

Abstract
Background & Aims
Southern Guangxi area is one of the endemic areas for hepatocellular carcinoma (HCC) in China. This study evaluates the roles of genetic variations of hepatitis B virus (HBV) and aflatoxin B1 (AFB1) exposure in the formation of HCC in this high-risk area.
Methods
The study recruited 60 HCC patients and 120 age-, gender-, residency-matched controls. HBV genotype and basic core promoter (BCP) mutations were determined by nested-PCR/direct sequencing. Serum AFB1-lysine adduct was measured by high performance liquid chromatography-fluorescence detection.
Results
HBV Genotype C was predominant in 75.0% of cases and 84.2% of controls. The 1762T/1764A double mutations, 1753V mutations, and 1752V mutations were associated with HCC risk evidenced by the adjusted odds ratio (OR) [95% confidence interval (95% CI)] of 3.89 (1.40–10.77), 2.87 (1.49–5.49), and 5.96 (1.75–20.25), respectively. The adjusted OR (95% CI) was 6.94 (1.68–27.78) for subjects with 1762T/1764A double mutations and high AFB1-lysine adduct level; 2.01 (0.24–14.29), for those with only 1762T/1764A double mutations; and 4.26 (1.16–15.38) for those with only high AFB1-lysine adduct level, respectively. The adjusted OR was 5.13 (1.79–14.71) for subjects with 1753V mutations and high AFB1-lysine adduct level; 1.20 (0.47–3.08) for those with only 1753V mutations, and 2.28 (1.01–5.31) for those with high AFB1-lysine adduct level, respectively.
Conclusions
These data confirmed the association of BCP mutations with HCC risk and the additive effects of 1762T/1764A double mutations and 1753V mutations with dietary AFB1 exposure in this high-risk area for HCC.
Keywords: hepatitis B virus, genotype, BCP mutations, aflatoxin-lysine adduct, hepatocellular carcinoma
Introduction
Chronic hepatitis B virus (HBV) infection is the major cause of hepatocellular carcinoma (HCC) worldwide [1]. Although the mechanism is not well defined, variations in the viral genome, including specific genotypes and mutations, are thought to contribute to the formation of HCC [2,3]. To date, eight HBV genotypes are classified (A–H) based on an inter-group divergence of equal or greater than 8% in the complete nucleotide sequence [4]. Infection with the genotype C was associated with greater severity of liver diseases compared to genotype B, although this is still controversial [2,5]. The basic core promoter (BCP) resides in the overlapping HBV functional X gene domain and controls the transcription activity of procore RNA [6]. Mutations in this region were anticipated to influence HBV-induced chronic infection and hepatocarcinogenesis [7,8]. The 1762T/1764A double mutations (1762A-to-T and 1764G-to-A) in BCP region were commonly found to be borne by HCC patients in some high-risk populations and were thus suggested as potential biomarkers for hepatocarcinogenesis [3,9–13]. Also, the 1753V mutations (1753T-to-C/A/G) were associated with the progression of liver disease [14,15]; nevertheless, their role in HCC development, especially in high risk populations, remains controversial [9,14,16].
The Southern Guangxi area is one of the areas in China with endemic HCC, reporting an incidence and mortality rate of more than 50/100,000 people per year [17]. Chronic HBV infection and dietary aflatoxin (AF) exposure are the two major risk factors for HCC in this area [18], while hepatitis C virus (HCV) infection is extremely uncommon [19]. AFB1-lysine adduct in serum has been considered the most reliable biomarker in monitoring long-term human AFB1 exposure [20]. The association of this adduct with HCC has been well established in high-risk populations in China [21], while its application in assessing HCC risk in southern Guangxi high-risk population has not yet been reported.
To this end, a case-control study was conducted to assess the role of HBV genetic variations, including HBV genotypes and BCP region mutations, dietary AFB1 exposure measured by serum AFB1-lysine adduct level, and their interactions in HCC risk in the Southern Guangxi population.
Materials and Methods
Study subjects, data collection and sample collection
A total of 60 HCC patients were recruited at Guangxi Medical University Cancer Hospital from August 2004 to August 2005. The eligibility criteria for cases were: (1) permanent resident in southern Guangxi area; (2) confirmed diagnosis of HCC as primary tumor by histological (surgical biopsy) or non-histological criteria (positive imaging test results, serum α-fetoprotein greater than 400 ng/ml, and clinical features); (3) no previous diagnosis of other cancers; (4) positive HBV surface antigen (HBsAg) test result when admitted into the hospital, using an enzyme-linked immunosorbent assay (ELISA) kit (Kehua Bio-engineering, Shanghai, China), and (5) negative HCV infection demonstrated by anti-HCV test using a ELISA kit (Kehua Bio-engineering, Shanghai, China). The 120 population-based controls were recruited from an ongoing cohort study conducted in Southern Guangxi area and were frequency-matched to cases on gender, age (in 10-year age groups) and place of residence (township). The inclusion criteria for these controls were: (1) normal liver and renal function tests as well as electrocardiograms; (2) negative for serum α-fetoprotein; (3) no cancer history; (4) no use of prescribed medications; (5) no pregnancy or lactation for female volunteers; (6) positive HBsAg test, and (7) negative HCV infection by anti-HCV test. Demographic information of cases was obtained from the case history in the hospital archive. Demographic information of controls was collected through in-person interviews by trained interviewers who administered structured questionnaires. Additionally, whole blood samples (7 ml) were taken from cases at the time of admission to hospital and collected from controls at the time of recruitment. Serum samples were separated and stored frozen (−20°C) for shipping to the US laboratory for further analysis. The study protocol was approved by the Institutional Review Board for human subject protections at Texas Tech University and Guangxi Cancer Institute.
