

Abstract
Green tea polyphenols (GTP) have been shown to exert a spectrum of health benefits to animals and humans. It is plausible that the beneficial effects of GTP are a result of its interaction with the gut microbiota. This study evaluated the effect of long-term treatment with GTP on the gut microbiota of experimental rats and the potential linkage between changes of the gut microbiota with the beneficial effects of GTP. Six-month-old Sprague-Dawley rats were randomly allocated into three dosing regimens (0, 0.5%, and 1.5% of GTP) and followed for 6 months. At the end of month 3 or month 6, half of the animals from each group were sacrificed and their colon contents were collected for microbiome analysis using 16S ribosomal RNA and shotgun metagenomic community sequencing. GTP treatment significantly decreased the biodiversity and modified the microbial community in a dose-dependent manner; similar patterns were observed at both sampling times. Multiple operational taxonomic units and phylotypes were modified: the phylotypes Bacteroidetes and Oscillospira, previously linked to the lean phenotype in human and animal studies, were enriched; and Peptostreptococcaceae previously linked to colorectal cancer phenotype was depleted in GTP treated groups in a dose-dependent manner. Several microbial gene orthologs were modified, among which genes related to energy production and conversion were consistently enriched in samples from month 6 in a dose-dependent manner. This study showed that long-term treatment with GTP induced a dose-dependent modification of the gut microbiome in experimental rats, which might be linked to beneficial effects of GTP.
Keywords: Green Tea Polyphenol, Catechin, Gut microbiome, Metagenomics, 16S gene survey, Next generation sequencing
1. Introduction
Green tea polyphenols (GTP), a mixture of natural flavonoids found in brewed green tea, offer a spectrum of health benefits to animals and humans, including protections from cardiovascular diseases [1], cancers [2,3], neurodegenerative diseases [1], and improving bone health [4]. The mechanisms are mainly ascribed to anti-oxidative and anti-inflammatory properties of GTP [1,5]. However, abundant research has shown that major GTP constituents are poorly absorbed in animals and humans, and exist primarily as conjugates which may not be bioactive in the blood stream once absorbed [6–9]. In recent years, animal and human health research has witnessed an increase in efforts to understand gut microbiota, due to its potential in influencing host physiology and health [10–12]. Given that a significant portion of unabsorbed GTP and GTP metabolites excreted through bile acid would pass from small intestine to large intestine [6,13,14], the potential interactions between GTP and the gut microbiota might be important to impact the overall health of their hosts [8,15,16].
Four major catechins found in fresh tea leaves are (-)-epicatechin (EC), (-)-epigallocatechin (EGC), (-)-epicatechin gallate (ECG), and (-)-epigallocatechin gallate (EGCG) [1]. Studies in humans with ileostomy have shown that these catechins are primarily absorbed in the upper gastrointestinal tract (GIT) [6,7], whereas a considerable amount of unabsorbed EGCG and ECG (~ 50–60% of intake) and smaller portions of EC and EGC (~10–30% of intake) would pass to the large intestine without being absorbed [6,7]. Absorbed catechins mostly present in conjugated forms (i.e. glucuronidated, sulfated, and methylated) in the blood stream, except for absorbed EGCG which primarily presents in unchanged form [14,17]. EGC and EC are mainly eliminated via urine in conjugated forms; EGCG, and possibly the other three catechins to a lesser extent, could be eliminated via bile acids [13,14,18], which then pass into the large intestine.
Many studies have evaluated the effects of the oral intake of potential beneficial agents, i.e. probiotics or prebiotics, on the gut microbiota in human and animals, including a variety of Lactobacillus, Enterococus, Bifidobacterium strains, or their mixtures [19,20], and certain polysaccharides [21,22]. It is unknown, however, whether these agents modify the gut microbiota, or whether the observed changes of microbiota are related to the beneficial effects of these agents. Large variations in doses administered and durations of treatment, inter-individual variation [19,23], lack of standard and appropriate experimental and data analysis protocols [24–26], and a lack of understanding of the mechanisms for beneficial effects may all contribute to the difficulty in reaching a consistent or plausible conclusion.
Given the relative low bioavailability of GTP and its direct contact with the gut microbiota, it is plausible that GTP would interact with the gut microbiota which might influence the overall health of the hosts. Few studies, however, have been conducted to evaluate interactions between GTP and the gut microbiota [16]. Two studies have shown that GTP increased beneficial Bifidobacteria and Bacteroidetes in the gut in two different mouse models [27,28]. However, another study did not find a significant effect of GTP on human gut microbiota in a 12-week randomized clinical trial [29]; the dosing scheme, the trial duration, and the selection of study subjects might make it difficult to reveal the effect of GTP in that study, especially considering the uncontrolled and diverse human diet. In the current study, a long-term repeated dosing experiment was conducted by administering GTP in drinking water for 6 months to female Sprague-Dawley rats. We investigated their functional genome profiles by shotgun metagenomic community sequencing (SMC-seq) and their microbial community composition by 16S ribosomal RNA (rRNA) amplicon sequencing (16S-seq). The study aims were to: (1) evaluate how GTP would modulate the gut microbiome in response to different doses, (2) evaluate the changes of gut microbiome across time, and (3) develop a reliable experimental and data analysis protocol.
2. Materials and Methods
2.1 Animals, Chemicals, and Experimental Design
This study was conducted concurrently with a recently published study of the chronic toxicity and tolerance of GTP extracts (decaffeinated) among middle-aged ovariectomized rats, in which the experimental design and procedures on chemicals and animal preparations were described [30]. This rat model was developed to evaluate the beneficial effects of long-term GTP supplementation to postmenopausal women. Briefly, after acclimation, female ovariectomized Sprague-Dawley (SD) rats (six months old, Harlan Laboratories, Indianapolis, IN) were randomly allocated into six groups (n=12/group) in three dosing regimens. The rats were fed a pelleted AIN-93M diet (Dyets, Bethlehem, PA) and had free access to either regular distilled water (as the control) or daily freshly prepared distilled water mixed with 0.5%, or 1.5% GTP (weight/volume or g/ml) (as two treatment groups) until the end of month 3 or month 6 after initiation of the experiment, when one group of animals from each dosing regimen were anesthetized with the isoflurane gas and euthanized by bilateral thoracotomy (experiment design was illustrated in Supplementary Figure S1). The typical duration for evaluating the chronic effects of a substance among rats is six months which roughly equivalent to twelve human years [31]. At each sampling time, colon contents were directly and immediately stored in −80°C freezer for further processing. Overnight fasting blood samples were also collected in the concurrent study for hematology and clinical chemistry analyses. Rats were housed in individual stainless-steel cages and examined daily. Individual animal body weight was recorded weekly, and food and water consumption were recorded daily. All procedures were approved by the Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee.
2.2 DNA Extraction, Library Preparation, and Sequencing
Microbial DNA was extracted from colon contents using Qiagen Fast DNA Stool Mini Kit (QIAGEN, Valencia, CA) following the protocol published previously [32] with one exception: manual homogenizing was replaced with a bead-beating step using MIBIO 0.1 mm glass beads tubes (MOBIO, Carlsbad, CA).
