

Chronic traumatic encephalopathy (CTE) is a neuropathologically defined tauopathy: CTE is closely related with repetitive, traumatic brain injury. In a most recent study of 202 deceased players of American football from a brain donation program, CTE was neuropathologically diagnosed in 177 players across all levels of play (87%), including 110 of 111 former National Football League players (99%) (Mez et al., 2017). Consistent with a progressive and neurodegenerative nature, the CTE presentation does not appear until midlife, usually decades after the repetitive brain trauma, and are not simply the aggravation of symptoms from earlier concussions or injuries. At early stage, patient may complain about headache and loss of attention, short-term memory difficulties, depression and impulse control problems. As the disease progresses, symptoms become more severe, with worsening memory impairment, worsening executive dysfunction, language difficulties, and motor disturbance. The patient would become eventually demented and more than 30% patients are suicidal at end stage. As the presentation differs in each individual and most symptoms are rather non-specific, the definitive diagnosis can only be made by postmortem neuropathologic examination. In 2016, the first National Institute of Neurological Disorders and Stroke/National Institute of Biomedical Engineering and Bioengineering (NINDS/NIBIB) consensus meeting defined CTE as a clearly distinct neurodegenerative disease consisting of p-tau aggregates in neurons, astrocytes, and cell processes around small vessels at the depths of the cortical sulci (McKee et al., 2016). It is critical to note that the pathognomonic change has only been found in individuals who were exposed to brain trauma, typically multiple episodes. This is important because neuronal and glial tau inclusions are the pathologic hall marker for other major neurodegenerative diseases, including Alzheimer's disease (AD) and various tauopathies of frontotemporal lobar degeneration (FTLD-tau) (Sun and Chen, 2015). Nevertheless, like in AD and primary tauopathies, what triggers the pathological conversion of the physiologically microtubule-associated protein to misfold and aggregate in CTE is still elusive.
Pathologic aggregation of tau and TAR DNA-binding protein 43 (TDP-43): Intriguingly, phosphorylated TDP-43, previously identified as the common pathologic proteinopathy linking frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) (Neumann et al., 2006), is found in more than 80% CTE cases (McKee et al., 2016), raising the question whether there is a “pathologic synergy” between these two proteins. TDP-43 is a ubiquitously expressed nuclear protein and has a high-binding affinity for either TG repeats in DNA or UG repeats in RNA through its N-terminal RNA recognition motifs (Ling et al., 2015). In contrast, the glycine rich, prion-like C-terminus harbors most of the mutations associated with ALS/FTD and mediates the protein-protein interactions important for transcription, pre-mRNA splicing, mRNA stability and transport (Ling et al., 2015). In ALS/FTD patients, TDP-43 is truncated, phosphorylated, ubiquitinated, and aggregated in the cytoplasm of neuron and glial cells, coinciding with the depletion of nuclear TDP-43 (Neumann et al., 2006). Recent studies has begun to shed some lights on the possible link between TDP-43 and tauopathy. Liu's group found that TDP-43 bound to two (UG)n elements at the 3’-UTR of tau mRNA and promoted its instability, resulting in a negative regulation of tau expression (Gu et al., 2017b). Interestingly in AD cerebellum where it is known to be free of tau pathology, TDP-43 was maintained at a higher level and the tau expression was decreased compared with control cerebellum. Importantly, both full-length and truncated TDP-43 were decreased in the frontal cortices in AD brains, whereas the total tau level was increased, suggesting a downregulation of TDP-43 may contribute to tau accumulation through decreased tau mRNA instability in AD cases. In another study, the same group found that TDP-43 bound intron 9 of tau pre-mRNA and promoted tau exon 10 inclusion, leading to an increase of the more self-assembly prone 4R-tau expression (Gu et al., 2017a). Both the N- and C-termini were required to carry out this function as deletion of the last 108 or the first 99 amino acids abolished the ability of TDP-43 in promoting tau exon 10 inclusion.