Determination of HBV genotypes and mutations
HBV DNA was extracted from 100 μl serum using a commercial viral extraction kit (Qiagen, Valencia, CA). A nested polymerase chain reaction (nPCR) was designed to amplify the major part of HBV X gene including BCP region. First round amplification was carried out using the sense primer H1 5′ - GCT TTC ACT TTC TCG CCA AC -3′ and antisense primer RH1 5′ - TGG AGG CTT GAA CAG TAG GAC -3′ with 767bp of PCR product. Second round amplification used first round PCR product as a template and was processed using the sense primer HBX1 5′ - GCC AAG TGT TTG CTG ACG C -3′ and antisense primer RHBX1 5′ - AAA GTT GCA TGG GGC TGG TG -3′ with 648 bp of PCR product. HBV amplicons were cleaned-up by a commercial gel purification kit (Qiagen, Valencia, CA) and sequenced subsequently in the ABI 3730XL sequencers (MCLAB, South San Francisco, CA). HBV genotypes were determined by the web based program-Viral Genotyping Tool [22]. For mutation analysis, the sequences generated were compared with non-mutational reference sequence (AY641563) using the BLAST program.
Measurement of serum AFB1-lysine adduct
Levels of serum AFB1-lysine adduct were measured by a high performance liquid chromatography (HPLC)-fluorescence detection [23] with modifications. In short, 150 μl of the serum sample was digested by Pronase (Calbiochem, San Diego, CA) for 3 hours at 37 °C and purified with Oasis Max cartridge (Water Co., Milford, MA). After elution with 2% formic acid in methanol, the eluents containing AFB1-lysine adduct were evaporated until dry and reconstituted with 10% methanol in PBS.
HPLC analysis was carried out on an 1100 liquid chromatography system (Agilent, Wilmington, DE). Chromatographic separation was performed on an Agilent C18 column (5 μM, 250 × 4.6 mm). The mobile phase consisted of 20 nm ammonium phosphate monobasic (pH 7.2) and methanol in a linear gradient profile. The concentration of AFB1-lysine adduct was monitored at the wavelength 405 nm (excitation) and 470 nm (emission). The typical retention time of authentic AFB1-lysine adduct was 12.7 minutes. The results of AFB1-lysine adduct concentration was adjusted for serum albumin level. The limit of detection was 0.5 pg/mg albumin.
Statistical Analysis
Fisher’s exact test, Chi-square test, t-test, or Wilcoxon test were used as appropriate to examine difference between cases and controls. Odds ratio (OR) and 95 percent confidence intervals (95% CI) for HCC risk were calculated using unconditional logistic regression, and a full assessment of potential confounding factors was conducted. First-degree family history of cancer was added as a confounding factor into the final logistic regression model since it was unevenly distributed in cases and controls (Table 1). Age and gender were fitted into the final model too. Other variables were included in the final model if they changed the target odds ratio by 10 percent or more when added to the unadjusted model [24]. Stratified analyses were performed on HBV genotypes, BCP mutations, and AFB1-lysine adduct levels to evaluate whether HCC risk differed with these factors. Considering the distribution frequency in each subgroup, the mean level of AFB1-lysine adduct in the control group was used as a cut-off point in the stratification of low or high AFB1-lysine levels. A p-value of less than 0.05 (two-tailed) was considered statistically significant. All data were analyzed by using SAS software version 9.1.5 (SAS Institute Inc., Cary, NC, USA).
Table 1.
Demographic characteristics of HCC cases and controls.
| Factor | Cases | Controls | p value |
|---|---|---|---|
| Age (years) (mean ± SD*) | 41±9 | 42±7 | 0.300 |
| Gender [n (%)] | |||
| Male | 54(90.0) | 108(90.0) | |
| Female | 6(10.0) | 12(10.0) | 1.000 |
| Current cigarette smoker [n (%)] | |||
| Yes | 26(43.3) | 63(52.5) | |
| No | 34(56.7) | 57(47.5) | 0.246 |
| Total pack-years of smoking (mean ± SD) | 16.6±9.0 | 23.2±14.1 | 0.059 |
| Alcohol drinker [n (%)] | |||
| Yes | 20(33.3) | 36(30.0) | |
| No | 40(66.7) | 84(70.0) | 0.649 |
| First-degree family history of cancer [n (%)] | |||
| Yes | 14(23.3) | 13(10.8) | |
| No | 46(76.7) | 107(89.2)** | 0.027 |
SD, standard deviation
p <0.05 from chi-square test
Results
As shown in Table 1, the cases and controls, taking into account the frequency-matched age, gender and residency, had similar demographic characteristics except for their first-degree familial history of cancer. Among 14 cases with first-degree family history of cancer, there were 12 liver cancers (85.7%), one esophageal cancer (7.1%), and one rectum cancer (7.1%). Among 13 controls with first-degree family history of cancer, there were 10 liver cancers (76.9%), one esophageal cancer (7.7%), one lung cancer (7.7%), and one gastric cancer (7.7%), respectively.
