

Abstract
Conventional drug discovery efforts at the β2-adrenoceptor (β2AR) have led to the development of ligands that bind almost exclusively to the receptor’s hormone-binding orthosteric site. However, targeting the largely unexplored and evolutionarily unique allosteric sites has potential for developing more specific drugs with fewer side effects than orthosteric ligands. Using our recently developed approach for screening G protein–coupled receptors (GPCRs) with DNA-encoded small-molecule libraries, we have discovered and characterized the first β2AR small-molecule positive allosteric modulators (PAMs)—compound (Cmpd)-6 [(R)-N-(4-amino-1-(4-(tert-butyl)phenyl)-4-oxobutan-2-yl)-5-(N-isopropyl-N-methylsulfamoyl)-2-((4-methoxyphenyl)thio)benzamide] and its analogs. We used purified human β2ARs, occupied by a high-affinity agonist, for the affinity-based screening of over 500 million distinct library compounds, which yielded Cmpd-6. It exhibits a low micro-molar affinity for the agonist-occupied β2AR and displays positive cooperativity with orthosteric agonists, thereby enhancing their binding to the receptor and ability to stabilize its active state. Cmpd-6 is cooperative with G protein and β-arrestin1 (a.k.a. arrestin2) to stabilize high-affinity, agonist-bound active states of the β2AR and potentiates downstream cAMP production and receptor recruitment of β-arrestin2 (a.k.a. arrestin3). Cmpd-6 is specific for the β2AR compared with the closely related β1AR. Structure–activity studies of select Cmpd-6 analogs defined the chemical groups that are critical for its biologic activity. We thus introduce the first small-molecule PAMs for the β2AR, which may serve as a lead molecule for the development of novel therapeutics. The approach described in this work establishes a broadly applicable proof-of-concept strategy for affinity-based discovery of small-molecule allosteric compounds targeting unique conformational states of GPCRs.
Introduction
The modulation of G protein–coupled receptor (GPCR) activity plays an integral role in the treatment of a wide range of diseases. As such, GPCRs have become the target for over one third of current pharmaceuticals, the vast majority of which bind to the orthosteric site of the receptors. This region is defined as the site to which the endogenous ligand(s) for the receptor binds, such as adrenaline for the adrenoceptors or histamine for the histamine receptors (Lefkowitz, 2007; Whalen et al., 2011; Wacker et al., 2017). Most clinically used antagonists are orthosteric binders and exert their effects by competitive inhibition. Recently, however, an increasing number of negative and positive allosteric modulators [negative allosteric modulators (NAMs) and positive allosteric modulators (PAMs), respectively] for GPCRs has been described (Gentry et al., 2015), although to date only two have reached the clinic (Dorr et al., 2005; Lindberg et al., 2005). Rather than directly stimulating or inhibiting biologic effects on their own, these allosteric compounds exert their effects by modulating receptor responsiveness to endogenous agonists. Such allosteric ligands offer a number of potential advantages as drugs, including greater specificity among closely related receptor subtypes, and maximum or ceiling effects that can reduce adverse actions, among others (Wootten et al., 2013; Christopoulos, 2014). Such allosteric modulators can also serve as valuable reagents in the research laboratory, where, by means of their cooperative interactions with orthosteric ligands, they can help to freeze or lock specific receptor conformations so that they can be studied by biophysical techniques (Christopoulos, 2014; Wacker et al., 2017).
Selection of allosteric modulators for GPCRs using the usual cell-based functional assays such as those for cAMP generation or β-arrestin2 (a.k.a. arrestin3) recruitment (Rajagopal et al., 2010) has a number of disadvantages. These include that they can be quite laborious and difficult to interpret because one is looking for modulation of a response rather than the on or off responses that such assays are better suited to measure. Such assays are also subject to a variety of artifacts and have relatively limited compounds throughput of ∼103–106. In contrast, interaction or affinity-based methods, in which large libraries of self-encoding potential binders are screened against a target protein molecule, circumvent these shortcomings. A particularly powerful approach is the use of DNA-encoded small-molecule libraries (DELs) potentially containing billions of compounds. Each molecule in such a library is covalently linked to a small stretch of nucleotides, which serves as a barcode that is used to identify target binders by next-generation sequencing (NGS) (Franzini and Randolph, 2016; Goodnow et al., 2017). Such approaches work well when applied to soluble protein targets but have been much more difficult to adapt to membrane proteins such as GPCRs. However, using this approach, we recently described isolation of the first NAM for the β2-adrenoceptor (allosteric β-blocker) (Ahn et al., 2017) and identified its intracellular binding site on the receptor by X-ray crystallography (Liu et al., 2017). This molecule, compound (Cmpd)-15, was isolated by panning DELs against the inactive receptor, in which the orthosteric site was unoccupied.
An advantage of affinity-based screening methods is that one can bias the selections toward isolation of molecules with a particular functional profile by including one or another orthosteric ligand or even allosteric transducer protein molecules, e.g., G protein or β-arrestin, in complex with the receptor. In this work, we report our successful isolation of the first PAMs of the β2-adrenoceptor (β2AR) by panning DELs against the purified receptor occupied by the high-affinity agonist BI-167107 (Rasmussen et al., 2011). We present a detailed pharmacological characterization of these molecules with the receptor and illustrate their potential utility as laboratory tools for interrogating biophysical properties of the receptors, as well as molecules for a new type of therapeutic agent.
Materials and Methods
Materials.
Cmpd-6 and its analogs were synthesized using the methods described below. With the exception of BI-167107, which was synthesized as described previously (Wang et al., 2013), all of the orthosteric β2AR ligands used were purchased from Sigma-Aldrich (St. Louis, MO) and sourced at a 95% or greater purity. Nuevolution provided the DNA-encoded libraries used for screening. The [3H](R,R′)-4-methoxyfenoterol used in the radioligand-binding studies was provided by Irving Wainer (Laboratory of Clinical Investigation, National Institute on Aging Intramural Research Program, Bethesda, MD). The β2AR-Gsα and β2 vasopressin 2 receptor (V2R)-β-arrestin1 fusion clones containing an N-terminal hemagglutinin signal sequence followed by a FLAG epitope tag for the receptor were generated in pcDNA3.1 by standard polymerase chain reaction (PCR) amplification and cloning procedure. β-arrestin1 cDNA was amplified by standard PCR methods and cloned in-frame with the C terminus of β2V2R essentially as before (Strachan et al., 2014). For the Gs fusion construct, the coding sequence for the short splice variant of human Gsα subunit was used from a plasmid obtained from cDNA Resource Center (Bloomsburg, PA). Both Gsα and β2AR sequences were PCR amplified separately, and the amplified fragments were assembled into a tetracycline-inducible pcDNA3.1 plasmid by using HiFi DNA assembly (NEB, Ipswich, MA) to finally generate the Gsα fusion at the C terminus of the receptor. Both fusion constructs were sequence verified, and aliquots of maxi-prepared DNA were used for transfections. Previous purification methods were used to obtain rat β-arrestin1 (a nonvisual arrestin, a.k.a. arrestin2) and heterotrimeric Gs protein (Shukla et al., 2013; Staus et al., 2016).
