

Abstract
Sulindac is a non-steroidal anti-inflammatory drug (NSAID) that has shown significant anticancer activity. Sulindac sulfide amide (1) possessing greatly reduced COX-related inhibition relative to sulindac displayed in vivo antitumor activity that was comparable to sulindac in a human colon tumor xenograft model. Inspired by these observations, a panel of diverse sulindac amide derivatives have been synthesized and their activity probed against three cancer cell lines (prostate, colon and breast). A neutral analog, compound 79 was identified with comparable potency relative to lead 1 and activity against a panel of lymphoblastic leukemia cell lines. Several new series also show good activity relative to the parent (1), including five analogs that also possess nanomolar inhibitory potencies against acute lymphoblastic leukemia cells. Several new analogs identified may serve as anticancer lead candidates for further development.
Keywords: NSAIDs, sulindac, amides, cancer
Graphical Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) are a chemically diverse family of drugs commonly used clinically to treat a variety of inflammatory conditions including pain associated with arthritis. A number of these drugs possess antipyretic activity in addition to having analgesic and anti-inflammatory action, and thus have utility in the treatment of pain and fever. These are widely known to be cyclooxygenase (COX) inhibitors. The COX enzymes participate in the metabolism of prostaglandins. For example, COX-1 enzymes produce prostaglandins that are important for the stomach lining and kidney function. COX-2 enzymes are crucial to anti-inflammatory reactions in the body.1 Many currently marketed NSAIDs have both COX-1 and COX-2 inhibitory effects. Although the class is highly used both clinically and over the counter, chronic COX inhibition is associated with gastrointestinal, renal, and cardiovascular side effects.2–6 Epidemiological, preclinical, and clinical studies suggest that NSAIDs and COX-2 inhibitors display striking cancer chemopreventive efficacy by reducing cancer incidence in the general population.1,7,8 Although the mechanism responsible for the anticancer activity of NSAIDs is still unknown, it is commonly attributed to COX-2 mediation.9–11 Studies have shown that several human tumors contain increased levels of COX-2 as compared to normal tissues. It has been determined that excess expression of COX-2 in cancer cells correlates to increased tumorigenic potential. Experiments have demonstrated that COX-2 specific NSAIDs restore apoptotic responses, or programmed cell death, in tumor cells. By selective enzyme inhibition and restoration of normal apoptotic responses, research data support the hypothesis that the anti-cancer mechanism is COX-2 dependent. Several COX-2 independent mechanisms have also been postulated for the antineoplastic properties of NSAIDs including activation of apoptosis, inhibition of angiogenesis, or direct inhibition of cancer cell growth by blocking signal transduction pathways responsible for cell proliferation.12–15 A number of molecular targets have been implicated in the cyclooxygenase independent pathways of anti-cancer effects of NSAIDs, as reviewed recently,16,17 and it was our intention to develop potential probes for these activities through ready variations in the scaffold. Sulindac, or Clinoril, is a substituted indene-3-acetic acid, and is a clinically used NSAID with established chemopreventive activity.13
Sulindac contains a chiral center (the sulfoxide), although the commercial drug is sold as a racemic mixture. In the body, the drug exists primarily in three active forms (see Figure 1) due to oxidation-reduction cycling. This cycling scrambles chirality at the SO bond and oxidation to the sulfone is irreversible.13 The sulfide metabolite generated through bioreduction in vivo is a potent, non-selective COX enzyme inhibitor and shows compelling anticancer activity as compared with other NSAIDS.
Figure 1.

In vivo metabolism (oxidation/reduction cycling) of the –SOCH3 center
Based on our and others’ data, it is clear that the NSAIDs have multifarious actions ranging from anti-inflammatory and anti-proliferative effects to selective anticancer cytotoxic activity.1,18 The fact that NSAIDs are relatively safe and orally effective suggests that there is an opportunity through simple, specific alterations of the diverse scaffolds to modify the basic structure to probe these various activities, and, at the same time, produce derivations that are more likely to yield good biological probes, animal activity, and drug leads. As an example, the preparation of neutral carboxamides is known to bias the NSAID scaffold towards selective COX-2 activity. As well, and supported by our preliminary results that replacement of the carboxylate functionality in sulindac in the form of sulindac sulfide amide (SSA) (1) can virtually abolish COX-related activity and toxicity while potentiating anticancer activity in vitro and yielding in vivo xenograft activity.18 These results are promising in that sulindac derivatives lacking the carboxylate functionality may show attenuated toxicity (gastric, renal, and cardiovascular) associated with COX inhibition. The presented chemical modifications have begun to explore, and possibly improve on, other NSAID activity space relating to selective cancer prevention and therapy.
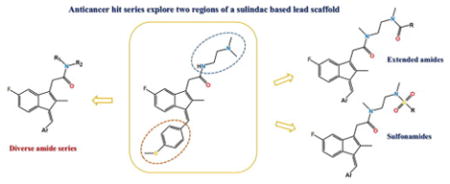
Herein, we describe the synthesis and anti-cancer activity of a series of sulindac analogs. Compound 1 (SSA) was considered the lead compound, and for this study two modification sites were chosen, the methylthiophenyl ring, and the acetamide linker (Figure 2). Various aryl groups were introduced in the indene core ring and a series of amides and sulfonamides were synthesized. All compounds were screened against three cancer cell lines (prostate, colon and breast) and selected compounds against a panel of additional cancer cell lines (five leukemia cancer cell lines and choroid plexus carcinoma cell line).
Figure 2.

Outline of structural modifications on the lead compound 1
Chemistry
Compounds 1–86 were synthesized by the coupling of different sulindac derivatives with various amines using either HBTU19 or HATU20 as the coupling agent (Scheme 1). Sulindac derivatives, if not commercially available, were synthesized from 2-(5-fluoro-2-methyl-1H-inden-3-yl)acetic acid and the corresponding aryl aldehydes in the presence of NaOMe.21
Scheme 1.