Extracted DNA from all samples was normalized to ~20 ng DNA/μl in PCR water for DNA library preparation for SMC-seq and 16S-seq. One aliquot for SMC-seq was fragmented using a Bioruptor Plus (Diagenode, Denville, NJ) targeting a dominant fragment size of ~500 base pairs (bp), which was checked by gel electrophoresis, and subsequently processed using KAPA LTP library preparation kit (Kapa Biosystems, Inc., Boston, MA) according to manufacturer’s protocol, but with iTru adapters and primers [33]. Another aliquot of the extracted DNA from selected samples for 16S-seq was processed using a 2-step Quadruple-index PCR procedure as previously described [32] in which fragments spanning V3 and V4 regions of 16S rRNA gene were amplified. Three replicate PCR reactions per sample were performed in the primary PCR reaction and pooled for the secondary limited-cycle PCR to reduce potential PCR bias [34].
Genomic DNAs from two standard mock microbial communities (HM-276D and HM-277D, BEI Resources, Manassas, VA) were processed using the same two-step Quadruple-index PCR procedure to gauge 16S-seq library preparation and data analysis procedure. HM-276D consists of 20 bacterial species with equal rRNA operon counts and HM-277D consists of the same 20 bacterial species with varying rRNA operon counts [35,36].
Prepared SMC-seq libraries were sequenced using Illumina NextSeq instruments to obtain ~2 million Paired-End (PE) reads of 150 bp per sample; and 16S-seq libraries were sequenced using Illumina MiSeq platform to obtain ~100,000 PE reads of 300 bp per sample, at the Georgia Genomic Facility (GGF, University of Georgia, Athens, GA). The sequencing depth was selected based on previous experimental results to optimize cost-benefit.
2.3 Sequencing Data Analysis
Metagenomic analysis was performed using the sequencing reads obtained from SMC-seq, following a similar strategy in Li et al. [37] and Nielson et al. [38]. Raw paired-end, de-multiplexed sequencing reads from each sample were quality filtered using Trimmomatic-v0.3.6 [39], and fed to MEGAHIT-v1.0.2 [40] for metagenome assembly. The proportion of quality filtered reads that could be mapped to MEGAHIT assemblies was checked using Bowtie2 [41]. MEGAHIT assemblies for each sample were processed by Prodigal-v2.6.3 [42] for microbial gene prediction. Predicted genes from each sample that were longer than 100 bp were pooled, de-replicated according to sequence prefix, and clustered with 99% identity using vsearch-v1.10.1 [43], producing a non-redundant microbial meta-gene catalog (MGC). The corresponding amino acid sequences for MGC were annotated according to Kyoto Encyclopaedia of Genes and Genomes (KEGG) database (genus_prokaryotes section) using GhostKOALA [44], and Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups (eggNOG) v4.5 database [45] (bacteria section, bactNOG) using in-house scripts, into gene orthologs (GOs). Both KEGG and eggNOG database group the GOs into functional categories with slightly different descriptions. This study used the single letter functional categories as in eggNOG database to assure consistency. The quality filtered reads from each sample were subsequently mapped to the annotated MGC using Bowtie2 to generate gene abundances which were further normalized using the gene length [46]. Normalized gene abundance tables for each sample were the products of metagenomic analysis and subjected to further statistical comparisons.
The procedure for analyzing 16S-seq data was slightly modified based on our previous report [32] due to an unexpected number of low-quality reads in this batch of samples. Consequently, a trimming procedure was performed on the 3′ end of each PE read initially, so that remaining reads did not contain more than three low quality (Q<20) or ambiguous nucleotides. Reads less than 200 bp in length were also filtered out. These procedures were executed in Geneious R8.1.8 (Biomatters Inc, San Francisco, California). The remaining quality trimming procedures and community analysis using QIIME pipeline [47] were similar to those we previously reported [32]. The final OTU abundance table was filtered according to results from analysis of standard mock communities and subject to statistical comparisons.
2.4 Statistical Methods
Changes in body weight, food and water intake for each individual were calculated by comparing respective endpoints at either month 3 or month 6 to those at the end of 1- week, and one-way ANOVA test was used with a post-hoc Duncan’s test to evaluate the treatment effect.
The comparisons of gene abundances in metagenomic analysis and OTU abundances in 16S-seq in samples from different dosing groups were performed using LEfSe (Linear discriminant analysis Effect Size)-v1.0 [48]. LEfSe incorporated a combination of univariate [Kruskal-Wallis rank sum test (Kruskal-Wallis test) and Wilcoxon rank-sum test (Wilcoxon test)] and multivariate (Linear Discrimination Analysis, LDA) statistical methods to determine the features (e.g. OTUs, Genes) that discriminate different classes (e.g. Dosing regimens) and provide a measure of effect size (i.e. LDA score) [48]. The LEfSe analyses were performed in samples from two sampling dates separately, and the significance level was set to be 0.01.
The comparisons of biodiversity of the gut microbial community (α-diversity) in samples from different dosing groups were performed using the ANOVA test, with the diversity indexes as the response variable, and dose and sampling date as the predictive variables. The β-diversity [49] between samples was measured using a weighted-Unifrac distance index, which integrated both the abundance and phylogenetic relationship of each OTU [50]. The weighted-Unifrac distance matrix constructed from each pair of samples were analyzed using a principal coordinate analysis (PCoA) and ADONIS tests [51] to evaluate the effects of predictive variables.
The availability of a gene abundance table, an OTU abundance table, as well as the concurrent GTP tolerance experiment which measured multiple clinical endpoints of the tested animals, enabled the exploration of potential correlations between the gut microbiome with host physiology. Hierarchical All-against All Association testing (HAllA)-v0.7.11 was used to find the multi-resolution associations among these three high-dimensional datasets [52]. HAllA employs a hierarchical clustering of features (e.g. genes, OTUs, or clinical measurements) in each dataset, dimensionality reduction, and iterative testing of associations between clusters, to provide well-powered association discoveries [52]. A false discovery rate of 0.1 using Benjamini–Hochberg–Yekutieli (BHY) correction was used to screen for candidate correlations.
Significance level for all tests in this study were set as 0.05 unless otherwise noted.
3. Results
3.1 Weekly monitoring of experimental rats
The body weight of experimental rats from all groups increased steadily during 6 months experiment. At each sampling time, on average, the body weight of the rats increased by 39.8, 41.2, and 49.4 grams in control, 0.5%, and 1.5% groups respectively after 3 months (Table 1), but no significant dosing effect was observed. After 6 months, 55.5, 53.2, and 55.9 grams increase in body weight was observed in control, 0.5%, and 1.5% groups respectively without a significant dosing effect. The food intake was decreased in both control and 0.5% dosing groups at the two sampling times (Table 1). Rats from 1.5% dosing group had a decrease in food intake at month 3 but the decrease was significantly less than the other two groups; at month 6, the food intake was slightly increased and significantly different from the other two groups. The water intake of the rats from all groups increased as compared to baseline during the experiment (Table 1). However, the two GTP treated groups have significantly less increase of water intake as compared to controls at both sampling times. No abnormal treatment-related findings in clinical observations were noted.
Table 1.