Cell Culture and Transfection.
HEK-293 and HEK-293T cells were cultured at 37°C and at 5% CO2 in a humidified condition. Cells were cultured in standard minimum Eagle’s growth media supplemented with 10% fetal bovine serum and penicillin/streptomycin. HEK-293 cell lines stably expressing the GloSensor (Promega, Madison, WI) cAMP reporter (Nobles et al., 2011) and HEK-293T cells for the Tango assay (Barnea et al., 2008) were maintained, as described before. The HEK-293 cell line stably expressing the GloSensor cAMP reporter together with the β2AR was created by transfecting a hygromycin B–resistant plasmid expressing the GloSensor reporter into cells stably overexpressing the β2AR (Shenoy et al., 2006), followed by selection with 150 μg/ml hygromycin B (Invitrogen, Carlsbad, CA). The clonal line with the greatest fold over basal ratio and highest sensitivity was selected and maintained with 150 μg/ml G418 (Sigma-Aldrich) and 100 μg/ml hygromycin B. Transient transfections were performed using FuGENE 6 (Promega), according to the manufacturer’s instructions, and all assays were done ∼48 hours post-transfection. The β2AR-Gs and β2V2R-β-arrestin 1 fusion proteins were transfected into Expi293F cells (Invitrogen) using Expifectamine (Invitrogen), as described by the manufacturer.
Expression, Purification, and High-Density Lipoprotein Reconstitution of the β2AR.
As previously described (Kobilka, 1995), BestBac Baculovirus Expression System was used to express the full-length human β2AR containing an amino-terminal FLAG epitope tag, carboxyl-terminal His-tag, and a N187E glycosylation mutation in Sf9 insect cells. In brief, cells were infected at a density of 3 × 106 cells/ml and harvested 67 hours thereafter. The cells were then solubilized in a buffer containing 1% n-dodecyl-β-d-maltoside (Anatrace, Maumee, OH), 20 mM HEPES, pH 7.4, 150 mM NaCl, and protease inhibitors. Functional β2AR was purified, as previously described (Kobilka, 1995; Kahsai et al., 2016), using FLAG-M1 antibody and alprenolol affinity chromatography, followed by size exclusion chromatography using a Superdex 200 (16/600 prep grade) column. The monomeric receptor peak from the size exclusion chromatography was pooled and concentrated to 1–2 mg/ml. Purified functional β2AR was then reconstituted into high-density lipoprotein (HDL) particles using previously published methods (Whorton et al., 2007; Staus et al., 2016). In brief, FLAG-β2AR was incubated with a 50-fold molar excess of biotinylated membrane scaffold protein 1 Apo A1 and 8 mM POPC:POPG (3:2 molar ratio; 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-[1′-rac-glycerol]) lipids (Avanti Polar Lipids, Alabaster, AL) for 1 hour at 4°C. Detergent was removed using BioBeads SM-2 (Bio-Rad, Hercules, CA) by incubating overnight at 4°C. Then, receptor-containing HDL particles were isolated using FLAG-M1 affinity chromatography and size exclusion chromatography.
DNA-Encoded Small-Molecule Library.
The DNA-encoded small-molecule libraries used for screening were created using a tagged-split-and-pool chemistry approach (Chemetics) at Nuevolution, as previously described (Kontijevskis, 2017).
Affinity Selection.
Figure 1A schematically illustrates the library selection process. More specifically, 30 μg biotinylated β2AR-HDL particles were immobilized on 25 μl NeutrAvidin beads (Thermo Fisher Scientific, Waltham, MA). The β2ARs bound to NeutrAvidin beads were then incubated with library molecules in 50 μl binding buffer (20 mM HEPES, pH 7.4, 100 mM NaCl) supplemented with 20 μM BI-167107 (BI) and 1 mg/ml sheared salmon sperm DNA (ssDNA; Ambion, Waltham, MA) for 45 minutes at room temperature, while intensely shaken. Prior to this incubation, 1 μl library molecules were allocated for later quantitative PCR (qPCR). Following this incubation, the beads were transferred to a micro-column connected to a vacuum apparatus and subsequently washed three times with 100 μl ice-cold binding buffer containing 10 μM BI. During each of the washing steps, excess liquid was removed via vacuum suction. To elute off the bound compounds, the beads were incubated twice with 50 μl water containing 1.5% Fos-choline (Avanti Polar Lipids) at 37°C for 15 minutes and subsequently at 72.5°C for 15 minutes. Following this incubation, the solution was separated from the beads by centrifugation at 1000 × g for 1 minute. After addition of 1 μl 10 mg/ml ssDNA, the combined supernatant was applied to a nucleotide removal kit (Qiagen, Hilden, Germany) to remove denatured protein and lipid molecules. The mixture of the bound compounds was then eluted with 50 μl water from the nucleotide removal column, and 1 μl purified material was allocated for later qPCR quantification. The remaining purified sample was then either used for the next round of selection with fresh β2AR-HDLs, or applied to NGS.
Fig. 1.

Hit compounds from DEL screening with the agonist-occupied β2AR in HDL particles. (A) Cartoon for DEL screening. Purified human β2ARs were reconstituted in HDL particles (β2AR Nanodiscs) and then occupied by BI-167107 (BI). DNA-encoded library molecules were mixed with the BI-occupied β2AR Nanodiscs immobilized on NeutrAvidin beads through biotin–avidin interaction of biotinylated membrane scaffolding protein ApoA1. Three rounds of iterative selection were performed with each library. (B) Structures of the Cmpd-6 and six other primary hits. These compounds have varied chemical scaffolds in a common region, designated as R1. The different chemical structures in the R1 region of each analog are illustrated. (C) Analysis of Cmpd-6 for its physical interaction with the agonist-bound, active β2AR by ITC. The thermogram (insert) and binding isotherm with the best titration curve fit are shown. One site model was used to fit the data. Data are representative of three independent experiments. The values summarizing binding affinity (KD), stoichiometry (N), enthalpy (ΔH), and entropy (ΔS) are shown in box below the graph.
Quantitative PCR.