Synthetic pathway to analogs 1–86. Reagents: (a) HBTU or HATU, TEA or DIEA, MeCN
N,N′-Dimethylaminoethyl derivatives of various sulindac analogs (10, 22, 31) were treated with Boc-L-valine under the same coupling reaction conditions followed by hydrolysis for the removal of Boc protecting group to produce 87–89 in good yields. Sulfonamides 90–103 were also synthesized from amides, 10 and 22 by reacting with suitable sulfonyl chlorides in pyridine and N-methylimidazole (Scheme 2).
Scheme 2.

87–103 Reagents and conditions: (a) HBTU, TEA, Boc-L-valine, MeCN (b) H+ (c) RSO2Cl, NMI, Pyridine, 0 °C
Screening Results
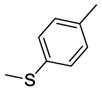
Using quantitative high-throughput screening all reported compounds were evaluated against PC3 prostate, HT29 colorectal carcinoma and MDA-MB-231 breast cancer cell lines. Description of the assays and screening method is provided in Appendix A, supplementary materials. SSA (1) was identified earlier as a lead compound for our structure-activity relationship (SAR) study with modification at the methylthiophenyl ring, and the acetamide linker. Amides shown in Scheme 1 were categorized as acyclic basic amides (Table 1), cyclic basic amides (Table 2), aromatic basic amides (Table 3) and neutral amides (Table 4). Screening data for the acyclic basic amides with modifications at the 4-methylthiobenzylidene ring at the C-1 position are shown in Table 1. First, the effect of various acyclic basic amide linkers at the C-3 position was explored (2–14) while retaining the 4-thiomethylbenzylidene group at the C-1 position.
Table 1.
Screening data for control 1 and basic acyclic amides 2–37.
| Ar | Cmpd | R1 & R2 | CC50 (μM)
|
||
|---|---|---|---|---|---|
| HT29 | PC3 | MDA-MB-231 | |||
|
1 | R1 = H, R2 = -CH2CH2N(CH3)2 | 0.65±0.03 | 3.12±0.15 | 2.67±0.08 |
| 2 | R1 = H, R2 = -CH2CH2CH2N(CH3)2 | 2.46±0.14 | 6.99±0.19 | 5.18±0.46 | |
| 3 | R1 = H, R2 = -CH2CH2N(CH2CH3)2 | 1.54±0.05 | 5.45±0.35 | 4.20±0.17 | |
| 4 | R1 = H, R2 = -CH2CH2CH2N(CH2CH3)2 | 1.53±3.33 | 4.33±0.22 | 3.19±0.08 | |
| 5 | R1 = CH3, R2 = -CH2CH2N(CH3)2 | 1.83±0.04 | 5.63±0.26 | 3.98±0.45 | |
| 6 | R1 = CH3, R2 = -CH2CH2CH2N(CH3)2 | 2.18±0.03 | 6.98±0.27 | 5.41±0.42 | |
| 7 | R1 = CH2CH3, R2 = -CH2CH2N(CH2CH3)2 | 1.83±0.04 | 6.21±0.44 | 5.22±0.20 | |
| 8 | R1 = CH3, R2 = -CH2CH2N(CH2CH3)2 | 1.49±0.02 | 5.65±0.26 | 3.77±0.35 | |
| 9 | R1 = CH2CH3, R2 = -CH2CH2N(CH3)2 | 1.36±0.05 | 5.07±0.31 | 3.96±1.46 | |
| 10 | R1 = CH3, R2 = -CH2CH2N(CH3)H | 2.80±0.07 | 6.75±0.24 | 5.84±0.35 | |
| 11 | R1 = CH3, R2 = -CH2CH2CH2N(CH3)H | 2.90±0.05 | 7.65±0.29 | 7.66±0.49 | |
| 12 | R1 = CH2CH3, R2 = -CH2CH2N(CH2CH3)H | 2.54±0.10 | 6.56±0.43 | 6.65±0.22 | |
| 13 | R1 = CH2CH3, R2 = -CH2CH2CH2N(CH2CH3)H | 1.38±0.05 | 3.92±0.29 | 3.03±0.19 | |
| 14 | R1 = CH(CH3)2, R2 = -CH2CH2CH2N(CH(CH3)2)H | 2.30±0.07 | 4.90±0.23 | 3.92±0.39 | |
|
| |||||
|
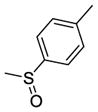
15 | R1 = H, R2 = -CH2CH2CH2N(CH3)2 | 33.27±2.92 | >50.00 | >50.00 |
| 16 | R1 = H, R2 = -CH2CH2N(CH2CH3)2 | 19.78±0.77 | >50.00 | >50.00 | |
| 17 | R1 = H, R2 = -CH2CH2CH2N(CH2CH3)2 | 27.12±2.0 | >50.00 | >50.00 | |
| 18 | R1 = CH3, R2 = -CH2CH2CH2N(CH3)2 | 38.92±7.98 | >50.00 | >50.00 | |
| 19 | R1 = CH2CH3, R2 = -CH2CH2N(CH2CH3)2 | 18.40±0.78 | >50.00 | >50.00 | |
| 20 | R1 = CH3, R2 = -CH2CH2N(CH2CH3)2 | 23.97±1.51 | >50.00 | >50.00 | |
| 21 | R1 = CH2CH3, R2 = -CH2CH2N(CH3)2 | 18.15±0.94 | >50.00 | >50.00 | |