The changes in body weight, food intake, and water intake at two sampling times as compared to the end of first weeka.
| Sampling Time | Control | 0.5 % | 1.5 % | |
|---|---|---|---|---|
| Changes in body weight (gram) | ||||
| Month 3 | 39.8 ± 3.2 | 41.2 ± 3.0 | 49.4 ± 3.8 | p-value=0.095b |
| Month 6 | 55.5 ± 4.6 | 53.2 ± 4.5 | 55.9 ± 7.3 | p-value=0.933 |
| Changes in food intake (gram) | ||||
| Month 3 | −5.46 ± 0.55A | −5.69 ± 0.30A | −0.30 ± 0.96B | p-value<0.001 |
| Month 6 | −1.87 ± 0.68A | −2.30 ± 0.78A | 2.02 ± 1.67B | p-value=0.021 |
| Changes in water intake (gram) | ||||
| Month 3 | 10.94 ± 0.97A | 3.6 ± 0.71B | 3.13 ± 0.73B | p-value<0.001 |
| Month 6 | 9.53± 1.74A | 4.52 ± 0.80B | 4.43 ± 1.26B | p-value=0.014 |
Values are Mean ± S.E.M. N equals to 24 for all groups at month 3, and equals to 12 for all groups at month 6.
One-way ANOVA tests were performed to compare measurements from different groups at each sampling time. Data in the same line with different letters are significantly different (α<0.05) identified by post-hoc Duncan’s test.
3.2 SMC and 16S amplicon sequencing data processing
Extracted DNA from a total of 72 samples from six groups (three dose groups at two sampling times) were processed for SMC-seq and metagenomic analysis. A total of 303,071,358 PE reads of 150 bp in length were obtained, among which 205,905,205 PE reads survived the stringent quality filtering process, making an average (Standard Deviation, SD) of 2,859,795 (1,019,602) PE reads per sample (average survival rate, 67.8%) for downstream analysis. Metagenome assembly was performed on each sample individually using MEGAHIT, achieving an average (SD) N50 length of 1,237 (314) nucleotides (nt), average maximum contig length of 126,871 (44,232) nt, and average contig counts of 87,730 (31,462), across all samples. The average mapping rate of quality filtered PE reads to the contigs assembled by MEGAHIT for each sample is 69.9% (7.6%) (range: 40.7% to 82.1%). Prodigal predicted an average of 139,878 (47,330) microbial genes per sample. After de-replication and clustering, the MGC constructed contained 2,367,134 genes, among which 829,036 and 1,748,024 genes respectively matched to GOs in the KEGG and eggNOG v4.5 databases; for a combined total of 1,760,017 genes assigned a GO, constituting 74.3% of the MGC. The mapping rate of quality filtered PE reads from each sample to MGC was on average 76.0% (0.05%).
The original genomic DNAs from both standard mock communities were diluted so that equal amounts of 50,000 16S rRNA gene operons from the 20 species in HM-276D (1:20 dilution), or 1,000 to 1,000,000 operons from the same 20 species in HM-277D (1:10 dilution) were input into the primary PCR (1 μl input DNA). A total of 24,723 and 25,507 quality filtered reads were available for HM-276D and HM-277D respectively, among them 197 OTUs were identified in HM-276D, including all of the 20 known species (abundances ranging from 0.76% to 11.87%), and 153 OTUs were identified in HM-277D, but only including 18 of the 20 known species (abundances ranged from 0.0039% to 49.68%). Given the results from the two standard mock communities, an abundance of 0.1% was selected based on a receiver operating characteristic (ROC) analysis to filter OTUs in the original 36 samples in GTP dosing experiments to prevent any spurious findings (Supplementary Figure S2). When using 0.1% as the minimum cut-off value, 25 OTUs including all 20 known species were captured in HM-276D and 16 OTUs including 11 known species were captured in HM-277D.
Six samples from each GTP dosing group were randomly selected for 16S-seq. A total of 5,855,866 PE reads from 36 samples were obtained, among which 2,683,283 PE reads survived quality filtering (average survival rate, 47.0%). A total of 2,500,350 of sequences remained in the final OTU abundance table after abundance filtering on the individual sample in GTP dosing experiment, making an average (SD) of 69,454 (24,460) sequences per sample for analysis.
3.3 Long-term treatment with GTP modified the abundance of several gene orthologs
LEfSe was performed to detect group discriminating GOs in samples collected at month 3 and month 6 separately. At month 3, 10 GOs were identified: 6 GOs decreased, and 4 GOs increased with increasing GTP doses (Table 2). Genes that involve “Cell wall/membrane/envelope biogenesis” (category [M]) had 2 GOs identified, both decreased; genes that involve “Replication, recombination and repair” (category [L]) had 3 GOs identified with 2 decreased (Figure 1A). Figure 1B presented the Log10 scaled normalized abundance for GOs identified by LEfSe in the order of decreasing LDA score.
Table 2.
Gene orthologs (GO) significantly modified by green tea polyphenols.
| GOa | Category | Function |
|---|---|---|
| 3 months treatment | ||
| Decreased | ||
| ENOG4105D70 | L | Integrase catalytic subunit |
| ENOG4105F5W | L | ATP-binding protein |
| ENOG4105EGG | V | ABC transporter |
| ENOG4105EXR | S | Unknown |
| ENOG4105CK0 | M | Membrane bound o-acyl transferase |
| ENOG4108SJI | M | Hydrolase, family 25 |
| Increased | ||
| K01869 | J | Leucyl-tRNA synthetase |
| ENOG4105WBW | S | Relaxase mobilization nuclease domain protein |
| ENOG4105EUN | L | Transposase |
| ENOG4105CCY | C | NADH flavin oxidoreductase, NADH oxidase |
| 6 months treatment | ||
| Decreased | ||
| ENOG4105D2F | G | Ig domain protein, group 2 domain protein |
| ENOG4105CDX | S | Unknown |
| ENOG4105XN2 | K | DNA-binding helix-turn-helix protein |
| ENOG4105C19 | V | ABC transporter |
| ENOG4108KGH | K | Transcriptional regulator |
| ENOG410667J | S | Hydrolase |
| ENOG4108SJI | M | Hydrolase, family 25 |
| ENOG4108VXD | M | Carboxypeptidase |
| ENOG4108UUM | L | Single-stranded DNA-binding protein |
| ENOG4105EF7 | K | Transcriptional regulator |
| ENOG4105DAV | K | RNA polymerase sigma factor RpoD |
| ENOG4108M4B | M | Domain protein |
| ENOG4105DZ1 | M | Carboxypeptidase |
| ENOG4105C5C | M | N-acetylmuramoyl-L-alanine amidase |
| Increased | ||
| ENOG4105F42 | S | AAA-ATPase |
| ENOG4105K8S | S | Unknown |
| ENOG4108J2R | C | Molybdopterin oxidoreductase |
| ENOG4105FAP | L | Transposase |
| ENOG4107WWN | L | Transposase |
| ENOG41063CS | L | Replication Protein |
| ENOG4105FGY | P | TonB-dependent receptor, Plug Domain |
| K01869 | J | Leucyl-tRNA synthetase |
| ENOG4105DAB | C | Fumarate reductase/succinate dehydrogenase, Flavoprotein |
| domain protein | ||
| ENOG4105CGS | G | Alpha-glucosidase |
| ENOG4105D3S | C | 4Fe-4S ferredoxin, iron-sulfur binding domain protein |
| ENOG4107VV4 | S | Virulence-associated protein e |
| ENOG4105X7U | K | Transcriptional regulator, ARAC family |
| ENOG4106DHI | S | Cleaved adhesin domain protein |
| ENOG4105CCY | C | NADH flavin oxidoreductase, NADH oxidase |
| ENOG4105WBW | S | Relaxase mobilization nuclease domain protein |
The gene ortholog ID, category, and function were assigned based on eggNOG v4.5 database except for K01869, which was assigned based on KEGG database.