Library DNA was quantified using qPCR at the end of each round of affinity selection. Briefly, the DNA samples were either directly amplified, or amplified after being diluted in solution containing 0.1% Tween 20, 20 mg/mL ssDNA using JumpStart Taq ReadyMix (Sigma-Aldrich), according to manufacturer’s guidelines. The samples from the libraries before selection were evaluated using at least four different concentrations on a log scale. This provides a standard to determine the DNA copy number of the samples from each iterative round of selection. The reaction was done with the primer set, including the universal forward primer (5′-CAAGTCACCAAGAATTCATG-3′) and a unique reverse primer for each library, and FAM/TAMRA probe 5′-CAGACGACCTAGGATCACC-3′ using a StepOnePlus (Applied Biosystems, Waltham, MA).
NGS and Analysis.
To increase the yield and then append the required sequencing adapters for emulsion PCR, the affinity-selected materials were amplified by two rounds of PCR. The first round of PCR was done with the same oligonucleotide primer set as used for qPCR. The second round of PCR was performed with oligonucleotide primers made from fusing the Ion Torrent adapter sequences to the universal forward primer, to which a sorting code was inserted to allow for sample pooling, and a unique reverse primer for each library. The unique sequence of the reverse primer provides precise sample tracking and a distinct identifier for each library. Following gel purification, the final PCR products were subjected to single-direction amplicon sequencing using the Ion Torrent platform (PrimBio, Exton, PA). Sequences having significant copy numbers (high signal-to-noise ratio), determined by analyzing the NGS output using a custom-built algorithm, were deconvoluted to their corresponding chemical structures from the database.
Isothermal Titration Calorimetry.
Isothermal titration calorimetry (ITC) experiments were performed with maltose neopentyl glycol (MNG; Anatrace)–solubilized β2AR on a MicroCal Auto-iTC200 system (Malvern, Malvern, UK), according to the previously reported method (Ahn et al., 2017). Dialysis of the purified β2AR was carried out against a dialysis buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 0.01% MNG, and 0.001% cholesteryl hemisuccinate). Titrations were performed at 25°C, in which 40 μl 200 μM Cmpd-6 in the aforementioned dialysis buffer was loaded into the syringe, followed by an initial injection of 0.2 μl, and then subsequent 2 μl injections (0.4-second duration, 150-second spacing, and 5-second filter period) into the 200 μl sample cell containing β2AR (at 30 μM final) prestimulated with isoproterenol (ISO; 2 mM final). During the experiment, the reference power was set to 7 μcal × s−1 and sample cell was stirred continuously at a speed of 1000 rpm. ITC raw data were baseline corrected, peak area integrated, and fitted by using a one-site nonlinear least-squares fit model using the MicroCal Origin software program, to provide affinity constant (KD), stoichiometry (N), and thermodynamic parameters such as enthalpy (ΔH) and entropy (ΔS).
Radioligand Binding.
Binding experiments were done as previously detailed (Ahn et al., 2017). In brief, competition radioligand binding was done using the radiolabeled antagonist [125I]-cyanopindolol (CYP; 2200 Ci/mmol; PerkinElmer, Waltham, MA) at a concentration of 60 pM. The β2AR-HDL particles were used at ∼0.7 ng per reaction. Reactions consisted of the β2AR-HDL particles, 125I-CYP, Cmpd-6 at varying concentrations, and a serial dilution of a competitor β2AR agonist, most often ISO. All of the components were diluted in an assay buffer [20 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% bovine serum albumin (BSA), and 1 mM ascorbic acid]. Each reaction was allowed to reach equilibrium by incubating for 90 minutes at room temperature. Assays were then terminated by rapid filtration onto GF/B glass-fiber filters (Brandel, Gaithersburg, MD) treated with 0.3% polyethyleneimine and washed with 8 ml cold binding buffer (20 mM HEPES, pH 7.4, 100 mM NaCl) using a harvester (Brandel). 125I-CYP bound to the β2AR-HDL particles was measured using either a Packard Cobra Quantum gamma counter (Packard; GMI, Ramsey, MN) or a WIZARD2 2-Detector Gamma Counter (PerkinElmer). Data were expressed as specific binding.
For [3H]-methoxyfenoterol (3H-FEN) (Toll et al., 2012) binding, membrane preparations from Sf9 cells expressing either β2AR or β2V2R were used. For in cell phosphorylation of β2V2R, GRK2-CAAX was coexpressed and, prior to harvest, cells were stimulated with the agonist ISO (10 µM) for 20 minutes. Membranes for β2AR and phosphorylated β2V2R were essentially prepared, as described earlier (Strachan et al., 2014; Ahn et al., 2017). For 3H-FEN binding with Gs or nanobody-80 (Nb80), β2AR membranes were incubated in the G protein assay buffer (50 mM Tris-HCl, pH 7.4, 2 mM EDTA, 12.5 mM MgCl2). For binding assays containing rat β-arrestin1, phosphorylated β2V2R membranes were incubated in the β-arrestin assay buffer (50 mM Tris-HCL, pH 7.4, 50 mM potassium acetate, 5 mM MgCl2). Both assay buffers were supplemented with 0.05% BSA and 0.018% L-ascorbic acid. 3H-FEN (12.6 Ci/mmol) was used at its K-hi (4.3 nM) in binding assays testing for PAM activity of Cmpd-6 (and its analogs) and Cmpd-43. In 3H-FEN saturation-binding assays, testing for the cooperativity of Cmpd-6 (20 µM) with Gs (100 nM), βarr1 (1 µM), or Nb80 (1 µM), 3H-FEN was used in the range of 0.39–50 nM to saturate the high-affinity agonist binding sites in the receptor. All binding reactions were incubated to equilibrium (90 minutes) at room temperature and then harvested onto polyethyleneimine-soaked GF/B filters, followed by four rapid washes of 2 ml with prechilled G protein wash buffer (50 mM Tris-HCl, pH 7.4, 2 mM EDTA, 12.5 mM MgCl2) or β-arrestin wash buffer (50 mM Tris-HCL, pH 7.4, 50 mM potassium acetate). Bound [3H] was extracted overnight with 5 ml scintillation fluid and quantified using a Packard Cobra Quantum gamma counter (Packard; GMI). Nonspecific radioligand binding was assessed in reactions that contained the antagonist propranolol (20 µM).