| 22 | R1 = CH3, R2 = -CH2CH2N(CH3)H | 34.53±2.45 | >50.00 | >50.00 | |
| 23 | R1 = CH3, R2 = -CH2CH2CH2N(CH3)H | >50.00 | >50.00 | >50.00 | |
| 24 | R1 = CH2CH3, R2 = -CH2CH2N(CH2CH3)H | 18.18±1.40 | >50.00 | >50.00 | |
| 25 | R1 = CH2CH3, R2 = -CH2CH2CH2N(CH2CH3)H | 17.48±1.14 | >50.00 | >50.00 | |
| 26 | R1 = CH(CH3)2, R2 = -CH2CH2CH2N(CH(CH3)2)H | 16.28±0.97 | 48.28±8.05 | >50.00 | |
|
| |||||
|
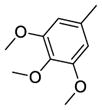
27 | R1 = H, R2 = -CH2CH2N(CH3)2 | 6.22±0.44 | 18.27±1.34 | 15.05±0.88 |
| 28 | R1 = H, R2 = -CH2CH2CH2N(CH3)2 | 6.08±0.30 | 13.03±0.74 | 10.79±0.45 | |
| 29 | R1 = H, R2 = -CH2CH2N(CH2CH3)2 | 5.42±0.38 | 13.88±0.78 | 12.26±0.34 | |
| 30 | R1 = CH3, R2 = -CH2CH2N(CH3)2 | 4.68±0.23 | 11.44±0.52 | 10.78±0.65 | |
| 31 | R1 = CH3, R2 = -CH2CH2N(CH3)H | 8.05±0.31 | 23.06±1.31 | 16.96±1.04 | |
|
| |||||
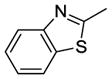
|
32 | R1 = H, R2 = -CH2CH2N(CH3)2 | 1.57±0.07 | 4.88±0.53 | 3.63±0.27 |
| 33 | R1 = H, R2 = -CH2CH2CH2N(CH3)2 | 1.05±0.05 | 3.25±0.13 | 1.93±0.13 | |
| 34 | R1 = H, R2 = -CH2CH2N(CH2CH3)2 | 1.12±0.05 | 4.48±0.32 | 3.54±0.24 | |
| 35 | R1 = CH3, R2 = -CH2CH2N(CH3)2 | 2.02±0.06 | 5.88±0.32 | 3.80±0.51 | |
|
| |||||
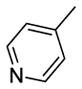
|
36 | R1 = H, R2 = -CH2CH2N(CH3)2 | 6.89±0.58 | 28.90±2.05 | 18.06±1.07 |
|
| |||||
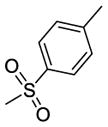
|
37 | R1 = H, R2 = -CH2CH2N(CH3)2 | 12.67±0.73 | >50.00 | 37.46±3.45 |
Table 2.
Screening data for basic cyclic amides 38–66.
| Ar | Cmpd | R1 & R2 | CC50 (μM)
|
||
|---|---|---|---|---|---|
| HT29 | PC3 | MDA-MB-231 | |||
|
|
38 |
|
1.81±0.06 | 5.27±0.32 | 3.78±0.26 |
| 39 |
|
1.65±0.14 | 5.36±0.18 | 5.42±0.24 | |
| 40 |
|
2.98±18.68 | 6.66±0.43 | 7.66±0.52 | |
| 41 |
|
1.41±0.04 | 4.33±0.23 | 3.75±0.23 | |
| 42 |
|
1.16±0.08 | 4.64±0.23 | 3.72±0.23 | |
| 43 |
|
1.55±0.06 | 5.10±0.36 | 4.16±0.29 | |
| 44 | R1, R2 = -(-CH2CH2-)2NCH2CH2CH2N(CH3)2 | 1.69±0.08 | 4.28±0.16 | 3.77±1.18 | |
| 45 | R1, R2 = -(-CH2CH2-)2NCH2CH2N(CH3)2 | 2.17±0.06 | 6.23±0.37 | 4.05±0.23 | |
| 46 | R1, R2 = -(-CH2CH2-)2N(pyridin-4-yl) | 1.13±0.07 | 2.76±0.19 | 3.08±0.07 | |
| 47 | R1, R2 = -(-CH2CH2-)2CH(pipyridin-1-yl) | 1.06±0.05 | 4.06±0.27 | 2.61±0.09 | |
| 48 |
|
1.64±0.07 | 4.45±0.16 | 4.10±0.19 | |
| 49 |
|
4.29±0.19 | 13.42±0.49 | 11.50±0.53 | |
| 50 |
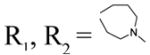
|
2.94±0.16 | 7.35±0.32 | 7.21±0.22 | |
| 51 |
|
2.47±0.67 | 6.50±0.84 | 3.69±0.17 | |
|
| |||||
|
|
52 | R1, R2 = -(-CH2CH2-)2NCH2CH2CH2N(CH3)2 | 23.75±1.48 | >50.00 | 46.18±2.63 |
| 53 | R1, R2 = -(-CH2CH2-)2N(Pyridin-4-yl) | 9.20±0.39 | 21.93±2.16 | 22.74±2.29 | |
| 54 | R1, R2 = -(-CH2CH2-)2CH(pipyridin-1-yl) | >50.00 | >50.00 | >50.00 | |
| 55 |
|
19.03±1.26 | >50.00 | >50.00 | |
| 56 |
|
20.16±1.06 | >50.00 | >50.00 | |
|
| |||||
|
|
57 |
|
13.39±0.71 | 46.49±5.98 | 41.09±3.78 |
| 58 |
|
4.18±0.19 | 9.62±0.66 | 10.38±0.59 | |
| 59 | R1, R2 = -(-CH2CH2-)2N(Pyridin-4-yl) | 2.79±0.13 | 5.45±0.68 | 6.50±0.77 | |
| 60 |
|
4.22±0.15 | 12.28±0.72 | 11.14±0.56 | |
|
| |||||
|
|
61 |
|
1.21±0.04 | 4.94±0.28 | 3.56±0.28 |
| 62 | R1, R2 = -(-CH2CH2-)2NCH2CH2CH2N(CH3)2 | 1.62±0.06 | 4.21±0.23 | 2.90±0.07 | |
| 63 | R1, R2 = -(-CH2CH2-)2N(Pyridin-4-yl) | 5.25±0.20 | 12.28±0.37 | 7.71±1.5 | |
| 64 |
|
1.66±0.04 | 7.72±0.45 | 5.58±0.33 | |
| 65 |
|
1.58±0.04 | 4.25±0.15 | 3.61±0.23 | |
|
| |||||
|
|
66 |
|
2.68±0.12 | 10.19±0.41 | 7.19±0.31 |
Table 3.
