K, Izumi F. J Appl Cryst. 2011;44:1272–1276. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.