Measurements of cAMP Production.
cAMP production, an indirect marker of Gs protein activation, was measured using the GloSensor (Promega), a chemiluminescence-based cAMP biosensor, as previously described (Ahn et al., 2017). In brief, HEK-293 cells stably expressing the GloSensor luciferase enzyme alone, or together with the βARs, were plated in 96-well, white clear-bottom plates at a density of ∼80,000 cells/well. Cells were given at least a 24-hour incubation to recover cell surface receptor expression before the assay was started. Cells were then treated with the GloSensor reagent (Promega) and incubated at 27°C and ∼100% relative humidity for ∼1 hour. Cells were then treated with either a varying dose of Cmpd-6 or a vehicle control [dimethylsulfoxide (DMSO)] diluted in Hanks’ balanced solution (Sigma-Aldrich), supplemented with 20 mM HEPES, pH 7.4, 0.05% BSA, and 3-isobutyl-1-methylxanthine (Sigma-Aldrich) at a final concentration of 100 μM. For most of the cAMP assays, cells were then incubated further for 20 minutes, before a serial dilution of the β-agonist was added. For the assays with HEK-293 cells stably overexpressing β2AR, Cmpd-6 and the β-agonist serial dilution were added to the cells simultaneously. Upon stimulation of the cells with the β-agonist, changes in luminescence were read using a NOVOstar microplate reader (BMG Labtech, Cary, NC) at various time points ranging from 5 to 35 minutes.
Measurement of β-Arrestin Recruitment.
β-arrestin2 recruitment to the receptor was measured using the previously described Tango assay (Barnea et al., 2008). HEK-293T cells stably expressing the β2V2R tethered to the tetracycline transactivator transcription factor by a tobacco etch virus protease cleavage site, the human β-arrestin2 protein fused to the tobacco etch virus protease, and the tetracycline transactivator–driven luciferase reporter were used for this assay. Cells were plated on a 96-well, white clear-bottom plate at a density of ∼50,000 cells/well and were given at least a 24-hour incubation at 37°C, 5% CO2, and ∼100% relative humidity to recover surface receptor expression. Cells were treated with either a varying dose of Cmpd-6 or a vehicle control (DMSO) diluted in Hanks’ balanced solution (Sigma-Aldrich), supplemented with 20 mM HEPES, pH 7.4, and 0.05% BSA, and then incubated at 37°C, 5% CO2, and ∼100% relative humidity for ∼20 minutes. After the incubation, a serial dilution of the β-agonist was added, following which the cells were incubated for 6 hours at 37°C and ∼100% relative humidity. At the end of the incubation, the plate was cooled to room temperature. After adding the Bright-Glo reagent (Promega), chemiluminescence signals were read using a NOVOstar microplate reader (BMG Labtech) at 5–10 minutes.
Bimane Assay.
The minimal cysteine β2AR (Yao et al., 2009) was used in bimane fluorescence experiments. For bimane labeling at cysteine-265 of the β2AR, threefold molar excess of monobromobimane (Sigma-Aldrich) was used, as previously described (Yao et al., 2009), and HDL reconstitution of the β2AR-bimane was carried out, as described above. Bimane-labeled β2AR-HDLs at 250 nM were incubated in black, solid-bottom 96-well microplates with the vehicle (DMSO) or 10 μM ISO, either alone or together with 1 μM Nb80 or 20 μM Cmpd-6 for 30 minutes at room temperature. All of the components were diluted in buffer comprised of 20 mM HEPES, pH 7.4, and 100 mM NaCl. A CLARIOstar plate reader (BMG Labtech) was used to collect fluorescence emission spectra using the top-read mode with excitation at 370 nm (16-nm bandpass) and emission scanning from 400 to 600 nm (10 nm bandpass) in 1-nm increments.
Nanobody Enzyme-Linked Immunosorbent Assay.
As previously described, the 6X-His tagged nanobodies, Nb80 (Rasmussen et al., 2011) and Nb6B9 (Ring et al., 2013), were purified from periplasmic extracts of Escherichia coli WK6 cells. Nanobodies were affinity purified using Ni-NTA agarose beads (Qiagen). Purified nanobodies were then dialyzed overnight in 20 mM HEPES, pH 7.4, 100 mM NaCl, followed by size-exclusion chromatography. Nb80 (10 µg/ml) was passively adsorbed onto Maxisorp (NUNC, Roskilde, Denmark) 96-well plates (Thermo Fisher Scientific, St. Louis, MO) in nanobody buffer (20 mM HEPES, pH 7.4, 100 mM NaCl), and plates were incubated overnight at 4°C. Enzyme-linked immunosorbent assay was performed essentially as described before (Staus et al., 2014). Purified β2AR was preincubated with 0.2% DMSO or 10 µM final of either ICI-118551 or BI-167107 ligands, and Cmpd-6 (20 µM) or Nb6B9 (1 µM) for 30 minutes in assay buffer (20 mM HEPES, pH 7.4, 100 mM NaCl) containing 0.01% MNG (Anatrace), 0.001% cholesteryl hemisuccinate (Sigma-Aldrich), and 0.5% BSA. The preincubated reactions were then overlaid on Nb80-adsorbed 96-well plates for 90 minutes at room temperature. Following incubation, the unbound material was washed with assay buffer, and the captured β2AR was detected using a horseradish peroxidase–conjugated anti-Flag (M2) antibody (1:5000) diluted in assay buffer. Following antibody incubation (1 hour, room temperature), plates were washed with assay buffer, and signal was developed using 100 µl Ultra-TMB (Pierce, Rockford, IL). The developed signal was quenched with 100 µl acidified assay buffer, and the absorbance was measured at 450 nm.
Synthesis and Characterization of Cmpd-6 and Its Derivatives.
Complete details of chemical syntheses are described in Supplemental Material.
Results
Screening and Identification of Primary Hits Including Cmpd-6 and -43.
Using our recently developed approach for screening DELs against GPCRs (Ahn et al., 2017), in this study we screened ∼500 million unique DNA-encoded small molecules (Supplemental Table 1) to obtain PAMs at the β2AR. To increase the chance of obtaining PAMs, the orthosteric site of the receptor was occupied by a high-affinity β-agonist BI-167107, which shifted the β2AR population toward active conformations (Rasmussen et al., 2011; Manglik et al., 2015) (Fig. 1A). Furthermore, purified human β2ARs were reconstituted in detergent-free HDL particles (Fig. 1A). The HDL reconstitutions were performed using a biotinylated version of the membrane scaffolding protein ApoA1 (Whorton et al., 2007). In addition to providing the receptor with a native-like membrane environment, the biotinylated HDL particles provide an excellent immobilization scheme that avoids any physical perturbations to the receptor during the screening process. The β2ARs in biotinylated HDL particles can be efficiently captured on NeutrAvidin beads (Supplemental Fig. 1A) and have a comparable affinity for antagonist binding to that of β2ARs in membrane preparations (Supplemental Fig. 1B). Furthermore, by competitive radioligand-binding assays, we show that β2ARs in HDL particles can functionally couple to heterotrimeric Gs (Supplemental Fig. 1C). G protein coupling to the β2AR substantially increases the affinity of the competing agonist—ISO. This high-affinity coupling of Gs to the β2AR can be completely blocked by the addition of GTPγS—a nonhydrolyzable GTP analog.