Role of TDP-43 in the pathogenesis of CTE: Perhaps the most revealing evidence concerning the role of TDP-43 in the pathogenesis of CTE came from a recent study exploring whether the neuritic plaque is necessary and sufficient for the development of tau pathology (Li et al., 2016). It is known in AD that there is a long pre-symptomatic phase during which time neuritic plaques accumulate without significant tau pathology and apparent cognitive impairment. Using a well-established β-amyloidosis mouse model, APPswe;PS1ΔE9, Li et al. (2016) convincingly demonstrated that the neuritic plaque is critical to facilitate the hyperphosphorylation of tau. However, this neuritic plaque-dependent biochemical alteration of tau failed to convert to tangle-like aggregates even in aged mice that were beyond 24 months. Similarly, a human 4R-tau fragment alone failed to drive the formation of tau tangle. However, when 4R-tau mice was crossbred with APPswe; PS1ΔE9 to generate compound Tau4R; APPswe; PS1ΔE9 (Tau4R-AP) mice, Tau4R-AP mice exhibited marked neuronal loss in forebrain and hippocampus together with the deposition of neurofibrillary tangles (NFTs). Taken together, their findings strongly argue that in addition to the neuritic plaque, a second-risk determinant is necessary and sufficient to drive the pathological conversion of wild-type tau. Thus, a combination of first- and second-risk determinants are required to initiate and complete the pathological conversion of the wild-type tau (Figure 1). This “two-hit” hypothesis could explain well the observation that those pre-symptomatic or mild cognitive impaired patients having substantial β-amyloid accumulation without tau pathology is simply because they fail to harbor the second-risk determinant that is required to drive the conversion of tau. Like the fragmentation of 4R-tau, loss of TDP-43 nuclear function is highly likely to act as a second-risk determinant in AD. Likewise, this model could also explain the pathogenesis of CTE as a multifactorial problem initiated by the trauma induced hyperphosphorylation of tau, followed by activation of a variety of potential second-risk determinants including the loss of TDP-43 nuclear function to drive the pathological conversion of tau (Figure 1). Indeed, transcriptome analyses of CTE brains has shown reduced expression of PPP3CA, a subunit of calcineurin which dephosphorylates tau (Seo et al., 2017). In addition, cleavage of TDP-43 due to activation of calpain and caspase-3 caused by traumatic brain injury can lead directly to diminished TDP-43 (Yang et al., 2014). Thus, both first- and second-risk determinants exist in CTE. On the other hand, it is known that ALS or FTLD-motor neuron disease (FTLD-MND) has negligible tau pathology in brain or spinal cord. Despite the predominant TDP-43 pathology in both ALS/FTLD-MND, it is very likely an alternative risk allele or factor is missing to cause tau hyperphosphorylation in the first place.
Figure 1.
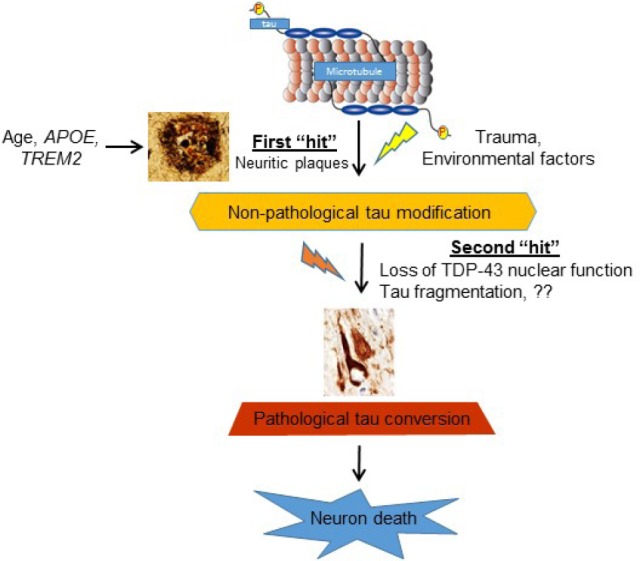
“Two-hit” model for pathologic conversion of tau.
The neuritic plaque is required, but insufficient for the pathological conversion of tau in Alzheimer's disease (AD). Similarly, traumatic brain injury in chronic traumatic encephalopathy (CTE) is necessary but insufficient to cause tau tangle formation. A second-risk determinant involving a variety of risk alleles/factors including loss of nuclear TAR DNA-binding protein 43 (TDP-43) function, is necessary to facilitate the pathological conversion of the wild-type tau that will eventually lead to neuronal loss and cognitive dysfunction.