Screening data for basic aromatic amides 67–71.
| Ar | Cmpd | R1 & R2 | CC50 (μM)
|
||
|---|---|---|---|---|---|
| HT29 | PC3 | MDA-MB-231 | |||
|
|
67 | R1 = H, R2 = 4-(N,N-dimethylamino)benzyl | >50.00 | >50.00 | >50.00 |
| 68 | R1 = H, R2 = 4-(N,N-dimethylamino)phenyl | >50.00 | >50.00 | 30.60±15.76 | |
| 69 | R1 = H, R2 = 4-pyridylmethyl | 4.88±0.18 | 5.83±1.18 | 6.46±0.93 | |
| 70 | R1 = H, R2 = 3-pyridylmethyl | 4.83±1.1 | 18.52±7.78 | 7.36±1.94 | |
| 71 | R1 = H, R2 = 2-pyridylmethyl | 10.31±0.79 | 13.64±2.10 | 13.69±1.85 | |
Table 4.
Screening data for neutral amides 72–86.
| Ar | Cmpd | R1 & R2 | CC50 (μM)
|
||
|---|---|---|---|---|---|
| HT29 | PC3 | MDA-MB-231 | |||
|
|
72 | R1 = H, R2 = benzyl | 1.52±0.39 | 16.36±8.55 | 3.61±1.87 |
| 73 | R1 = H, R2 = furan-2-ylmethyl | 2.08±0.82 | 12.98±6.83 | 3.40±1.07 | |
| 74 | R1 = H, R2 = thiophen-2-ylmethyl | 2.16±0.71 | 11.03±5.95 | 5.66±2.69 | |
| 75 | R1 = H, R2 = -CH2CH2CH(CH3)2 | >50.00 | >50.00 | >50.00 | |
| 76 | R1 = H, R2 = D-Alanine methyl ester | 16.22±2.61 | 44.18±7.38 | 31.99±2.33 | |
| 77 | R1 = H, R2 = L-Alanine methyl ester | 23.92±1.92 | >50.00 | 38.86±6.04 | |
| 78 | R1 = H, R2 = L-Histidine methyl ester | 23.01±3.09 | >50.00 | >50.00 | |
|
| |||||
|
|
79 | R1 = H, R2 = benzyl | 0.50±0.23 | 2.64±1.19 | 0.78±0.32 |
| 80 | R1 = H, R2 = furan-2-ylmethyl | 1.00±0.50 | >50.00 | 2.73±1.21 | |
| 81 | R1 = H, R2 = -CH2CH2CH(CH3)2 | 17.65±1.11 | 20.23±2.29 | 27.72±3.37 | |
|
| |||||
|
|
82 | R1 = H, R2 = -CH2CH2CH(CH3)2 | 5.24±1.18 | 8.20±1.92 | 8.73±1.97 |
|
| |||||
|
|
83 | R1 = H, R2 = benzyl | 23.56±2.04 | >50.00 | 19.63±2.98 |
| 84 | R1 = H, R2 = furan-2-ylmethyl | 5.48±1.70 | 26.44±15.80 | 6.70±2.23 | |
| 85 | R1 = H, R2 = -CH2CH2CH(CH3)2 | >50.00 | >50.00 | 24.20±5.35 | |
|
| |||||
|
|
86 | R1 = H, R2 = benzyl | 2.14±0.46 | 5.71±1.75 | 3.68±1.10 |
Among these 13 compounds, none was found to be more active compared to the lead agent 1 although many showed relatively comparable cell growth inhibition. Simple modifications of the N,N-dimethylaminoethylamino group of 1 reduced the activity by 2–5 fold. Compound 2, with a N,N-dimethylpropyl substitution at the acetamide linker, reduced the activity by 4-fold compared to the lead compound, SSA (1) in HT29 cells, and by 2-fold in PC3 and MDA-MB-231. Compounds 3 and 4 with a N,N-diethylamino group at the ethylene or propylene acetamide groups were more active than 2, but less potent than 1 in all three cell lines. Either an additional substituent at the acetamide nitrogen or removal of a substituent from the terminal nitrogen did not improve the activity (5–14) dramatically. While maintaining the 4-methylsulfinylbenzylidene at the C-1 position, we explored the SAR of selected basic acyclic acetamide linkers (15–26). All these analogs led to reduced, or a complete loss of potency. Selected basic acyclic acetamide linkers with a 3,4,5-trimethoxybenzylidene group at the C-1 position (27–31) showed better activity than their corresponding 4-methylsulfinylbenzylidene analogs, but were less active than corresponding 4-thiobenzylidene analogs. These compounds were less active than 1, but still were highly active in vitro in all three assays. We then expanded the SAR with selected basic acyclic acetamide linkers, while keeping the benzothiazol-2-ylmethylene group as a fixed unit (32–35). These analogs demonstrated significant activity similar to their 4-methylthiobenzylidene counterparts. We also explored the anticancer activity of pyridin-4-ylmethylene (36) and 4-sulfonylbenzylidene (37) analogs of 1. These compounds were modestly active against all three cell lines, but less potent than 1.