Using the BI-167107–occupied β2AR in HDL particles, we iteratively screened four different DELs (Kontijevskis, 2017), each of which comprised over 100 million unique compounds (Supplemental Table 1), to isolate molecules that specifically bound to the active state of the receptor. The total number of molecules in each library was ∼0.5–1 × 1014. Three rounds of iterative selection (Fig. 1A) were performed with each library until the total number of target-bound molecules was decreased to ∼1 × 106, which was monitored by qPCR. Following amplification of preserved DNA barcodes by PCR, the samples were subjected to NGS to identify compounds that outlasted the entire selection procedure. Sequences having significant copy numbers (i.e., high signal-to-noise ratio) were deconvoluted to their corresponding chemical structures from the database. Through this analysis, we determined 50 compounds as primary candidates that possibly bind to the β2AR (Supplemental Table 1) and named them Cmpd-1 to Cmpd-50. These 50 candidate compounds were synthesized on a small scale without their DNA barcodes to evaluate their activity as PAMs in secondary screens.
PAMs are expected to potentiate the binding of orthosteric agonists to GPCRs and even plausibly the coupling of transducer proteins, e.g., G protein and β-arrestin, to receptors (Wootten et al., 2013; Christopoulos, 2014). Accordingly, these 50 potential hits were tested for their ability to increase the binding of the radiolabeled agonist, 3H-FEN (Toll et al., 2012), to the β2AR in membrane preparations, both in the absence and presence of transducers (Supplemental Fig. 1D). Through this secondary screen, we identified seven structurally related compounds, as shown in Fig. 1B, including Cmpd-6, which showed the strongest PAM activity among the compounds (Supplemental Fig. 1E). These compounds not only increased 3H-FEN binding to the β2AR alone, but also, to varied extents, potentiated the transducer-induced high-affinity 3H-FEN binding at the receptor. Interestingly, between these compounds, only subtle structural differences were observed, which were confined to one variable region designated as R1 (blue colored in Fig. 1B). Two of the seven compounds (Cmpd-6 and -43) were chosen for further characterization of their PAM activity and were synthesized on a large scale.
To assess direct molecular interaction between Cmpd-6 and the agonist-bound, active β2AR, ITC was employed. The values summarizing binding affinity (KD), stoichiometry (N), and thermodynamic parameters are shown in Fig. 1C. ITC values indicate that the process of interaction between Cmpd-6 and active β2AR is exothermic, therefore enthalpically favored with a stoichiometry of ∼1 and KD of 5.2 ± 0.5 μM.
PAM Activity of Cmpd-6 and -43 in β2AR-Mediated Downstream Signaling.
To evaluate the PAM activity of Cmpd-6 and -43 in β2AR-mediated downstream functions, we monitored their effects on both agonist-induced Gs protein–cAMP production (Binkowski et al., 2011; Rajagopal et al., 2011) and β-arrestin2 recruitment to the receptor (Rajagopal et al., 2011; Bassoni et al., 2012) using cellular assays. An issue encountered in these functional assays is the differential levels of the signal produced by virtue of the high amplification process downstream of Gs protein activation, compared with the stoichiometric recruitment of β-arrestin2 to the receptor (Rajagopal et al., 2011). To circumvent this problem and achieve similar levels of the signal in the two cellular assays, we used the endogenously expressed β2AR in cAMP production assays, whereas β-arrestin2 recruitment was measured using cells stably overexpressing the β2V2R. This chimeric receptor has the V2R tail recombinantly appended at the C terminus of the β2AR, retaining the pharmacological traits of the native β2AR, but displaying a more stable interaction with β-arrestin, which is an advantage for β-arrestin recruitment assays (Tohgo et al., 2003). Both Cmpd-6 and -43 increased the ability of the agonist ISO to activate G protein–mediated cAMP production through the β2AR in a dose-dependent way (Fig. 2, A and B). We observed that Cmpd-6 (Fig. 2A) and Cmpd-43 (Fig. 2B) increased the maximal response induced by ISO, as well as potentiating the EC50 value of ISO, which was apparent in its left-shifted dose–response curve. In this assay, Cmpd-6 shows stronger activity than Cmpd-43, which is consistent with the preliminary data showing the extent of dose-dependent increases in 3H-FEN binding to the β2AR induced by these compounds, shown in Supplemental Fig. 1E. We also obtained a comparable pattern of agonist-induced β-arrestin2 recruitment to the β2V2R with Cmpd-6 (Fig. 2C) and Cmpd-43 (Fig. 2D), respectively.
Fig. 2.

Activity assays for G protein activation and β-arrestin2 recruitment in the presence of Cmpd-6 and -43. Either Cmpd-6 (A and C) or Cmpd-43 (B and D) was pretreated in assay cells for 15–20 minutes at various concentrations, as indicated, and then cells were stimulated with ISO in a dose-dependent manner. (A and B) The amount of cAMP production by the endogenously expressed β2AR was measured at 15–20 minutes after stimulation with ISO. (C and D) The level of β-arrestin2 recruitment to the stably overexpressed β2V2R was measured at 6 hours after stimulation with ISO. Curve fits were generated using the software GraphPad Prism with data points obtained from four (B–D) or five (A) independent experiments done in duplicate. Each data point was normalized to the maximal level of the ISO-induced activity in the vehicle (0.32% DMSO) control, expressed as a percentage, and represents mean ± S.D. The shift of curves was expressed as fold changes in EC50 and Bmax values. Statistical analyses for these shifts in each of the directions were performed using one-way analysis of variance, repeated (related) measures with Tukey’s multiple comparison post-tests. P values shown on each graph were for the curve obtained when compound was pretreated at the highest concentration, compared with the control DMSO curve. Adjusted **P < 0.01; ***P < 0.001; ****P < 0.0001.
Increases in the ISO-induced maximal response by Cmpd-6 and -43 in both assays suggest that ISO may act as a partial agonist, which does not reach the maximum response possible in these systems (Langmead, 2011), allowing Cmpd-6 and -43 to further increase the maximal agonist-induced response. To verify this, we monitored cAMP production by overexpressed β2AR (a system that has much higher amplification; Supplemental Fig. 2) in the presence or absence of Cmpd-6. We observed that Cmpd-6 led to dose-dependent leftward shifts of the ISO dose-response EC50 values, with increases in the basal activity, but did not increase the ISO-stimulated maximal response. This shows that even a full agonist such as ISO can act as a partial agonist depending on the assay system used, which would not have been suspected without the cooperativity displayed by these new PAMs. Overall, these results strongly demonstrate that Cmpd-6 and -43 have PAM activity for β2AR-mediated downstream functions, and that Cmpd-6 has stronger PAM activity than Cmpd-43.
Cmpd-6 and -43 Potentiate the Binding Affinity of Agonists for the β2AR.