Figure 1.
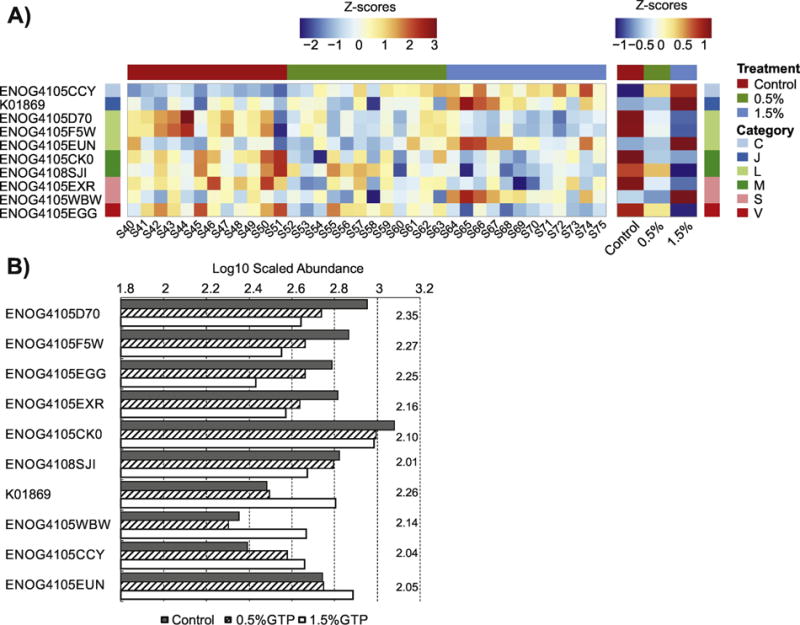
Comparisons of gene orthologs (GOs) significantly modified by 3 months treatment with GTP. (A) The heatmap of normalized abundance of GO with values for individual samples and group averages from left to right; (B) The scaled abundance of GO with LDA scores presented on the right of each column group.
At month 6, 30 GOs were identified, among which Genes from category [M] and [K] (“transcription”) were the most affected (5 GOs each), followed by category [L] and [C] (“Energy production and conversion”) (4 GOs each) (Table 2, Figure 2A–2C). It was surprising, however, that only four GOs were consistently identified at both of the sampling times, including K01869, ENOG4105WBW, and ENOG4105CCY, which were enriched; and ENOG4108SJI, which was depleted.
Figure 2.
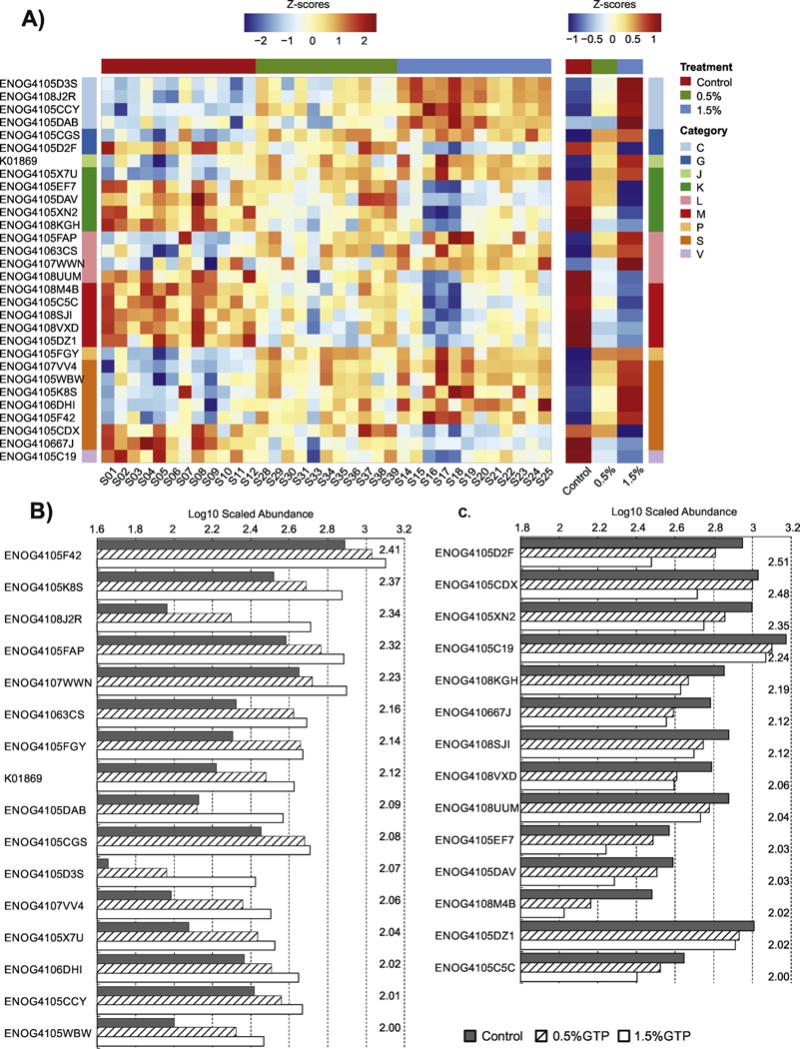
Comparisons of gene orthologs (GOs) significantly modified by 6 months treatment with GTP. (A) The heatmap of normalized abundance of GO with values for individual samples and group averages from left to right; (B) The scaled abundance of GO that were enriched with LDA scores presented on the right of each column group; (C) The scaled abundance of GO that were depleted with LDA scores presented on the right of each column group.
3.4 Long-term treatment with GTP modified the gut microbial community composition
Increasing doses of GTP among experimental rats decreased the gut community biodiversity as measured by Shannon index and Faith’s phylogenetic diversity (PD) [53] at the two sampling times, although an unexpected surge of biodiversity was observed at the dose of 0.5% GTP at month 6 (Figure 3). The two-way ANOVA test confirmed significant dosing effect (p-value = 0.0041 for Shannon, and p-value = 0.0250 for PD), but no significant effect from sampling time was found. A post-hoc Duncan’s test identified that samples from 1.5% GTP groups had significantly lower biodiversity indexes.
Figure 3.
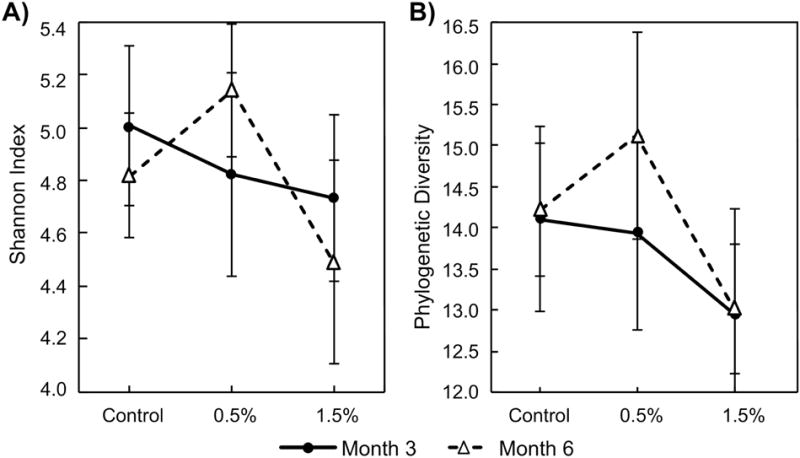
The alpha-diversity of samples collected at the end of 3 months and 6 months GTP treatment. Group average and standard error was presented. Two-way ANOVA tests were performed for each index, which was used a response variable with dose and sampling time as predictive variable. Significant dosing effect (p = 0.0041 for Shannon and 0.0250 for Faith’s phylogenetic diversity (PD)) but insignificant sampling effect (p > 0.05) were obtained. (A) Shannon index; (B) PD.