Screening results for basic cyclic amides 38–66 are summarized in Table 2. Compounds 38–51, with a 4-methylthiobenzylidene ring at the C-1 position and various basic cyclic acetamide linkers at the C-3 position showed significant activity against cancer similar to acyclic basic amide analogs 1–14. We also explored the SAR of selected aryl modifications at the C-1 position compared to selected basic cyclic acetamides (52–66). However, none of these series, except the benzothiazolyl analogs (61–65), demonstrated significant activity.
While maintaining the 4-methylthiobenzylidene at the C-1 position, we also examined the effects of basic aromatic acetamide linkers at the C-3 position (Table 3). Compounds 67 and 68 with a N,N-dimethylamino group at the phenyl group were inactive in all three assays. Compounds with 2, 3, and 4-pyridylmethyl acetamide (69, 70 and 71) showed inhibition but all with lesser potency than lead 1.
We next explored the anticancer activity of neutral amides (Table 4).
Compounds 72–78 have a 4-methylthiobenzylidene ring at the C-1 position. Compound 72, with a benzyl group at the acetamide linker, reduced the activity by 4-fold as compared to lead compound in HT29 and PC3 cells and in MDA-MB-231 cells by 1.5-fold. The aromatic heterocyclic compounds such as furan-2-yl analog 73 and thiophene-2-yl analog 74 had activity very similar to the corresponding phenyl analog 72 in all screening assays, but were less active than lead 1. Compound 75, where the terminal dimethylamino group of 1 was replaced by an isopropyl group, was not active. The amino acid analogs 76–78 were >25-fold less potent than the lead compound. By maintaining the 4-methylsulfinylbenzylidene at the C-1 position, we explored the SAR of selected acetamide linkers. Among these three compounds (79–81), compound 79 with a benzyl group at the acetamide linker showed excellent activity relative to lead 1. Furan-2-yl analog 80 also showed significant activity as compared to its methylthiobenzylidene analog 73, but was less active than 1. Selected acetamide linkers with a 3,4,5-trimethoxybenzylidene group, benzothiazol-2-ylmethylene group or a pyridin-4-ylmethylene group at the C-1 position (82–86) did not show improved activity.
Intestinal cells, cancer cells, and other cell types are known to have specific amino acid (AA) uptake mechanisms that have been utilized to increase drug uptake upon AA conjugation. For example, conjugation of the active HSV agent acyclovir and penciclovir as the valyl ester through the AA-COOH group results in blood levels 3–5 times that of the parent drug due to specific valine uptake mechanisms.22,23 Inspired by this concept, compounds 87–89 were synthesized from N-methyl-N-(2-(methylamino)ethyl)acetamide analogs 10, 22 and 31. All the three compounds, however, were less active than their corresponding parent compounds (Table 5) and potential gastric uptake remains untested.
Table 5.
Screening data for compounds 87–89.
| Cmpd | CC50 (μM)
|
||
|---|---|---|---|
| HT29 | PC3 | MDA-MB-231 | |
| 87 | 3.72±1.50 | 7.08±0.77 | 7.33±0.85 |
| 88 | 30.20±0.59 | >50.00 | >50.00 |
| 89 | 10.73±0.71 | 17.65±1.89 | 18.84±1.61 |
We also explored a sulfonamide linkage due to its inherent biostability and the opportunity to take advantage of further diversity generation using commercially available sulfonyl chlorides as well as the ease and typical high yields of sulfonamide formation. Further, certain sulfonamides have been reported to demonstrate anticancer activity.24–26 Hence, compounds 90–103 were synthesized from 10 and 22 and screened in the three cancer cell panel. A few analogs showed modest activity in these screens (Table 6).
Table 6.
Screening data for compounds 90–103.
| Cmpd | CC50 (μM)
|
||
|---|---|---|---|
| HT29 | PC3 | MDA-MB-231 | |
| 90 | 4.04±0.52 | 15.54±1.39 | 11.57±1.22 |
| 91 | 4.20±0.43 | 14.34±1.97 | 10.09±1.00 |
| 92 | 8.82±1.16 | >50.00 | >50.00 |
| 93 | 12.92±1.24 | >50.00 | >50.00 |
| 94 | 16.20±3.80 | >50.00 | >50.00 |
| 95 | 35.28±7.75 | >50.00 | >50.00 |
| 96 | >50.00 | >50.00 | >50.00 |
| 97 | 12.36±0.98 | 42.59±7.47 | 12.54±0.80 |
| 98 | 42.64±3.16 | >50.00 | >50.00 |
| 99 | 6.09±0.30 | 8.20±0.57 | 12.17±1.37 |
| 100 | 6.18±0.39 | 10.95±0.96 | 17.77±2.46 |
| 101 | 3.99±0.49 | 8.96±1.79 | 29.08±7.05 |
| 102 | 6.35±1.06 | 7.52±1.16 | 32.08±8.17 |
| 103 | 10.60±1.91 | 9.20±1.46 | >50.00 |
A selected set of analogs were additionally screened against a panel of six cancer cell lines at St. Jude Children’s Research Hospital consisting of four acute lymphoblastic leukemia (ALL) cell lines, one lymphoma line and a cell line derived from a mouse model of choroid plexus carcinoma (CPC), as described in Appendix A, supplementary materials. The selected compounds included the most potent series, namely, compounds 1–14, 32–35, 38–51, 59, 61–65 and 79. Out of the analog series presented in Table 4 5, – representative analogs 72–74, 79, 82, 84, 87 and 89 were also evaluated against this panel. Table 7 lists screening results for lead 1 and the analogs that show activity against at least one of these cancer cells. Compounds 5, 33, 35, 50, 62 and 89 stand out as nanomolar growth inhibitors of at least one type of acute lymphoblastic leukemia cancer cell line on this screening panel. Further, compound 79 shows low micromolar growth inhibition against all lymphoblastic leukemia cell lines on the screening panel while very weak activity against the choroid plexus carcinoma cell line. These analogs with the exception of 89 are also good inhibitors of the HT29, PC3 and MDA cancer cell lines. Interestingly, a related scaffold containing a 3,4,5-trimethoxyphenyl aryl moiety, the indomethacin analog of compound 27 was previously shown to inhibit potently proliferation of multiple cancer cell lines through interfering with tubulin polymerization.27 Thus, we identified a number of analogs potently inhibiting multiple types of cancer cell lines. These compounds may be pursued in future work as potential anticancer leads. Of note, the parent hit sulindac sulfide amide (SSA, 1) showed no activity against cancer cell lines in this panel (Table 7).