Genotypes B, C, A+C and B+C were detected with genotype C predominant in both cases and controls (75% and 84.2%) (Table 2). No significant difference was found in the distribution of genotypes between the cases and controls (p = 0.280).
Table 2.
HBV genotypes in HCC cases and controls.
| Groups | N | HBV genotypes [n (%)] | |||
|---|---|---|---|---|---|
| A | B | C | D | ||
| HCC Cases | 60 | 13(21.7) | 45(75.0) | 0(0.0) | 2(3.3) |
| Controls | 120 | 17(14.2) | 101(84.2) | 2(1.7) | 0(0.0) |
The distribution of BCP mutations is presented in Table 3. Detectable 1762T/1764A double mutations was found in 91.7% (55/60) cases and 72.5% (88/120) controls with significant difference (p = 0.007). The 1762T single mutation was rare and only found in one genotype C-control. When stratified by HBV genotypes, 1762T/1764A double mutations/1762T single mutation still showed significant difference between cases and controls. In genotype B-cases, the detectable rate of 1762T/1764A double mutations was 69.2% (9/13), which was significantly higher than the rate of 23.5% (4/17) in genotype B-controls after adjustment of age, gender, first-degree family history of cancer (p = 0.017). The detectable rate of 97.8% (44/45) in genotype C-cases was also significantly higher than the rate in genotype C-controls (83.2%, 84/101) after adjustment for age, gender, first-degree family history of cancer (p = 0.044). Of particular interest, a significantly higher rate of 1762T/1764A double mutations was found in genotype C-subjects compared to genotype B-subjects (case group: 97.8% vs. 69.2%, p = 0.007; control group: 82.2% vs. 23.5%, p <0.001).
Table 3.
HBV BCP mutations in HCC cases and controls.
| BCP Mutations | Controls (%) | Cases (%) | OR (95% CI)* | Adjusted OR (95%CI) | |
|---|---|---|---|---|---|
| 1762T/1764A mutations | No (1762A/1764G) | 31(25.8) | 5 (8.3) | 1.00 | 1.00 |
| Yes (1762T/1764G or 1762T/1764A)** | 89(74.1) | 55(91.7)*** | 3.83(1.41–10.41) | 3.89(1.40–10.77)# | |
| 1753V mutations | No (1753T) | 89 (74.2) | 30 (50.0) | 1.00 | 1.00 |
| Yes (1753CAG) | 31(25.8) | 30(50.0)*** | 2.87 (1.50–5.50) | 2.78(1.43–5.40)## | |
| 1752V mutations | No (1752A) | 116 (96.7) | 50(83.3) | 1.00 | 1.00 |
| Yes (1752TCG) | 4 (3.3) | 10(16.7)*** | 5.80(1.74–19.37) | 5.96(1.75–20.25)# |
OR, odds ratio; CI, confidence interval
1 single mutation (1762T/1764G) was found in control group, all others were 1762T/1764A double mutations.
p <0.05 from unconditional logistic regression in comparing the distribution of mutations in cases and controls
adjusted for age, gender, first-degree family history of cancer;
adjusted for age, gender, alcohol drinking status, first-degree family history of cancer.
The rate of 1753V mutations was more prevalent in cases than in controls (50.0% vs. 25.8%, p = 0.001) (Table 3). Subjects having 1753V mutations had an increased risk for HCC (adjusted OR = 2.78, 95% CI: 1.43–5.40). After stratifying by HBV genotype, the rates of 1753V mutations were significantly higher in genotype C-cases compared to genotype C-controls (60.0%, 27/45 vs. 29.7%, 30/101, p <0.001), whereas these types of mutations were rarely detected in genotype B-cases (2/13) and -controls (1/17), which showed no significant difference (p = 0.565). In all 1753V mutations, 1753C mutation was the major type detected in both cases (83.3%) and controls (90.3%). There were five other 1753G mutations (4 cases and 1 control) and three 1753A mutations (1 case and 2 controls).
Moreover, the co-existence of 1753V mutations and 1762T/1764A double mutations was usually detected. Among 30 cases with 1753V mutations, 29 (96.7%) had 1762T/1764A double mutations; and of the 31 controls with 1753V mutations, 29 (93.5%) had the double mutations. In addition, combinations of 1753C mutation and 1762T/1764A double mutations were detected in 40.0% (24/60) of cases and 22.5% (27/120) of controls (age-, gender-, family history of cancer-adjusted OR: 2.35, 95% CI: 1.18–4.70, p = 0.016).