A hallmark of PAMs is that they allosterically stabilize the agonist-bound active conformation of the receptor (Langmead, 2011), as do transducer proteins, G protein and β-arrestin, as illustrated in the GPCR ternary complex model (De Lean et al., 1980). Because PAM-mediated stabilization of active GPCR conformations leads to potentiation of agonist-binding affinity for the receptor, we next tested whether Cmpd-6 and -43 increase the binding of an agonist to the β2AR. For this, we monitored the competition binding of the orthosteric agonist ISO against the radiolabeled antagonist 125I-cyanopindolol (CYP) to the β2AR-HDL in the presence or absence of Cmpd-6 (Fig. 3A) and Cmpd-43 (Fig. 3B). As expected, both compounds potentiated the binding of ISO to the β2AR in a dose-dependent way, as evidenced by the robust left shifts in ISO competition curves. Consistent with the results obtained in the cellular assays (Fig. 2), at their highest concentration tested (Fig. 3, A and B), Cmpd-6 potentiated the IC50 value of ISO close to 50-fold, which was substantially more than the ∼30-fold change elicited by Cmpd-43. Additionally, we obtained comparable shifts in the ISO dose–response curve induced by Cmpd-6 and -43 in radioligand competition binding done with membranes prepared from β2AR-overexpressing cells (Supplemental Fig. 3, A and B).
Fig. 3.

Positive allosteric activity of Cmpd-6 and -43 for orthosteric ligand binding to the β2AR. (A and B) Cmpd-6– and Cmpd-43–mediated dose-dependent left shifts of the ISO competition curve against 125I-CYP binding to the β2AR. Binding of 125I-CYP against ISO in a dose-dependent manner was measured in the absence (DMSO) or the presence of various concentrations of Cmpd-6 (A) or Cmpd-43 (B), as indicated. Curve fits were plotted by a one-site competition-binding log IC50 curve fit (GraphPad Prism) with data sets obtained from four independent experiments done in duplicate. Each data point was normalized to the percentage of the maximal 125I-CYP binding level obtained from the control (0.64% DMSO-treated) curve and represents mean ± S.D. Statistical analyses for the shift of IC50 values were performed using one-way analysis of variance, repeated (related) measures with Tukey’s multiple comparison post-tests. P values were for the curve obtained when compound was pretreated at the highest concentration, compared with the control DMSO curve. Adjusted ****P < 0.0001. (C) Cmpd-6 and -43 dose-dependent increases in 3H-FEN binding to the β2AR. Curve fits were generated using the software GraphPad Prism with data points obtained from six independent experiments. Each data point was normalized to the maximal level obtained in the presence of Cmpd-6 and represents mean ± S.D.
Additionally, results shown in Fig. 3C further confirm the PAM activity of Cmpd-6 and -43 for increasing the binding of an orthosteric agonist to the β2AR. Cmpd-6 and -43 dose dependently increased the binding of the radiolabeled agonist 3H-FEN to the β2AR expressed in cell membranes, consistent with what we observed in our preliminary experiments with these compounds (Supplemental Fig. 1E). Again, Cmpd-6 is more efficacious than Cmpd-43 in increasing 3H-FEN binding to the β2AR. Furthermore, the low micro-molar affinity (EC50) value of Cmpd-6 obtained in this assay (Fig. 3C) is comparable to its KD value measured for its direct interaction with the β2AR by ITC analyses (Fig. 1C). We also observed another feature of allosteric molecules, the ceiling effect, with these compounds in both binding experiments (Fig. 3). The increases in the binding of both agonists, ISO (Fig. 3, A and B) and FEN (Fig. 3C), were saturated with increasing concentrations of these allosteric compounds.
Cmpd-6 Stabilizes the Agonist-Induced Active Conformation of the β2AR.
We further demonstrated that Cmpd-6 stabilizes active conformational ensembles of the β2AR through a biophysical assay. Agonist-induced activation of the β2AR causes the outward movement of transmembrane helix 6 (TM6), which can be detected by labeling of cysteine-265 at the intracellular base of TM6 with monobromobimane, an environmentally sensitive fluorescent label. Following receptor activation, the outward movement of TM6 leads to a decrease in fluorescence intensity with a concomitant increase in the maximum wavelength for emission (Rasmussen et al., 2011). Cmpd-6 alone induced decreases in overall fluorescence intensity, but not increases in the maximum wavelength for emission from the bimane-labeled β2AR (Fig. 4). In contrast, ISO decreased fluorescence to a similar extent, but also increased the maximum wavelength. This suggests that the conformational ensemble of the β2AR when bound to Cmpd-6 alone is similar to, but distinct from, that induced by orthosteric agonists. Interestingly, Cmpd-6 further potentiated ISO-induced decreases in the fluorescence intensity and increases in the maximum wavelength from the bimane-labeled β2AR (Fig. 4). Importantly, Cmpd-6–mediated potentiation of ISO effects was similar in magnitude to that observed with an allosteric nanobody (Nb80) that mimics the G protein–stabilized active conformation of the agonist-bound β2AR (Rasmussen et al., 2011). These data clearly demonstrate that Cmpd-6 stabilizes active conformations of the agonist-bound β2AR, engaging the outward movement of TM6 to an extent comparable to that mediated by transducers like G protein.
Fig. 4.

Positive allosteric activity of Cmpd-6 in agonist-induced β2AR bimane signals. (A) The fluorescence emission spectrum of monobromobimane-labeled β2AR in HDL particles. Data shown are representative of three independent experiments. (B) Bar graph summarizes the analyses, in which normalized peak fluorescence values are expressed relative to DMSO control (0.2%). Values indicate mean ± S.D. Statistical analysis for the results depicted as the bar graph was performed using one-way analysis of variance, repeated (related) measures with Tukey’s multiple comparison post-tests. Adjusted **P < 0.01; ***P < 0.001; ns, not significant.
Functional Cooperativity of Cmpd-6 with Transducers at the β2AR.