Table 7.
Cancer cell line screening data and cytotoxicity counter screen against BJ cells. Confidence intervals are shown in parentheses; NA: not active; ND: undetermined due to questionable curve fit.
| Cmpds | EC50 (μM)
|
||||||
|---|---|---|---|---|---|---|---|
| CPC300 | NALM16 | RAJI | JURKT | REH | 697 | BJ | |
| 1 | NA | NA | NA | NA | NA | NA | >7.57 |
| 4 | NA | ND | 21 (16–27) | NA | NA | NA | >7.57 |
| 5 | NA | 0.071 (0.059–0.1) | 0.83 (0.26–0.79) | NA | NA | NA | >7.57 |
| 33 | 65 (53–96) | 0.35 (0.27–0.53) | NA | NA | NA | NA | >7.57 |
| 35 | NA | 0.3 (0.16–0.74) | NA | NA | NA | NA | >7.57 |
| 43 | NA | ND | 76 (36–72) | NA | NA | NA | >7.57 |
| 44 | NA | ND | 76 (48–74) | NA | NA | NA | >7.57 |
| 45 | 27 (26–29) | 1.9 (1.4–2.5) | NA | NA | NA | NA | >7.57 |
| 46 | 50 (48–52) | NA | NA | NA | NA | NA | >7.57 |
| 50 | NA | 0.49 (0.13–1.3) | ND | NA | NA | NA | >7.57 |
| 59 | NA | NA | NA | NA | 27 (21–34) | NA | >7.57 |
| 61 | NA | NA | 47 (26–73) | NA | 18 (15–29) | NA | >7.57 |
| 62 | NA | 0.097 (0.067–0.19) | NA | NA | NA | NA | >7.57 |
| 63 | 72 (56–130) | NA | NA | NA | NA | NA | >7.57 |
| 79 | 120 (95–150) | 1.3 (1.2–1.3) | 2.6 (2.4–2.7) | 1.3 (1.2–1.4) | 3.3 (3.2–3.5) | 4.1 (4.0–4.1) | >7.57 |
| 89 | NA | NA | NA | NA | ND | 0.96 (0.26–1.2) | >7.57 |
Cytotoxicity considerations and computed physicochemical properties
Lipophilicity (in addition to molecular weight and other molecular characteristics) is a crucial physicochemical property to consider during lead optimization.28–30 Like the parent compound, SSA, most analogs presented contain a positively charged amine, which coupled with high lipophilicity raises concern that such compounds may accumulate in cell membranes interacting with non-polar lipid chains while the positively charged amine associates with negatively charged head groups of fatty acids. This may lead to cytotoxicity through membrane destabilization. Selected series and representative analogs have been counter-screened for cytotoxicity against BJ cells, a normal human foreskin fibroblast cell line31 where cytotoxicity was determined at 10 μM drug concentration. The following compounds were evaluated for cytotoxicity in this assay: compounds 1–14, 32–35, 38–51, 59, 61–66, 72–74, 79, 82, 84, 87 and 89 (Table 7). All compounds screened showed EC50 > 7.57 μM in the BJ cytotoxicity assay suggesting that these compounds are not overtly toxic to this “normal” fibroblast cell line (Table 7). Compound 79 lacks a charged amine, yet it has the highest efficacy against all three cancer cell lines among the presented analogs while also potently inhibiting five lymphoblastic leukemia cell lines. Replacement of the thiomethyl (SSA) with the polar methylsulfinyl in 79 decreases lipophilicity of this analog.
A study examining correlations between physicochemical properties and toxicological outcomes has shown that topological polar surface area (TPSA) and cLogP properties correlate with in vivo toxicity in a set of 245 preclinical Pfizer compounds for which animal toxicity and exposure data has been collected.32 The ‘least toxic’ group was reported to have TPSA > 75 Å2 and cLogP < 3. We computed physicochemical properties relevant to lipophilicity for selected representative analogs in the most potent series, including LogP, LogD, TPSA and lipophilic efficiency (LipE), as shown in Table A.1, Appendix A. While listed compounds have LogP > 3 and show only modest variations compared to lead 1 (LogP 5.03), TPSA values are higher for most analogs relative to SSA which may be considered an improvement compared to the parent compound. Analogs 32–34, 61 and 80 have the highest (close to 75 Å2) TPSA values compared to lead 1 (TPSA 57.64 Å2). Compounds 40, 41, 44, 61, 62 have LogD values less than 3, compared to 4.41 of lead 1.
Lipophilic efficiency (LipE) relates potency to lipophilicity and may be used to assess and optimize binding efficiency in the context of lipophilicity. LipE listed in Table A.1 (Appendix A) was calculated as LipE = pCC50 (HT29) – logD (at pH 7.4). While the optimal target range for LipE is considered between 5 and 7, these analogs show a trend of slight improvement compared to SSA (LipE 1.78), compounds 41, 61 and 62 possessing LipE > 3. It is important to note that the CC50 values utilized for computing LipE results for these compounds were determined using whole cell screening assays. Generally, we would expect more potent in vitro dose-response activities against a specific protein target compared to whole cell activities which then would reflect in calculated LipE values. Thus, it is not surprising that LipE values obtained for these compounds are lower than the accepted optimal norms assessed in enzymatic, target-based assays.