Moreover, several other mutations in BCP region were detected in study subjects at low frequencies. 1752V mutation (A-to-T/C/G) was found in 10 (16.7%) cases and 4 (3.3%) controls, showing a significant increase of HCC risk (p = 0.004) (Table 3). Mutation at position 1751 (G-to-A) was detected in 1 (1.7%) case. Mutation at position 1757 (G-to-A) was detected in 3 (5.0%) cases and 5 (4.2%) controls. Also, mutation at position 1768 (T-to-A) was found in 3 (5%) cases, and 1 of them was accompanied by a point mutation at position 1766 (C-to-T).
The distribution of serum AFB1-lysine adduct in HCC cases and controls was illustrated in Figure 1. The average level of serum AFB1-lysine adduct in cases was 11.10 ± 30.13 (mean ± SD) pg/mg albumin, with the median level of 4.84 pg/mg albumin (range: 0.54–227.13). The average level of AFB1-lysine adduct in matched control group was 6.46 ± 8.34 (mean ± SD) pg/mg albumin with a median level of 3.77 pg/mg albumin (range: 0.79–51.93). Although levels of AFB1-lysine adduct were higher in cases than controls, no statistically significant difference was found (p = 0.115).
Fig. 1. Serum AFB1-lysine adducts level in cases and controls.
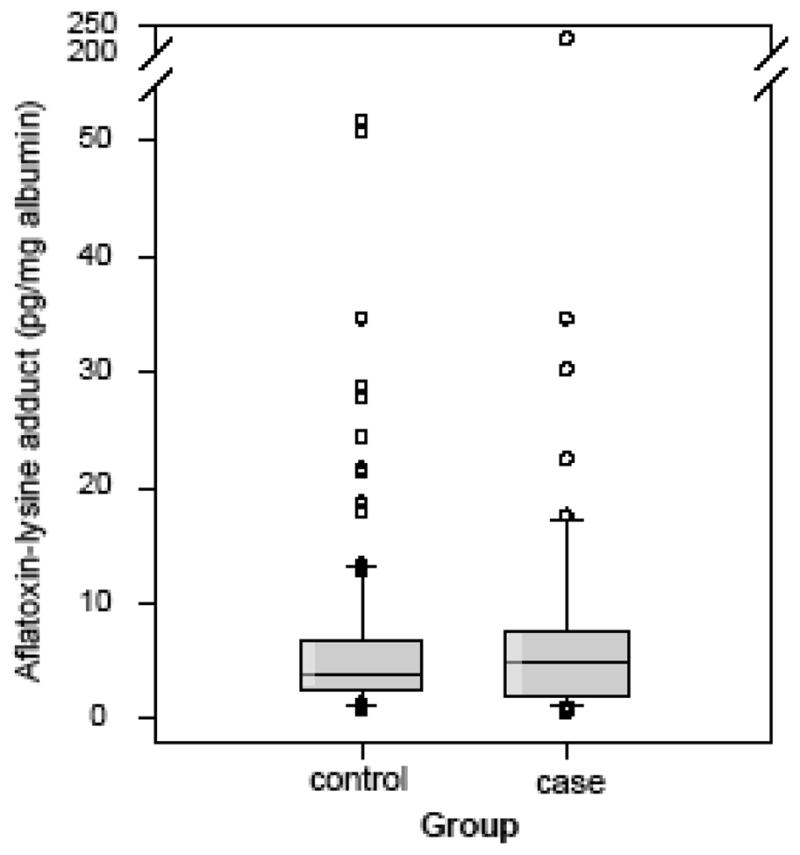
The median and interquartile range of percentages in cases and controls were 4.84 (1.88–7.65) pg/mg albumin and 3.77 (2.62–6.78) pg/mg albumin, respectively. The dots, error bars and upper and lower ends of the box represent outliers, spread, and first and third quartiles, respectively.
Data for both AFB1-lysine adduct levels and HBV BCP mutations on the risk of developing HCC are presented in Table 4. It was found that there was additive effects of 1762T/1764A double mutations and 1753V mutations, and dietary AFB1 exposure: the adjusted OR (95% CI) was 6.94 (1.68–27.78) for subjects with 1762T/1764A double mutations and high AFB1-lysine adduct level; it was 2.01 (0.24–14.29) for those with only high AFB1-lysine adduct level, and 4.26 (1.16–15.38) for those with only 1762T/1764A double mutations, respectively. The adjusted OR was 5.13 (1.79–14.71) for subjects with 1753V mutations and high AFB1-lysine adduct level; 1.20 (0.47–3.08) for those with only a high AFB1-lysine adduct level, and 2.28 (1.01–5.31) for those with only 1753V mutations, respectively.
Table 4.