Conclusions: Significant progress has been made over the past two decades on the pathogenesis of individual neurodegenerative diseases, including AD, CTE, as well as their distinct neurodegenerative processes. Apart from descriptive postmortem neuropathological studies, different neurodegenerative syndromes have been mainly studied mechanistically in isolation from one another. There has been a lack of concerted effort to ascertain whether and how these pathogenic processes may be linked to one another, a great unmet need in the field. TDP-43 proteinopathy, a pathology first linked to ALS and FTD, is one of the most common non-canonical pathologies observed in AD cases and is strongly associated with worsened neurodegeneration and cognition. Importantly, abnormal TDP-43 inclusions are also found in most CTE brains, suggesting a convergent mechanism of neurodegeneration. Animal models, including vertebrate and invertebrate models (Li et al., 2016; Sun and Chen, 2017) would be great assets to determine whether and how loss of TDP-43 function interacts with canonical tauopathy in AD and CTE. Recent study has demonstrated that TDP-43 is a splicing repressor and this function is compromised in ALS and FTD (Ling et al., 2015). We have recently extended the finding to include such defect in brains of AD cases with nuclear clearance of TDP-43, suggesting that loss of TDP-43 function from neurons, a common shared feature with ALS and FTD, could contribute to pathogenesis of AD (Sun et al., 2017). Remarkably, the observation of cryptic splicing defects in AD brains prior to TDP-43 inclusion formation suggests that these TDP-43 targets may represent early biomarkers of disease or tools to monitor the efficacy of treatment in clinical trials. It would be important to show that cryptic exon incorporation also exists in CTE brains. It will establish that loss of TDP-43-mediated splicing repression represents a key conserved mechanism underling multiple major neurodegenerative disorders. An AAV9-mediated gene delivery approach can be employed in animal models and clinical trials to deliver a chimeric protein comprised of the N-terminal RNA recognition domain of TDP-43 fused to an unrelated splicing repressor (RAVER1), an approached has been validated in mouse embryonic stem cells (Ling et al., 2015). As predicted by the “two-hit” model, therapies directed at the second-risk determinant long before the pathological conversion of tau would greatly slow disease progression by delaying the onset of tau pathology.
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- Gu J, Chen F, Iqbal K, Gong CX, Wang X, Liu F. Transactive response DNA-binding protein 43 (TDP-43) regulates alternative splicing of tau exon 10: Implications for the pathogenesis of tauopathies. J Biol Chem. 2017a;292:10600–10612. doi: 10.1074/jbc.M117.783498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Wu F, Xu W, Shi J, Hu W, Jin N, Qian W, Wang X, Iqbal K, Gong CX, Liu F. TDP-43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Res. 2017b;45:6177–6193. doi: 10.1093/nar/gkx175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Braunstein KE, Zhang J, Lau A, Sibener L, Deeble C, Wong PC. The neuritic plaque facilitates pathological conversion of tau in an Alzheimer's disease mouse model. Nat Commun. 2016;7:12082. doi: 10.1038/ncomms12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC, Wong PC. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349:650–655. doi: 10.1126/science.aab0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O’Connor K, Alvarez VE, Gordon WA TBI/CTE group. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131:75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, Alosco ML, Solomon TM, Nowinski CJ, McHale L, Cormier KA, Kubilus CA, Martin BM, Murphy L, Baugh CM, Montenigro PH, Chaisson CE, Tripodis Y, Kowall NW, Weuve J, et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA. 2017;318:360–370. doi: 10.1001/jama.2017.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Seo JS, Lee S, Shin JY, Hwang YJ, Cho H, Yoo SK, Kim Y, Lim S, Kim YK, Hwang EM, Kim SH, Kim CH, Hyeon SJ, Yun JY, Kim J, Kim Y, Alvarez VE, Stein TD, Lee J, Kim DJ, et al. Transcriptome analyses of chronic traumatic encephalopathy show alterations in protein phosphatase expression associated with tauopathy. Exp Mol Med. 2017;49:e333. doi: 10.1038/emm.2017.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Chen L. Studying tauopathies in Drosophila: A fruitful model. Exp Neurol. 2015;274:52–57. doi: 10.1016/j.expneurol.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Sun M, Chen LL. A novel method to model chronic traumatic encephalopathy in Drosophila. J Vis Exp. 2017 doi: 10.3791/55602. doi: 10.3791/55602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Bell W, LaClair KD, Ling JP, Han H, Kageyama Y, Pletnikova O, Troncoso JC, Wong PC, Chen LL. Cryptic exon incorporation occurs in Alzheimer's brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol. 2017;133:923–931. doi: 10.1007/s00401-017-1701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Lin F, Robertson CS, Wang KK. Dual vulnerability of TDP-43 to calpain and caspase-3 proteolysis after neurotoxic conditions and traumatic brain injury. J Cereb Blood Flow Metab. 2014;34:1444–1452. doi: 10.1038/jcbfm.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]