A number of analogs in the reported series were designed to improve metabolic stability, based on metabolic site liability predictions using the CYP3A4 metabolism model implemented in the StarDrop software. For example, introducing a benzothiazolyl group as in 32, 61 is predicted to remove metabolically labile sites associated with the 4-methylthiophenyl group in SSA (as shown in Figures A.2, A.4, compared to SSA in Figure A.1, Appendix A). In addition, metabolic vulnerability may be also reduced by cyclization of the amine as exemplified for compounds 40 and 61 (Figures A.3 and A.4 in Appendix A, respectively).
In summary, we report herein an extensive SAR study of sulindac amide analogs that show significant anticancer activity in HT29, PC3 and MDA-MB-231 proliferation assays. Our primary goal in this work was to expand diversity around positions of the basic indene structure for probing anticancer activities of diverse amide analogs of sulindac sulfide amide (SSA). The most potent compound identified, compound 79, shows significant activity and potency compared to the lead compound SSA (1). This analog also has potent growth inhibitory activity against the five lymphoblastic lecukemia cell lines screened, against which SSA is inactive. Compound 79 with a benzyl group at the acetamide linker and a 4-methylsulfinylbenzylidene group at the C-1 position is a neutral analog lacking the basic amine moiety of SSA. Introducing various heterocycles or substituents in place of the 4-methylthiobenzylidine and the dimethylethylamine in lead compound 1 resulted in analog series that maintain growth inhibition at low micromolar potency in all three cancer cell lines suggesting that there is fair tolerance for alterations in these regions while maintaining good anticancer activity. Screening against the panel of six additional cancer cell lines identified five new analogs, compounds 5, 33, 35, 50, 62, that also show highly potent growth inhibitory activity against acute lymphoblastic leukemia cell line(s) in which the parent compound 1 was inactive. While possessing comparable anticancer activity to lead 1, a number of analogs within the presented series show improved computed physicochemical properties compared to compound 1, namely, improvement of lipophilic efficiency (LipE) and/or topological polar surface area (TPSA) and/or LogD properties. Among potent analogs that show improvement of physicochemical properties are compounds 32–34 with a benzothiazol-2-ylmethylene at the C-1 position and acyclic basic amide linkers, the series 40, 41, 44 containing various basic cyclic amide groups, analogs 61, 62 combining benzothiazol-2-ylmethylene at the C-1 position with basic cyclic amides at C-3, while compounds 79, 80 containing a benzyl and furanylmethyl amide group are neutral analogs. Analogs screened in a BJ cell cytotoxicity assay showed no toxicity against this fibroblast cell line. Our original design parameters were based on generation of structural diversity and ease of synthesis while, for the most part, maintaining a basic group near the carboxamide generated during coupling. Our presumption, based on earlier modeling and testing of our lead agent SSA, has been that the basic amine moiety replacing carboxylate in sulindac would attenuate or abrogate COX binding due to charge-charge interactions of the protonated amine with positively charged amino acid side chains in the COX 1–2 active sites.13,18 Whether compound 79, a neutral analog of sulindac lacking a basic amine, has any COX activity is presently unknown. This hypothesis and the mechanism of action for the new compounds presented herein will require experimental confirmation, and such experiments will be the focus of future work. Selected active compounds may also serve as potential candidates for formulation and in vivo assessment for bioavailability and efficacy.
Supplementary Material
Appendix A. Biological studies: experimental methods, computed physicochemical properties and metabolic stability predictions
Appendix B. Synthetic experimental methods and analytical data.
Acknowledgments
This work was supported by grants from the National Institutes of Health NCI 1R01CA131378 (RCR PI) and DOD Era of Hope award W81XWH-07-1-0463 (RCR PI). We thank Lucile White, Lynn Rasmussen and Melinda Ingram for HTS support services. We would also like to acknowledge contributions by the High Throughput Biology Center in the Department of Chemical Biology and Therapeutics at St. Jude Children’s Research Hospital in Memphis, TN for BJ cell line data and additional anticancer cell line screens. The work at St. Jude was supported by American Lebanese Syrian Associated Charities (ALSAC).