Our data from the cellular assays (Fig. 2) strongly support the PAM activity of Cmpd-6 and suggest a functional cooperativity between the compound and the transducers Gs and β-arrestin. To confirm this cooperative property of Cmpd-6, we performed competition radioligand binding on membrane preparations expressing β2AR or its C-terminal fusions with the transducer, Gsα (Fig. 5, A and B) or β-arrestin1 (a.k.a arrestin2) (Fig. 5, C and D). Compared with β2AR alone, both transducer fusions revealed the expected high-affinity coupling to the receptor with a left shift in ISO dose–response curves. Importantly, addition of Cmpd-6 at β2AR fusions enhanced both Gsα- and β-arrestin1–mediated high-affinity coupling to the receptor and also resulted in a significant potentiation of ISO affinity compared with uncoupled receptor (Fig. 5, B and D). Additionally, we assessed the PAM activity of Cmpd-6 by measuring binding of the radiolabeled orthosteric agonist 3H-FEN aimed at saturating high-affinity sites on the β2AR (Fig. 5, E and F). Compared with no transducer controls, addition of Cmpd-6 or the exogenous transducers, heterotrimeric Gs (at β2AR membranes; Fig. 5E) and β-arrestin1 (at phosphorylated β2V2R membranes; Fig. 5F), robustly increased the high-affinity 3H-FEN binding to the receptor. Interestingly, addition of Cmpd-6 together with Gs or βarrestin1 further enhanced the maximal high-affinity 3H-FEN binding. Although there was noticeable cooperativity between Cmpd-6 and Gs, this potentiation in 3H-FEN binding was also prominent in the presence of the G protein mimic Nb80 (Rasmussen et al., 2011) (Supplemental Fig. 4A). Together with our findings from cellular assays, these binding studies clearly demonstrate a positive cooperativity between Cmpd-6 and transducers to modulate high-affinity state agonist binding to the β2AR.
Fig. 5.

Positive allosteric cooperativity of Cmpd-6 for transducer-induced activities at the β2AR. (A–D) Data showing positive cooperativity of Cmpd-6 (20 µM) assessed by 125I-CYP versus (ISO) competition binding at membrane preparations from HEK cells overexpressing β2AR or the transducer fusions β2AR-Gsα (A) and β2V2R-β-arrestin1 (βarr1) (B). Points on the curves represent normalized cpm values from three independent experiments, expressed as two-site curve fit with shared IC50Low (GraphPad Prism). Associated bar graphs show Cmpd-6–mediated fold changes in ISO affinity at β2AR-Gsα (B), and β2V2R-βarr1 (D) fusions, respectively, expressed as a ratio of IC50Low/IC50High. (E and F) 3H-FEN saturation-binding curves showing Cmpd-6–mediated potentiation 3H-FEN binding at Sf9 cell membranes expressing the β2AR (E) and in cells expressing the phosphorylated β2V2R (F). Points on the curves represent cpm values normalized to the maximal level mediated by Gs (E) or βarr1 (F), respectively. DMSO (0.2%) was included as vehicle control in respective experiments for conditions without Cmpd-6. Values indicate mean ± S.D. from at least three independent experiments. Statistical analyses for the results depicted as the bar graphs (B and D) as well as Bmax changes in (E and F) were performed using one-way analysis of variance, repeated (related) measures with Tukey’s multiple comparison post-tests. Adjusted *P < 0.05; **P < 0.01.
Of note, the data in Supplemental Fig. 4A also suggest that Cmpd-6 does not occlude transducer coupling to the β2AR and most likely binds to a potentially unique allosteric site in the receptor. Accordingly, to test whether Cmpd-6 physically competes for binding to the intracellular transducer-binding pocket, we performed an enzyme-linked immunosorbent assay to capture the β2AR with the G protein mimic Nb80 that recognizes agonist-bound active state of the receptor (Supplemental Fig. 4B). In the presence of the high-affinity agonist BI-167107, and compared with DMSO or the antagonist ICI-118551, there was a marked increase in receptor capture by Nb80. This receptor capture was significantly inhibited in the presence of saturating amounts of a competing nanobody Nb6B9 (Ring et al., 2013), which is an affinity-matured version of Nb80 and thus competes for a common binding epitope on the β2AR. Interestingly, and in contrast to Nb6B9, under these experimental conditions the addition of a saturating concentration of Cmpd-6 did not alter the capture of β2AR by Nb80. These data suggest that presence of Cmpd-6 does not interfere with transducer coupling to the β2AR, which further establishes positive cooperativity between transducers and the compound.
The PAM Activity of Cmpd-6 Is Specific for the β2AR.
The specificity of Cmpd-6 for the β2AR was first evaluated through in vitro competition radioligand (125I-CYP) binding at the β1AR, the most closely related subtype of the adrenoceptor. In this assay, Cmpd-6 induces a minimal left shift of the ISO competition curve for binding to the β1AR (Supplemental Fig. 5A) unlike the robust ISO curve shift by Cmpd-6 observed with the β2AR (Fig. 3A). This shows that Cmpd-6 specifically induces the high-affinity binding of the orthosteric agonist ISO to the β2AR, but not to the β1AR. Furthermore, we observed only marginal changes promoted by Cmpd-6 in the ISO dose–response pattern of β1AR-mediated cAMP production (Supplemental Fig. 5B), which is markedly different from that of the β2AR-mediated response (Supplemental Fig. 2). We also detected minimal allosteric effects of Cmpd-6 on cAMP production mediated by other receptors. These are the transiently overexpressed V2R (Supplemental Fig. 5C), as well as the prostaglandin E2 (Supplemental Fig. 5D) and the vasoactive intestinal peptide (Supplemental Fig. 5E) receptors, both endogenously expressed in the assay cells. These findings clearly demonstrate that the PAM activity of Cmpd-6 is specific for the β2AR relative to the β1AR and other receptors tested in this work.
PAM Activity of Cmpd-6 When the β2AR Is Stimulated with a Range of Different Agonists.
Some allosteric modulators show differential activity depending on the orthosteric agonist stimulating the receptor, a phenomenon known as probe dependence (Wootten et al., 2013; Christopoulos, 2014). We examined whether Cmpd-6 displays such differential activity when the orthosteric site of the β2AR is occupied with a range of agonists, namely epinephrine (EPI) and fenoterol (FEN), which are very strong partial agonists compared with ISO, and clenbuterol (CLEN), which is a weak partial agonist (Rajagopal et al., 2011). We first evaluated the extent of the dose–response curve (IC50 value) shift induced by Cmpd-6 in radioligand (125I-CYP) competition binding to the β2AR (Supplemental Fig. 6, A–D). This allowed us to test the allosteric activity of Cmpd-6 solely for binding of an agonist in the absence of transducer coupling to the receptor. We observed that the extent of the curve shift in the presence of Cmpd-6 in this assay essentially followed the efficacy of the tested agonists to induce downstream signaling.
We next compared the PAM activity of Cmpd-6 for downstream signaling of the β2AR when stimulated with these four agonists using cell-based functional assays, monitoring cAMP accumulation (Supplemental Fig. 6, E–H) and β-arrestin2 recruitment to the receptor (Supplemental Fig. 6, I–L). In general, full and strong partial agonists show greater affinity (EC50 value) shifts by Cmpd-6 compared with that observed with the weak partial agonist CLEN. However, CLEN displayed a substantially greater Cmpd-6–mediated increase in maximal response than did the full agonists. Interestingly, no direct relationship between the extent of the EC50 shift by Cmpd-6 and the efficacy of ISO, EPI, and FEN was seen. In functional assays, Cmpd-6 induced a noticeably greater shift with EPI (Supplemental Fig. 6, F and J) than with ISO (Supplemental Fig. 6, E and I) and FEN (Supplemental Fig. 6, G and K), whereas the fold increases by Cmpd-6 in the maximal response induced by these agonists were comparable. Thus, we did not observe any unique probe dependence of Cmpd-6 with this small panel of agonists.