Correspondingly, the PCoA analysis of beta diversity using weighted Unifrac index revealed that different doses of GTP but not sampling time differentiated the gut microbial community (Figure 4A). An ADONIS test [51] based on the weighted UniFrac distance matrix suggested a significant dosing effect, which could explain 29% (R2) of overall variation (p-value<0.001). Plotting PC1 coordinates of each sample against the GTP doses showed a steady shift of the gut microbial community while increasing doses (Figure 4B).
Figure 4.
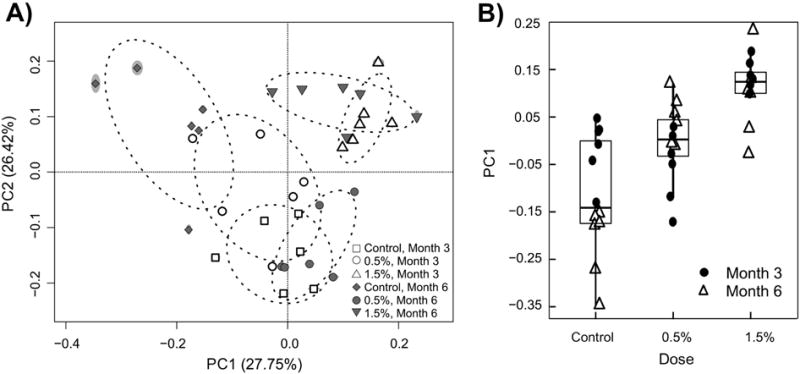
Principle Coordinate Analysis of beta-diversity measured by weighted UniFrac index. ADONIS test of dosing and sampling effect identified a significant dosing effect (p<0.001, R2=0.29) but not sampling effect. (A) PC1 vs PC2; (B) PC1 with increasing doses.
LEfSe was used to detect class discriminating OTUs or bacterial phylotypes (from phylum to family level) in different GTP dosing groups. Again, the two sampling times were analyzed separately. Manual filtering was performed so that only features (either OTUs or phylotypes) that had at least 2.5-fold change of overall abundance in different dosing groups were identified. The threshold fold change was determined based on our analysis of the standard mock community. Overall, all the OTUs or phylotypes selected had relatively high LDA scores ranging from 3.39 to 5.10 with an average of 4.08.
At month 3, 10 OTUs were identified: 6 OTUs from Clostridiales of Firmicutes increased, and 2 OTUs from Bacteroidales of Bacteroidetes decreased with increasing GTP doses (Figure 5A, Table 3). However, family Paraprevotellaceae of Bacteroidetes was the only phylotype that was identified as discriminating different dosing groups, which decreased with increasing GTP doses, probably due to the decrease of the genus CF231 as one of the identified decreasing OTUs from Bacteroidales (Table 3). Neither Clostridiales nor Bacteroidales, when testing as an overall order, was discriminating, suggesting that the changes of individual OTUs did not translate to the whole bacterial order.
Figure 5.
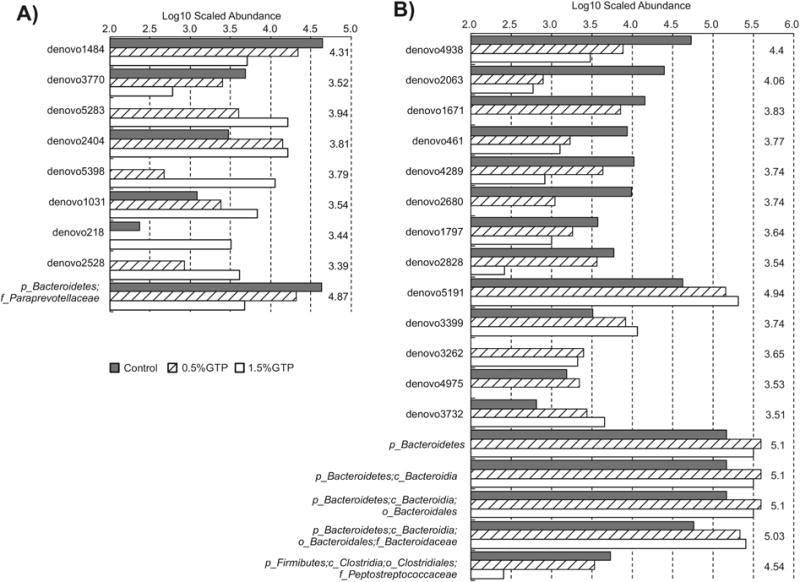
The abundance of operational taxonomic units that were significantly modified by GTP treatment: (A) month 3; (B) month 6.
Table 3.
OTUs, with their taxonomic assignments, significantly modified by green tea polyphenols.
| OTUa | Phylum | Class | Order | Family | Genus |
|---|---|---|---|---|---|
| 3 months treatment | |||||
| Decreased | |||||
| denovo1484 | Bacteroidetes | Bacteroidia | Bacteroidales | [Paraprevotellaceae] | CF231 |
| denovo3770b | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides |
| Increased | |||||
| denovo5283 | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Ruminococcus |
| denovo2404 | Firmicutes | Clostridia | Clostridiales | ||
| denovo5398 | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Blautia |
| denovo1031 | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | |
| denovo218 | Firmicutes | Clostridia | Clostridiales | ||
| denovo2528 | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | |
| 6 months treatment | |||||
| Decreased | |||||
| denovo4938 | Firmicutes | Erysipelotrichi | Erysipelotrichales | Erysipelotrichaceae | Allobaculum |
| denovo2063 | Firmicutes | Clostridia | Clostridiales | ||
| denovo1671 | Firmicutes | Erysipelotrichi | Erysipelotrichales | Erysipelotrichaceae | Allobaculum |
| denovo461 | Firmicutes | Clostridia | Clostridiales | ||
| denovo4289 | Bacteroidetes | Bacteroidia | Bacteroidales | S24-7 | |
| denovo2680 | Firmicutes | Clostridia | Clostridiales | ||
| denovo1797 | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | |
| denovo2828 | Firmicutes | Clostridia | Clostridiales | Peptostreptococcaceae | |
| Increased | |||||
| denovo5191 | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides |
| denovo3399 | Bacteroidetes | Bacteroidia | Bacteroidales | S24-7 | |
| denovo3262 | Proteobacteria | Deltaproteobacteria | Desulfovibrionales | Desulfovibrionaceae | |
| denovo4975 | Bacteroidetes | Bacteroidia | Bacteroidales | S24-7 | |
| denovo3732 | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Oscillospira |
OTU refers to operational taxonomic unit;
devono3770 was the only OTU with a species assignment as Bacteroides acidifaciens.