Abbreviations
- NSAID
non-steroidal anti-inflammatory drug
- COX
cyclooxygenase
- SSA
sulindac sulfide amide
- SAR
structure-activity relationship
- AA
amino acid
- ALL
acute lymphoblastic leukemia
- CPC
choroid plexus carcinoma
- TPSA
topological polar surface area
- LipE
lipophilic efficiency
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002;3:166–174. doi: 10.1016/s1470-2045(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 2.Cannon CP, Cannon PJ. Physiology. COX-2 inhibitors and cardiovascular risk. Science. 2012;336:1386–1387. doi: 10.1126/science.1224398. [DOI] [PubMed] [Google Scholar]
- 3.Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, et al. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4:132ra154. doi: 10.1126/scitranslmed.3003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vane JR, Botting RM. Mechanism of action of antiinflammatory drugs. Int J Tissue React. 1998;20:3–15. [PubMed] [Google Scholar]
- 5.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee D. Selective cyclooxygenase-2 (COX-2) inhibitors and potential risk of cardiovascular events. Biochem Pharmacol. 2002;63:817–821. doi: 10.1016/s0006-2952(02)00842-0. [DOI] [PubMed] [Google Scholar]
- 7.Marx J. Cancer research. Anti-inflammatories inhibit cancer growth--but how? Science. 2001;291:581–582. doi: 10.1126/science.291.5504.581. [DOI] [PubMed] [Google Scholar]
- 8.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 9.Brown JR, DuBois RN. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 2005;23:2840–2855. doi: 10.1200/JCO.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 10.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 11.Husain SS, Szabo IL, Tamawski AS. NSAID inhibition of GI cancer growth: clinical implications and molecular mechanisms of action. Am J Gastroenterol. 2002;97:542–553. doi: 10.1111/j.1572-0241.2002.05528.x. [DOI] [PubMed] [Google Scholar]
- 12.Alberts DS, Hixson L, Ahnen D, Bogert C, Einspahr J, et al. Do NSAIDs exert their colon cancer chemoprevention activities through the inhibition of mucosal prostaglandin synthetase? J Cell Biochem. 1995;(Suppl 22):18–23. doi: 10.1002/jcb.240590804. [DOI] [PubMed] [Google Scholar]
- 13.Piazza GA, Keeton AB, Tinsley HN, Whitt JD, Gary BD, et al. NSAIDs: old drugs reveal new anticancer targets. Pharmaceuticals. 2010;3:1652–1667. doi: 10.3390/ph3051652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, et al. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochemical Pharmacology. 1996;52:237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 15.Elder DJ, Halton DE, Hague A, Paraskeva C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin Cancer Res. 1997;3:1679–1683. [PubMed] [Google Scholar]
- 16.Liggett JL, Zhang X, Eling TE, Baek SJ. Anti-tumor activity of non-steroidal anti-inflammatory drugs: Cyclooxygenase-independent targets. Cancer Letters (NY, US) 2014;346:217–224. doi: 10.1016/j.canlet.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurpinar E, Grizzle WE, Piazza GA. NSAIDs Inhibit Tumorigenesis, but How? Clinical Cancer Research. 2013;20:1104–1113. doi: 10.1158/1078-0432.CCR-13-1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piazza GA, Keeton AB, Tinsley HN, Gary BD, Whitt JD, et al. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev Res (Phila) 2009;2:572–580. doi: 10.1158/1940-6207.CAPR-09-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dourtoglou V, Gross B, Lambropoulou V, Ziodrou C. O-Benzotriazolyl-N,N,N′,N′-tetramethyluronium hexafluorophosphate as coupling reagent for the synthesis of peptides of biological interest. Synthesis. 1984;7:572–574. [Google Scholar]
- 20.Carpino LA. 1-Hydroxy-7-Azabenzotriazole - an Efficient Peptide Coupling Additive. Journal of the American Chemical Society. 1993;115:4397–4398. [Google Scholar]
- 21.Sperl G, Gross P, Brendel K, Pamukcu R, Piazza G. Indenyl hydroxamic acids, (hydroxy) ureas and urethanes for treating patients with precancerous lesions. US6300346 B1. US Patent. 2000
- 22.Kim DK, Lee N, Kim YW, Chang K, Im GJ, et al. Synthesis and evaluation of amino acid esters of 6-deoxypenciclovir as potential prodrugs of penciclovir. Bioorg Med Chem. 1999;7:419–424. doi: 10.1016/s0968-0896(98)00235-1. [DOI] [PubMed] [Google Scholar]
- 23.Han HK, Oh DM, Amidon GL. Cellular uptake mechanism of amino acid ester prodrugs in Caco-2/hPEPT1 cells overexpressing a human peptide transporter. Pharm Res. 1998;15:1382–1386. doi: 10.1023/a:1011945420235. [DOI] [PubMed] [Google Scholar]
- 24.Abbate F, Casini A, Owa T, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: E7070, a sulfonamide anticancer agent, potently inhibits cytosolic isozymes I and II, and transmembrane, tumor-associated isozyme IX. Bioorg Med Chem Lett. 2004;14:217–223. doi: 10.1016/j.bmcl.2003.09.062. [DOI] [PubMed] [Google Scholar]
- 25.Rostom SA. Synthesis and in vitro antitumor evaluation of some indeno[1,2-c]pyrazol(in)es substituted with sulfonamide, sulfonylurea(-thiourea) pharmacophores, and some derived thiazole ring systems. Bioorg Med Chem. 2006;14:6475–6485. doi: 10.1016/j.bmc.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 26.Lee SK, Park SM, Im C. Cytotoxicities and Quantitative Structure Activity Relationships of B13 Sulfonamides in HT-29 and A549 Cells. Korean J Physiol Pharmacol. 2011;15:423–429. doi: 10.4196/kjpp.2011.15.6.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chennamaneni S, Zhong B, Lama R, Su B. COX inhibitors Indomethacin and Sulindac derivatives as antiproliferative agents: Synthesis, biological evaluation, and mechanism investigation. Eur J Med Chem. 2012;56:17–29. doi: 10.1016/j.ejmech.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Waring MJ. Defining optimum lipophilicity and molecular weight ranges for drug candidates-Molecular weight dependent lower logD limits based on permeability. Bioorg Med Chem Lett. 2009;19:2844–2851. doi: 10.1016/j.bmcl.2009.03.109. [DOI] [PubMed] [Google Scholar]
- 29.Johnson TW, Dress KR, Edwards M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg Med Chem Lett. 2009;19:5560–5564. doi: 10.1016/j.bmcl.2009.08.045. [DOI] [PubMed] [Google Scholar]
- 30.Meanwell NA. Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem Res Toxicol. 2011;24:1420–1456. doi: 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- 31.Mathew B, Hobrath JV, Ross L, Connelly MC, Lofton H, Rajagopalan M, Guy RK, Reynolds RC. Screening and development of new inhibitors of FtsZ from M. tuberculosis. PLoS ONE. 2016;11(10):e0164100. doi: 10.1371/journal.pone.0164100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes JD, Blagg J, Price DA, Bailey S, Decrescenzo GA, et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg Med Chem Lett. 2008;18:4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix A. Biological studies: experimental methods, computed physicochemical properties and metabolic stability predictions
Appendix B. Synthetic experimental methods and analytical data.