HCC risks in relation to HBV mutations and AFB1 exposure in cases and controls.
| BCP Mutations | AFB1-lysine adduct | Controls (%) | Cases (%) | OR (95% CI) | Adjusted OR (95%CI)* |
|---|---|---|---|---|---|
| 1762T/1764A double mutations or 1762T single mutations | Concentration (pgrfmg alb.) | ||||
| No | <6.46 | 22(18.3) | 3 (5.0) | 1.00 | 1.00 |
| ≥6.46 | 9 (7.5) | 2 (3.3) | 1.63 (0.23–11.45) | 2.01 (0.24–14.49) | |
| Yes | <6.46 | 66 (55.0) | 36 (60.0)** | 4.00(1.12–14.27) | 4.26 (1.16–15.38) |
| ≥6.46 | 23(19.2) | 19(31.7)** | 6.06(1.57–23.37) | 6.94 (1.68–27.78) | |
| 1752V mutations | Concentration (pg/mg alb.) | ||||
| No | <6.46 | 65 (54.2) | 21(35.0) | 1.00 | 1.00 |
| ≥6.46 | 24 (20.0) | 9 (15.0) | 1.16(0.47–2.89) | 1.20 (0.47–3.08) | |
| Yes | <6.46 | 23(19.2) | 18(30.0)** | 2.42(1.10–5.33) | 2.28(1.01–5.31) |
| ≥6.46 | 8 (6.7) | 12(20.0)** | 4.65(1.67–12.89) | 5.13 (1.79–14.71) |
adjusted for age, gender, first-degree family history of cancer
p <0.05 from unconditional logistic regression in comparison with baseline group (no mutation and lower than average AFB1-lysin adduct)
Discussion
Our study showed a significant association of increased HCC risk with HBV 1762T/1764A double mutations, 1753V mutations, and 1752V mutations in the Southern Guangxi area, a high-risk area for HCC in China. In addition, we found additive effects of HBV BCP mutations (1762T/1764A double mutations and 1753V mutations) and high serum AFB1-lysine adduct level on the risk of developing HCC. To our knowledge, there has been no previously published data exploring the interactions between HBV genomic variations (HBV genotypes, BCP mutations) and AFB1 exposure biomarkers on HCC risks in the literature.
HBV genotype B and C are the two common genotypes in China [25–27] and Vietnam [28,29], a neighboring country of Southern Guangxi, where more than 80% of subjects have genotypes B or C. A previous study in Guangxi area reported 100% of genotype C in 20 asymptomatic HBV carriers [30], which was higher but still comparable to 84.2% of genotype C in our controls.
Several studies reported that genotype C was associated with a higher HCC risk as compared to genotype B [2,5]. However, the lack of such association was also reported by several other studies. A cohort study following up HBV infected children for 15 years in Taiwan found no increased HCC risk in genotype C-children, and instead, genotype B was the predominant type (74%) in HCC patients [31]. Also, in Qidong, another HCC high-risk area in China, genotype C was found to be predominant but with no difference between HCC patients and controls [32]. Similarly, our study showed this lack of association. Additionally, we found a significantly higher frequency of 1762T/1764A double mutations in genotype C-subjects than genotype B-subjects, which is consistent with previous studies [32,33] suggesting a possible linkage between 1762T/1764A double mutations and genotype C.
The 1762T/1764A double mutations and 1753V mutations are the two most common mutations in the HBV BCP region. Although the pathogenic mechanism remains unclear, an in vitro study found that 1762T/1764A double mutations and 1753V mutations could suppress the expression of HBeAg and induce a modest increase in viral replication rate [7,8]. The association of 1762T/1764A double mutations with the increased HCC risk was well established in high-risk populations in China and South Africa [9,10,34,35]. The relationship between 1753V mutations and HCC development is less studied [14], and remains controversial, especially in high risk populations [9,14,16]. Our study supports the association of both 1762T/1764A double mutations and 1753V mutations with HCC risk. The noteworthy fact is that 1753V mutations presented at a lower frequency in both cases (50.0%) and controls (25.8%) than 1762T/1764A double mutations (91.7% and 74.1%, respectively), and always occurred along with the double mutations. Therefore, it is reasonable to hypothesize that mutations in the BCP region may appear in a time-dependent manner: 1762T/1764A double mutations may be involved in the early stage of hepatocarcinogenesis and 1753V mutations may occur at a later stage. A previous study found that 1762T/1764A double mutations were persistently detected in patients over a period of four years before the diagnosis of HCC [3]. Although there is no such longitudinal study for 1753V mutations, a study reported that the prevalence rate of 1753V mutations dramatically increased with the severity of liver diseases: 0% in asymptomatic HBV carriers, 9% in chronic hepatitis patients, and 47% in liver cirrhosis or HCC patients [15]. Therefore, 1762T/1764A double mutations and 1753V mutations may serve as valuable biomarkers for HCC risk among individuals with HBV chronic infection, especially in high risk areas.
Besides, our study found that 1752V mutations in BCP region were also associated to HCC risk. There is no previously published data indicating such an association. Based on the low frequency of 1752V mutations detected in study subjects (3.3%–16.7%), it seems that these mutations appear later after 1753v mutations, which suggests these mutations as possible biomarkers for monitoring high-risk population. In a previous study conducted in Qidong, China, 1766T or 1768A mutation accompanying 1762T/1764A double mutations was associated with increased HCC risk [32]. However, the point mutation at position 1766 and 1768 was rarely found in our study subjects.