At month 6, 26 OTUs were identified including 5 Clostridiales and 2 Erysipelotrichales of Firmicutes, 1 Bacteroidales decreased with GTP doses, and 3 Bacteroidales, 1 Clostridiales (Oscillospira), 1 Desulfovibrionales of Proteobacteria increased (Figure 5B, Table 3). The phylum Bacteroidetes and its descendant phylotype, Bacteroidia, Bacteroidales, and family Bacteroidaceae, were all enriched with increasing GTP doses. One family Peptostreptococcaceae of Firmicutes decreased, which may be due to the decreasing of an OTU (denovo 2828) from this family recognized previously.
3.5 Correlations between the gut microbiome with clinical measurements
Information for 105 OTUs, 31,222 GOs, and 51 clinical endpoints were available in 36 samples from 3 different dosing regimens and 2 sampling times. The relationships between OTU abundance and clinical measurements, and between GO abundance and the clinical measurements, were checked using HAllA after filtering features that contain low information (entropy threshold was set to 1.5 in HAllA).
The HAllA testing suggested 12 possible correlations between the abundance of specific OTUs and clinical measurements (Supplemental Table S1). Three of the OTUs were among those significantly altered by GTP doses at month 6: denovo5191, which was increased by GTP, was negatively correlated to mean corpuscular volume (MCV); denovo2828 (assigned to family Peptostreptococcaceae of Firmicutes), which was decreased by GTP, was positively correlated to a cluster of hematologic parameters consisted of total cholesterol, total bilirubin, and triglycerides concentrations; and denovo1797 (assigned to family Ruminococcaceae of Firmicutes), which was decreased by GTP, was positively correlated to blood total cholesterol concentrations. However, HAllA did not identify any correlations between specific GOs and clinical measurements, even though a substantial amount of OTUs were correlated to GOs (data not shown).
4. Discussion
The potential health effects of long-term GTP consumption via drinking water by middle-aged ovariectomized SD rats were evaluated. Results on the chronic safety and clinical observations of the rats supplemented with GTP has been reported recently [30]. This study, focused on the gut microbiota, successfully evaluated both functional genomic and microbial compositional alterations of gut microbiota at two different sampling times, using next-generation sequencing (NGS) technologies. We found that 3 months of repeated dosing of 1.5% GTP in drinking water induced significant changes in gut microbial composition as shown by both α-diversity and β-diversity analysis, which was persistent until the end of this experiment at month 6. Several microbial species and functional genes were altered in accordance to GTP doses at month 3, but many more species and genes, and more profound changes were observed in a GTP dose-dependent manner at month 6.
Several microbial species or phylotypes were impacted by GTP after 6 months of treatment. Intriguingly, the phylum Bacteroidetes was increased in response to GTP in a dose-dependent manner. Bacteroidetes is a major phylum colonizing all different parts of GIT in mammals regardless of pH, nutrients, oxygen availability [54]. Among many critical roles of Bacteroidetes in host physiology, their main biological function is to degrade resistant biopolymers in the large intestine that mammals cannot digest by themselves [54]. Recent advances in understanding the interactions between gut microbiota and obesity have shown that Bacteroidetes were heavily depleted in both obese mice and obese humans [55–57], but fat-restricted or carbohydrate-restricted diet therapy could increase the abundance of Bacteroidetes, which was correlated with the loss of body weight among obese human subjects [56]. A metagenomic analysis of obese and lean twin pairs showed that a significant portion of microbial genes that were enriched in the lean phenotype were from Bacteroidetes [58]. In addition to OTUs from Bacteroidetes, one OTU that was assigned to genus Oscillospira from Firmicutes was also increased in a dose-dependent manner. Oscillospira could not be grown in pure culture but was consistently captured by the 16S gene survey of the human microbiome [59]. Several recent studies have shown a positive association between Oscillospira abundance and leanness [60–63]. Oscillospira is thought to be a bile-resistant bacterium that may be able to degrade host glycans and contribute to the formation of secondary bile acids [59]. The only phylotype which decreased in a dose-dependent manner was the family Peptostreptococcaceae of Fimicutes, which was previously reported to be enriched in colorectal cancer patients in one study [64].
All identified GOs from category [C] were increased in response to GTP dosing at both sampling times. Category [C] includes GOs that are responsible for energy production and conversion. An GO, ENOG4105CCY from [C] with a function description of “NADH Flavin oxidoreductase, NADH oxidase”, was identified in both sampling times. NADH is a key co-enzyme in the catabolic pathways for energy production: NADH Flavin oxidoreductase catalyzes the reduction of NAD+ to NADH and oxidative degradation of complex organic compounds; NADH oxidase then catalyses the oxidation of NADH typically using O2 as the ultimate electron acceptor [65]. These results readily correspond to the enrichment of Bacteroidetes as mentioned previously.
A decrease of community biodiversity was correlated with an increase of GTP doses, which may be counter-intuitive assuming beneficial effects of GTP. However, the consistent supply of GTP to the gut microbial ecosystem could serve as an outside input to the ecosystem, increasing the abundance of beneficial species and improve the microbial functions, at the same time decrease the potential pathogenic species, as shown in this study, which compensates potential negative impacts due to the decrease of biodiversity. Furthermore, it is common to observe a decrease in biodiversity in gut microbiota when treated with potential probiotic or prebiotic compounds. For example, Martin et al. noted that administering two Lactobacillus strains to female germ-free C3H mice inoculated with bacteria isolated from an infant significantly lowered the CFU counts of several interesting bacterial species [66]. In a recent study employing 16S rRNA-based community composition analyses, Uronis et al. showed that a commercial probiotic mixture decreased the colonic microbial community diversity of colitis rats, which was significantly correlated with reduced clinical colitis scores [67].
The modifications of the gut microbiome as observed in this study, e.g. the enrichment of Bacteroidetes and genes related to energy metabolism, may explain the beneficial effects of GTP especially for its weight management effects [68]. These results were further supported by the weekly monitoring which showed that rats from 1.5% dosing group maintained a similar increase of body weight while taking significant more foods as compared to the other two groups. It is worth mentioning that several previous studies have shown that rat models with ovariectomy would increase rats’ body weight and lead to elevated body weights as compared to sham controls [69–71]. However, conflicting results were reported on how ovariectomy impacted the composition of the rat gut microbiota. One study showed more than 30% lower of Bacteroidetes in OVX rats than sham controls [71]. Our experiment comparing OVX rats receiving different doses of GTP to OVX controls showed a similar increase of body weights across experiment duration and a possible restoration of Bacteroidetes in GTP treated OVX rats. Furthermore, many mechanisms have been suggested for GTP’s beneficial effects, among which anti-oxidative properties were the most prominent and widely studied [5,72]. However, the potential value of these mechanisms to explain the health benefits of GTP is limited due to the low bioavailability and fast metabolism (mostly conjugated) of GTP constituents in the human physiological system [72–74]. Consequently, this study shows that the hypothesis that GTP benefit host physiological system by modifying the gut microbiome is appealing and worth further verification, given the modifiable nature of the gut microbiome.
Supplementary Material
Acknowledgments
Special thanks are due to Troy Kieran for sequencing assistance; Interdisciplinary Toxicology Program at the University of Georgia for stipend supports.
Financial supports: ECG-A-00-07-00001-00, from USAID via Peanut CRSP at University of Georgia; 1R24TW009489, from NIH Fogarty International Center; and U01AT006691 from NCCIH, NIH.
Funding
This work was supported by USAID via Peanut CRSP at University of Georgia [grant number ECG-A-00-07-00001-00]; the National Institutes of Health Fogarty International Center [grant number 1R24TW009489] and National Center for Complementary and Integrative Health [grant number U01AT006691].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Higdon J, Frei B. Tea catechins and polyphenols: health effects, metabolism, and antioxidant functions. Crit Rev Food Sci Nutr. 2003;43:89–143. doi: 10.1080/10408690390826464. [DOI] [PubMed] [Google Scholar]
- 2.Luo H, Tang L, Tang M, Billam M, Huang T, Yu J, et al. Phase IIa chemoprevention trial of green tea polyphenols in high-risk individuals of liver cancer: modulation of urinary excretion of green tea polyphenols and 8-hydroxydeoxyguanosine. Carcinogenesis. 2006;27:262–8. doi: 10.1093/carcin/bgi147. [DOI] [PubMed] [Google Scholar]
- 3.Tang L, Tang M, Xu L, Luo H, Huang T, Yu J, et al. Modulation of aflatoxin biomarkers in human blood and urine by green tea polyphenols intervention. Carcinogenesis. 2008;29:411–7. doi: 10.1093/carcin/bgn008. [DOI] [PubMed] [Google Scholar]
- 4.Shen C-L, Chyu M-C, Wang JS. Tea and bone health: steps forward in translational nutrition. Am J Clin Nutr. 2013;98:1694S–1699S. doi: 10.3945/ajcn.113.058255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qian G, Xue K, Tang L, Wang F, Song X, Chyu M-C, et al. Mitigation of Oxidative Damage by Green Tea Polyphenols and Tai Chi Exercise in Postmenopausal Women with Osteopenia. PLOS ONE. 2012;7:e48090. doi: 10.1371/journal.pone.0048090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auger C, Mullen W, Hara Y, Crozier A. Bioavailability of polyphenon E flavan-3-ols in humans with an ileostomy. J Nutr. 2008;138:1535S–1542S. doi: 10.1093/jn/138.8.1535S. [DOI] [PubMed] [Google Scholar]
- 7.Stalmach A, Mullen W, Steiling H, Williamson G, Lean MEJ, Crozier A. Absorption, metabolism, and excretion of green tea flavan-3-ols in humans with an ileostomy. Mol Nutr Food Res. 2010;54:323–34. doi: 10.1002/mnfr.200900194. [DOI] [PubMed] [Google Scholar]
- 8.Del Rio D, Costa LG, Lean MEJ, Crozier A. Polyphenols and health: what compounds are involved? . Nutr Metab Cardiovasc Dis. 2010;20:1–6. doi: 10.1016/j.numecd.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 9.Clifford MN, Hooft JJ, Crozier A. Human studies on the absorption, distribution, metabolism, and excretion of tea polyphenols. Am J Clin Nutr. 2013;98:1619S–1630S. doi: 10.3945/ajcn.113.058958. [DOI] [PubMed] [Google Scholar]
- 10.Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–8. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 11.Leser TD, Mølbak L. Better living through microbial action: the benefits of the mammalian gastrointestinal microbiota on the host. Environ Microbiol. 2009;11:2194–206. doi: 10.1111/j.1462-2920.2009.01941.x. [DOI] [PubMed] [Google Scholar]
- 12.Clemente JC, Ursell LK, Parfrey LW, Knight R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell. 2012;148:1258–70. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L, Lee M-J, Li HE, Yang CS. Absorption, distribution, and elimination of tea polyphenols in rats. Drug Metab Dispos. 1997;25:1045–1050. [PubMed] [Google Scholar]
- 14.Clifford MN, van der Hooft JJ, Crozier A. Human studies on the absorption, distribution, metabolism, and excretion of tea polyphenols. Am J Clin Nutr. 2013;98:1619S–1630S. doi: 10.3945/ajcn.113.058958. [DOI] [PubMed] [Google Scholar]
- 15.Visioli F, Lastra CADL, Andres-Lacueva C, Aviram M, Calhau C, Cassano A, et al. Polyphenols and Human Health: A Prospectus. Crit Rev Food Sci Nutr. 2011;51:524–46. doi: 10.1080/10408391003698677. [DOI] [PubMed] [Google Scholar]
- 16.Cardona F, Andrés-Lacueva C, Tulipani S, Tinahones FJ, Queipo-Ortuño MI. Benefits of polyphenols on gut microbiota and implications in human health. J Nutr Biochem. 2013;24:1415–22. doi: 10.1016/j.jnutbio.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Lee M-J, Wang Z-Y, Li H, Chen L, Sun Y, Gobbo S, et al. Analysis of plasma and urinary tea polyphenols in human subjects. Cancer Epidemiol Prev Biomark. 1995;4:393–9. [PubMed] [Google Scholar]
- 18.Kida K, Suzuki M, Matsumoto N, Nanjo F, Hara Y. Identification of Biliary Metabolites of (−)-Epigallocatechin Gallate in Rats. J Agric Food Chem. 2000;48:4151–5. doi: 10.1021/jf000386x. [DOI] [PubMed] [Google Scholar]
- 19.Eloe-Fadrosh EA, Brady A, Crabtree J, Drabek EF, Ma B, Mahurkar A, et al. Functional dynamics of the gut microbiome in elderly people during probiotic consumption. MBio. 2015;6:e00231–15. doi: 10.1128/mBio.00231-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci Transl Med. 2011;3:106ra106. doi: 10.1126/scitranslmed.3002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Everard A, Lazarevic V, Gaïa N, Johansson M, St\a ahlman M, Backhed F, et al. Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J. 2014;8:2116–2130. doi: 10.1038/ismej.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sohail MU, Hume ME, Byrd JA, Nisbet DJ, Shabbir MZ, Ijaz A, et al. Molecular analysis of the caecal and tracheal microbiome of heat-stressed broilers supplemented with prebiotic and probiotic. Avian Pathol. 2015;44:67–74. doi: 10.1080/03079457.2015.1004622. [DOI] [PubMed] [Google Scholar]
- 23.del Campo R, Garriga M, Pérez-Aragón A, Guallarte P, Lamas A, Máiz L, et al. Improvement of digestive health and reduction in proteobacterial populations in the gut microbiota of cystic fibrosis patients using a Lactobacillus reuteri probiotic preparation: A double blind prospective study. J Cyst Fibros. 2014;13:716–22. doi: 10.1016/j.jcf.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Kuczynski J, Liu Z, Lozupone C, McDonald D, Fierer N, Knight R. Microbial community resemblance methods differ in their ability to detect biologically relevant patter. Nat Methods. 2010;7:813–9. doi: 10.1038/nmeth.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huttenhower C, Knight R, Brown CT, Caporaso JG, Clemente JC, Gevers D, et al. Advancing the microbiome research community. Cell. 2014;159:227–30. doi: 10.1016/j.cell.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha R, Abnet CC, White O, Knight R, Huttenhower C. The microbiome quality control project: baseline study design and future directions. Genome Biol. 2015;16:276. doi: 10.1186/s13059-015-0841-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao Z-L, Zeng B-H, Wang W, Li G-H, Wu F, Wang L, et al. Impact of the Consumption of Tea Polyphenols on Early Atherosclerotic Lesion Formation and Intestinal Bifidobacteria in High-Fat-Fed ApoE−/− Mice. Front Nutr. 2016;3 doi: 10.3389/fnut.2016.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng M, Zhang X, Miao Y, Cao J, Wu Z, Weng P. The modulatory effect of (-)-epigallocatechin 3-O-(3-O-methyl) gallate (EGCG3″Me) on intestinal microbiota of high fat diet-induced obesity mice model. Food Res Int. 2017;92:9–16. doi: 10.1016/j.foodres.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 29.Janssens PLHR, Penders J, Hursel R, Budding AE, Savelkoul PHM, Westerterp-Plantenga MS. Long-Term Green Tea Supplementation Does Not Change the Human Gut Microbiota. PloS One. 2016;11:e0153134. doi: 10.1371/journal.pone.0153134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen C-L, Brackee G, Song X, Tomison MD, Finckbone V, Mitchell KT, et al. Safety Evaluation of Green Tea Polyphenols Consumption in Middle-aged Ovariectomized Rat Model. J Food Sci. 2017 doi: 10.1111/1750-3841.13745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.International Conference on Harmonisation. S4 Toxicity Tesing: Duration of chronic toxicity testing in animals (rodent and non rodent toxicity testing) 1998 [Google Scholar]