The contributing role of AFB1 to HCC risk is confirmative in many populations [36]. Combined dietary AF exposure and chronic HBV infection were correlated with HCC risk in high-risk areas in China. In a nested case-control study from a Shanghai cohort, the relative risk for HCC in HBsAg and urinary AFB1 metabolites positive subjects was found 59 times higher than those negative in HBsAg and urinary AFB1 metabolites [37]. Two other studies in Taiwan confirmed the combinative effect of HBV infection and AFB1 exposure on HCC risk [38, 39]. As proposed [40], it is possible that chronic HBV infection could fix and enhance DNA damage and oxidative stress induced by AFB1, and vice versa. Since BCP mutations could alter HBV function/toxicity [7,8], we anticipated that the interaction between HBV and AF on HCC could be aggravated by BCP mutations. Our study indicated the additive effects of serum AFB1-lysine adduct level with both 1762T/1764A double mutations and 1753V mutation on the risk of developing HCC.
Exposure to AF-contaminated food in Southern Guangxi residents was repeatedly reported over the past several decades [17,18,41,42]. This study confirmed the high AF-exposure in both cases and controls, which were matched with residence to ensure similar dietary intake background. Also, to avoid temporal variation as a modifying factor, the blood samples were collected within the same season of the year in case and control groups. Thus, overwhelming dietary exposure to AF may result in no significant difference of AFB1-lysine adduct levels between cases and controls in this study.
In summary, this study confirmed the association of BCP mutations with HCC development; based on data from this study and available evidence, the accumulation of 1762T/1764A double mutations, 1753V mutations, and 1752V mutations may occur at different stages of hepatocarcinogenesis. Limitations of this study include the nature of the case-control design and the limited sample size. Further evaluation and confirmation will be valuable in cohort studies with a larger sample size.
Acknowledgments
Funding
National Cancer Institute (RO1 90997) to J.-S W.
Abbreviations
- HBV
hepatitis B virus
- AFB1
aflatoxin B1
- HCC
hepatocellular carcinoma
- BCP
basic core promoter
- OR
odds ratio
- CI
confidence interval
- AF
aflatoxin; hepatitis C virus (HCV)
- IARC
International Agency for Research on Cancer
- HBsAg
HBV surface antigen
- HPLC
high performance liquid chromatography
- nPCR
nested polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Garcia M, Jemal A, Ward EM, Center MM, Hao Y, et al. Global Cancer Facts & Figures 2007. Atlanta, GA: American Cancer Society; 2007. [Google Scholar]
- 2.Kao JH. Hepatitis B viral genotypes: clinical relevance and molecular characteristics. J Gastroenterol Hepatol. 2002;17:643–650. doi: 10.1046/j.1440-1746.2002.02737.x. [DOI] [PubMed] [Google Scholar]
- 3.Kuang SY, Jackson PE, Wang JB, Lu PX, Munoz A, Qian GS, et al. Specific mutations of hepatitis B virus in plasma predict liver cancer development. Proc Natl Acad Sci U S A. 2004;101:3575–3580. doi: 10.1073/pnas.0308232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyakawa Y, Mizokami M. Classifying hepatitis B virus genotypes. Intervirology. 2003;46:329–338. doi: 10.1159/000074988. [DOI] [PubMed] [Google Scholar]
- 5.Chan HL, Hui AY, Wong ML, Tse AM, Hung LC, Wong VW, et al. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut. 2004;53:1494–1498. doi: 10.1136/gut.2003.033324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrisani OM, Barnabas S. The transcriptional function of the hepatitis B virus X protein and its role in hepatocarcinogenesis (Review) Int J Oncol. 1999;15:373–379. doi: 10.3892/ijo.15.2.373. [DOI] [PubMed] [Google Scholar]
- 7.Gunther S, Piwon N, Will H. Wild-type levels of pregenomic RNA and replication but reduced pre-C RNA and e-antigen synthesis of hepatitis B virus with C(1653) --> T, A(1762) --> T and G(1764) --> A mutations in the core promoter. J Gen Virol. 1998;79:375–380. doi: 10.1099/0022-1317-79-2-375. [DOI] [PubMed] [Google Scholar]
- 8.Jammeh S, Tavner F, Watson R, Thomas HC, Karayiannis P. Effect of basal core promoter and pre-core mutations on hepatitis B virus replication. J Gen Virol. 2008;89:901–909. doi: 10.1099/vir.0.83468-0. [DOI] [PubMed] [Google Scholar]