- 32.Wang J, Tang L, Glenn TC, Wang JS. Aflatoxin B1 induced compositional changes in gut microbial communities of male F344 rats. Toxicol Sci. 2016;150:54–63. doi: 10.1093/toxsci/kfv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glenn TC, Nilsen R, Kieran TJ, Finger JW, Pierson TW, Bentley KE, et al. Adapterama I: Universal stubs and primers for thousands of dual-indexed Illumina libraries (iTru & iNext) BioRxiv. 2016;049114 doi: 10.1101/049114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nocker A, Burr M, Camper AK. Genotypic microbial community profiling: a critical technical review. Microb Ecol. 2007;54:276–89. doi: 10.1007/s00248-006-9199-5. [DOI] [PubMed] [Google Scholar]
- 35.The Human Microbiome Project Consortium. A framework for human microbiome research. Nature. 2012;486:215–21. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jumpstart Consortium Human Microbiome Project Data Generation Working Group. Evaluation of 16S rDNA-Based Community Profiling for Human Microbiome Research. PLOS ONE. 2012;7:e39315. doi: 10.1371/journal.pone.0039315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–41. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 38.Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 2014;32:822–8. doi: 10.1038/nbt.2939. [DOI] [PubMed] [Google Scholar]
- 39.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li D, Luo R, Liu C-M, Leung C-M, Ting H-F, Sadakane K, et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 2016;102:3–11. doi: 10.1016/j.ymeth.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 41.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:1. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.VSEARCH. n.d.
- 44.Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–31. doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 45.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–93. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 47.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lozupone CA, Knight R. Species divergence and the measurement of microbial diversity. FEMS Microbiol Rev. 2008;32:557–78. doi: 10.1111/j.1574-6976.2008.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and Qualitative Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Appl Environ Microbiol. 2007;73:1576–85. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
- 52.High-sensitivity pattern discovery in large multi’omic datasets. n.d.
- 53.Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61:1–10. [Google Scholar]
- 54.Thomas F, Hehemann J-H, Rebuffet E, Czjzek M, Michel G. Environmental and gut bacteroidetes: the food connection. Front Microbiol. 2011;2 doi: 10.3389/fmicb.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–3. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 57.Tilg H. Obesity, metabolic syndrome, and microbiota: multiple interactions. J Clin Gastroenterol. 2010;44:S16–S18. doi: 10.1097/MCG.0b013e3181dd8b64. [DOI] [PubMed] [Google Scholar]
- 58.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Konikoff T, Gophna U. Oscillospira: a Central, Enigmatic Component of the Human Gut Microbiota. Trends Microbiol. 2016;24:523–4. doi: 10.1016/j.tim.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 60.Tims S, Derom C, Jonkers DM, Vlietinck R, Saris WH, Kleerebezem M, et al. Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J. 2013;7:707–17. doi: 10.1038/ismej.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW, et al. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity. 2013;21:E607–15. doi: 10.1002/oby.20466. [DOI] [PubMed] [Google Scholar]
- 62.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human Genetics Shape the Gut Microbiome. Cell. 2014;159:789–99. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Escobar JS, Klotz B, Valdes BE, Agudelo GM. The gut microbiota of Colombians differs from that of Americans, Europeans and Asians. BMC Microbiol. 2014;14:311. doi: 10.1186/s12866-014-0311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, et al. Human Gut Microbiome and Risk for Colorectal Cancer. J Natl Cancer Inst. 2013;105:1907–11. doi: 10.1093/jnci/djt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garrett RH, Grisham CM. Biochemistry. Belmont, CA: Brooks/Cole, Cengage Learning; 2010. [Google Scholar]
- 66.Martin F-PJ, Wang Y, Sprenger N, Yap IKS, Lundstedt T, Lek P, et al. Probiotic modulation of symbiotic gut microbial–host metabolic interactions in a humanized microbiome mouse model. Mol Syst Biol. 2008;4 doi: 10.1038/msb4100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Uronis JM, Arthur JC, Keku T, Fodor A, Carroll IM, Cruz ML, et al. Gut microbial diversity is reduced by the probiotic VSL#3 and correlates with decreased TNBS-induced colitis. Inflamm Bowel Dis. 2011;17:289–97. doi: 10.1002/ibd.21366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen C-L, Cao JJ, Dagda RY, Chanjaplammootil S, Lu C, Chyu M-C, et al. Green tea polyphenols benefits body composition and improves bone quality in long-term high-fat diet–induced obese rats. Nutr Res. 2012;32:448–57. doi: 10.1016/j.nutres.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 69.Cox‐York KA, Sheflin AM, Foster MT, Gentile CL, Kahl A, Koch LG, et al. Ovariectomy results in differential shifts in gut microbiota in low versus high aerobic capacity rats. Physiol Rep. 2015;3:e12488. doi: 10.14814/phy2.12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scholz-Ahrens KE, Adolphi B, Rochat F, Barclay DV, de Vrese M, Açil Y, et al. Effects of probiotics, prebiotics, and synbiotics on mineral metabolism in ovariectomized rats — impact of bacterial mass, intestinal absorptive area and reduction of bone turn-over. NFS J. 2016;3:41–50. doi: 10.1016/j.nfs.2016.03.001. [DOI] [Google Scholar]
- 71.Zhang Z, Chen Y, Xiang L, Wang Z, Xiao GG, Hu J. Effect of Curcumin on the Diversity of Gut Microbiota in Ovariectomized Rats. Nutrients. 2017;9:1146. doi: 10.3390/nu9101146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scalbert A, Manach C, Morand C, Rémésy C, Jiménez L. Dietary Polyphenols and the Prevention of Diseases. Crit Rev Food Sci Nutr. 2005;45:287–306. doi: 10.1080/1040869059096. [DOI] [PubMed] [Google Scholar]
- 73.Manach C, Williamson G, Morand C, Scalbert A, Rémésy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr. 2005;81:230S–242S. doi: 10.1093/ajcn/81.1.230S. [DOI] [PubMed] [Google Scholar]
- 74.Wang J, Tang L, Wang JS. Biomarkers of dietary polyphenols in cancer studies: current evidence and beyond. Oxid Med Cell Longev. 2015;2015:1–14. doi: 10.1155/2015/732302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.