- 9.Fang ZL, Sabin CA, Dong BQ, Ge LY, Wei SC, Chen QY, et al. HBV A1762T, G1764A mutations are a valuable biomarker for identifying a subset of male HBsAg carriers at extremely high risk of hepatocellular carcinoma: a prospective study. Am J Gastroenterol. 2008;103:2254–2262. doi: 10.1111/j.1572-0241.2008.01974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan JM, Ambinder A, Fan Y, Gao YT, Yu MC, Groopman JD. Prospective evaluation of hepatitis B 1762(T)/1764(A) mutations on hepatocellular carcinoma development in Shanghai, China. Cancer Epidemiol Biomarkers Prev. 2009;18:590–594. doi: 10.1158/1055-9965.EPI-08-0966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JG, Kuang SY, Egner PA, Lu JH, Zhu YR, Wang JB, et al. Acceleration to death from liver cancer in people with hepatitis B viral mutations detected in plasma by mass spectrometry. Cancer Epidemiol Biomarkers Prev. 2007;16:1213–1218. doi: 10.1158/1055-9965.EPI-06-0905. [DOI] [PubMed] [Google Scholar]
- 12.Kuang SY, Lekawanvijit S, Maneekarn N, Thongsawat S, Brodovicz K, Nelson K, et al. Hepatitis B 1762T/1764A mutations, hepatitis C infection, and codon 249 p53 mutations in hepatocellular carcinomas from Thailand. Cancer Epidemiol Biomarkers Prev. 2005;14:380–384. doi: 10.1158/1055-9965.EPI-04-0380. [DOI] [PubMed] [Google Scholar]
- 13.Hou J, Lau GK, Cheng J, Cheng CC, Luo K, Carman WF. T1762/A1764 variants of the basal core promoter of hepatitis B virus; serological and clinical correlations in Chinese patients. Liver. 1999;19:411–417. doi: 10.1111/j.1478-3231.1999.tb00070.x. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka Y, Mukaide M, Orito E, Yuen MF, Ito K, Kurbanov F, et al. Specific mutations in enhancer II/core promoter of hepatitis B virus subgenotypes C1/C2 increase the risk of hepatocellular carcinoma. J Hepatol. 2006;45:646–653. doi: 10.1016/j.jhep.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Ohta Y, Kanai K, Akahane Y, Iwasa Y, Hino K, et al. Clinical implications of mutations C-to-T1653 and T-to-C/A/G1753 of hepatitis B virus genotype C genome in chronic liver disease. Arch Virol. 1999;144:1299–1308. doi: 10.1007/s007050050588. [DOI] [PubMed] [Google Scholar]
- 16.Yuen MF, Tanaka Y, Shinkai N, Poon RT, But DY, Fong DY, et al. Risk for hepatocellular carcinoma with respect to hepatitis B virus genotypes B/C, specific mutations of enhancer II/core promoter/precore regions and HBV DNA levels. Gut. 2008;57:98–102. doi: 10.1136/gut.2007.119859. [DOI] [PubMed] [Google Scholar]
- 17.Yeh FS, Mo CC, Yen RC. Risk factors for hepatocellular carcinoma in Guangxi, People’s Republic of China. Natl Cancer Inst Monogr. 1985;69:47–48. [PubMed] [Google Scholar]
- 18.Yeh FS, Yu MC, Mo CC, Luo S, Tong MJ, Henderson BE. Hepatitis B virus, aflatoxins, and hepatocellular carcinoma in southern Guangxi, China. Cancer Res. 1989;49:2506–2509. [PubMed] [Google Scholar]
- 19.Yuan JM, Govindarajan S, Henderson BE, Yu MC. Low prevalence of hepatitis C infection in hepatocellular carcinoma (HCC) cases and population controls in Guangxi, a hyperendemic region for HCC in the People’s Republic of China. British journal of cancer. 1996;74:491–493. doi: 10.1038/bjc.1996.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makarananda K, Pengpan U, Srisakulthong M, Yoovathaworn K, Sriwatanakul K. Monitoring of aflatoxin exposure by biomarkers. J Toxicol Sci. 1998;23 (Suppl 2):155–159. doi: 10.2131/jts.23.supplementii_155. [DOI] [PubMed] [Google Scholar]
- 21.Groopman JD, Johnson D, Kensler TW. Aflatoxin and hepatitis B virus biomarkers: a paradigm for complex environmental exposures and cancer risk. Cancer Biomark. 2005;1:5–14. doi: 10.3233/cbm-2005-1103. [DOI] [PubMed] [Google Scholar]
- 22.Rozanov M, Plikat U, Chappey C, Kochergin A, Tatusova T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004;32:W654–659. doi: 10.1093/nar/gkh419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang JS, Qian GS, Zarba A, He X, Zhu YR, Zhang BC, et al. Temporal patterns of aflatoxin-albumin adducts in hepatitis B surface antigen-positive and antigen-negative residents of Daxin, Qidong County, People’s Republic of China. Cancer Epidemiol Biomarkers Prev. 1996;5:253–261. [PubMed] [Google Scholar]
- 24.Mickey RM, Greenland S. The impact of confounder selection criteria on effect estimation. Am J Epidemiol. 1989;129:125–137. doi: 10.1093/oxfordjournals.aje.a115101. [DOI] [PubMed] [Google Scholar]
- 25.Yin J, Zhang H, Li C, Gao C, He Y, Zhai Y, et al. Role of hepatitis B virus genotype mixture, subgenotypes C2 and B2 on hepatocellular carcinoma: compared with chronic hepatitis B and asymptomatic carrier state in the same area. Carcinogenesis. 2008;29:1685–1691. doi: 10.1093/carcin/bgm301. [DOI] [PubMed] [Google Scholar]
- 26.Zeng G, Wang Z, Wen S, Jiang J, Wang L, Cheng J, et al. Geographic distribution, virologic and clinical characteristics of hepatitis B virus genotypes in China. J Viral Hepat. 2005;12:609–617. doi: 10.1111/j.1365-2893.2005.00657.x. [DOI] [PubMed] [Google Scholar]
- 27.Dong Y, Liu SL, Zhai XJ, Zhu FC, Pan H, Yu JX, et al. A serological and molecular survey of hepatitis B in children 15 years after inception of the national hepatitis B vaccination program in eastern China. J Med Virol. 2009;81:1517–1524. doi: 10.1002/jmv.21522. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi K, Katano Y, Chuong TX, Takeda Y, Ishigami M, Itoh A, et al. Prevalence of hepatitis B virus subgenotypes and basal core promoter, precore variants in patients with acute hepatitis B in central Vietnam. Intervirology. 2009;52:22–28. doi: 10.1159/000210835. [DOI] [PubMed] [Google Scholar]
- 29.Truong BX, Seo Y, Yano Y, Ho PT, Phuong TM, Long DV, et al. Genotype and variations in core promoter and pre-core regions are related to progression of disease in HBV-infected patients from Northern Vietnam. Int J Mol Med. 2007;19:293–299. [PubMed] [Google Scholar]
- 30.Fang ZL, Zhuang H, Wang XY, Ge XM, Harrison TJ. Hepatitis B virus genotypes, phylogeny and occult infection in a region with a high incidence of hepatocellular carcinoma in China. World J Gastroenterol. 2004;10:3264–3268. doi: 10.3748/wjg.v10.i22.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ni YH, Chang MH, Wang KJ, Hsu HY, Chen HL, Kao JH, et al. Clinical relevance of hepatitis B virus genotype in children with chronic infection and hepatocellular carcinoma. Gastroenterology. 2004;127:1733–1738. doi: 10.1053/j.gastro.2004.09.048. [DOI] [PubMed] [Google Scholar]
- 32.Guo X, Jin Y, Qian G, Tu H. Sequential accumulation of the mutations in core promoter of hepatitis B virus is associated with the development of hepatocellular carcinoma in Qidong, China. J Hepatol. 2008;49:718–725. doi: 10.1016/j.jhep.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 33.Orito E, Mizokami M, Sakugawa H, Michitaka K, Ishikawa K, Ichida T, et al. A case-control study for clinical and molecular biological differences between hepatitis B viruses of genotypes B and C. Japan HBV Genotype Research Group Hepatology. 2001;33:218–223. doi: 10.1053/jhep.2001.20532. [DOI] [PubMed] [Google Scholar]
- 34.Yang HI, Yeh SH, Chen PJ, Iloeje UH, Jen CL, Su J, et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:1134–1143. doi: 10.1093/jnci/djn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baptista M, Kramvis A, Kew MC. High prevalence of 1762(T) 1764(A) mutations in the basic core promoter of hepatitis B virus isolated from black Africans with hepatocellular carcinoma compared with asymptomatic carriers. Hepatology. 1999;29:946–953. doi: 10.1002/hep.510290336. [DOI] [PubMed] [Google Scholar]
- 36.IARC. Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins. 1993. pp. 245–395. [Google Scholar]
- 37.Ross RK, Yuan JM, Yu MC, Wogan GN, Qian GS, Tu JT, et al. Urinary aflatoxin biomarkers and risk of hepatocellular carcinoma. Lancet. 1992;339:943–946. doi: 10.1016/0140-6736(92)91528-g. [DOI] [PubMed] [Google Scholar]
- 38.Wang LY, Hatch M, Chen CJ, Levin B, You SL, Lu SN, et al. Aflatoxin exposure and risk of hepatocellular carcinoma in Taiwan. Int J Cancer. 1996;67:620–625. doi: 10.1002/(SICI)1097-0215(19960904)67:5<620::AID-IJC5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 39.Chen CJ, Wang LY, Lu SN, Wu MH, You SL, Zhang YJ, et al. Elevated aflatoxin exposure and increased risk of hepatocellular carcinoma. Hepatology. 1996;24:38–42. doi: 10.1002/hep.510240108. [DOI] [PubMed] [Google Scholar]
- 40.Wild CP, Montesano R. A model of interaction: aflatoxins and hepatitis viruses in liver cancer aetiology and prevention. Cancer Lett. 2009;286:22–28. doi: 10.1016/j.canlet.2009.02.053. [DOI] [PubMed] [Google Scholar]
- 41.Groopman JD, Zhu JQ, Donahue PR, Pikul A, Zhang LS, Chen JS, et al. Molecular dosimetry of urinary aflatoxin-DNA adducts in people living in Guangxi Autonomous Region, People’s Republic of China. Cancer Res. 1992;52:45–52. [PubMed] [Google Scholar]
- 42.Wang JS, Huang T, Su J, Liang F, Wei Z, Liang Y, et al. Hepatocellular carcinoma and aflatoxin exposure in Zhuqing Village, Fusui County, People’s Republic of China. Cancer Epidemiol Biomarkers Prev. 2001;10:143–146. [PubMed] [Google Scholar]