Structure–Activity Relationships of Cmpd-6 Analogs.
To determine structure–activity relationships (SAR) around Cmpd-6, a rational design approach and synthesis of a series of Cmpd-6 analogs (Table 1) were carried out. We evaluated the allosteric effect of these derivatives on orthosteric agonist 3H-FEN binding to the β2AR in the absence and presence of transducers, either trimeric Gs protein or β-arrestin1. We also tested them for their allosteric activity in ISO-stimulated β2AR downstream signaling, that is G protein–mediated cAMP production and β-arrestin2 recruitment to the activated receptor. For convenience of presenting the SAR analyses, Cmpd-6 and its analogs have been broken down into a common core scaffold, 5-hydrosulfonyl-2-mercaptobenzaldehyde (black), and three diverse substituents (R): N-methylpropan-2-amine (R1, blue), 4-methoxy-benzene (R2, red), and (R)-3-amino-4-(4-(tert-butyl)phenyl) butanamide (R3, purple). The R1 region is common in this subset of analogs although varying in the initial analogs from the screening, as shown in Fig. 1B. The main focus of this initial analog series was to probe the importance of modifications at the R2 and R3 positions of Cmpd-6; these modifications are shown in Table 1. When the whole R3 portion of Cmpd-6 was replaced with a much smaller piperidine moiety as in analog A3, the PAM activity of Cmpd-6 was abolished, indicating that the bulkiness imparted by the tert-butyl at this region of the molecule is critical for the PAM activity of the compound. Further exploration of the relationship between activity and structure at R3, achieved by replacing the 4-tert-butyl on the benzene ring with 4-OH, and the amide tail with carboxyl as in analog A6, as well as with 4-H as in analog A7, revealed the importance of the large lipophilic ring system for the activity of Cmpd-6. This was more evident with the highly polar analog A6, which was considerably less active than the original compound, whereas the relatively small, but lipophilic, analog A7 showed moderate activity. SAR around R2 also showed significant decreases in the PAM activity of Cmpd-6, when its electron-donating group 4-OCH3 was replaced with an electron-withdrawing 4-OCF3 group at its para position to yield analog A4. Interestingly, when the para-OCH3 group in Cmpd-6 was replaced with a meta-OCH3 substitution, as in A5, there was a moderate decrease in the PAM activity of Cmpd-6. The results from these two analogs suggest that there may be a polar interaction, like H-bonding type, located in the area surrounding this 4-position of R2, which interacts with the putative binding site on the β2AR.
TABLE 1.
Structure–activity relationships of Cmpd-6 analogs
Different chemical scaffolds in the R2 and R3 regions between Cmpd-6 and its analogs are illustrated. Changes in the Vmax value by Cmpd-6 or each analog at 32 μM are expressed as percentages of the maximal level of the ISO-induced activity in the vehicle (DMSO) control in each assay. Changes in the EC50 value are expressed as fold shifts compared with the control value obtained in the vehicle (DMSO)-treated curve in each assay. Every value represents mean ± S.D. obtained from four independent experiments done in duplicate. Statistical analyses were performed using one-way analysis of variance with Dunnett’s multiple comparison post-tests compared with the control Cmpd-6–treated value in each assay. Adjusted *P < 0.05; **P < 0.01; ***P < 0.001.
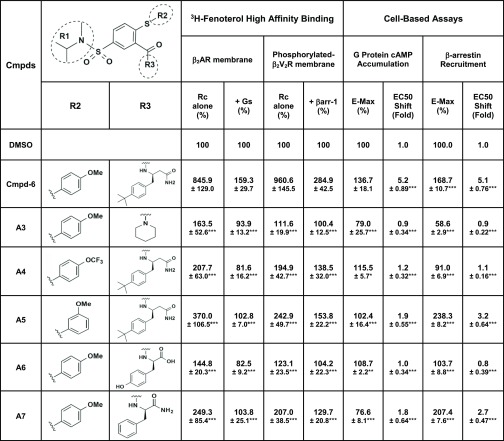 |
Rc, receptor.
Discussion
In the present study, DEL screening with the agonist-occupied β2AR in HDL particles has yielded the first small-molecule PAMs for the β2AR. We had isolated a small-molecule β2AR NAM, Cmpd-15, from a previous screening (Ahn et al., 2017), but until now no PAM small molecule for this receptor had been described. Among these isolated PAM molecules, the most efficacious one, Cmpd-6, was characterized in detail through multiple assays. Cmpd-6 has a low micro-molar binding affinity for the agonist-occupied β2AR and displays potent PAM activity for this receptor. Cmpd-6 positively cooperates with β2AR agonists to enhance downstream signaling responses such as cAMP production and β-arrestin recruitment to the receptor. It not only potentiates the affinity (EC50 values) of agonists for these responses, but also increases the agonist-induced maximal level of the responses. This is in agreement with pharmacological studies wherein Cmpd-6 shows positive cooperativity with transducers in mediating the agonist-bound high-affinity state of the β2AR. Furthermore, Cmpd-6 stabilizes the agonist-induced active conformation of the β2AR, leading to potentiation of the agonist-binding affinity for the receptor, which is a hallmark of PAMs (Langmead, 2011; Wootten et al., 2013; Christopoulos, 2014). Although Cmpd-6 does not show unique probe dependence or obvious biased activity toward either G protein or β-arrestin signaling, it clearly displays a ceiling effect for its activity, another pharmacological characteristic of PAMs (Wootten et al., 2013; Christopoulos, 2014). As expected for a PAM, Cmpd-6 also shows strong specificity for the β2AR relative to the β1AR and other receptors tested in this study. It only minimally potentiates the binding affinity of agonists to the β1AR, as well as β agonist–induced downstream functional activity.
Through a set of Cmpd-6 derivatives, we were able to discern the SAR patterns of the positive allosteric modulation of β2AR agonist activities and where the potential pharmacophore regions of the compound might be. As is apparent from our SAR studies, the N-isopropyl-N-methyl group is the preferred substituent at the R1 position attached to the common core chemical scaffold, 5-hydrosulfonyl-2-mercaptobenzaldehyde. At this region, bulkier groups such as N-cyclopentyl and N-phenyl result in increasingly poor PAM activity. At the R2 position, an electron-donating methoxy group, in the para position on the phenyl ring, is favored. This suggests that there may be a polar interaction involving the methoxy functional group at the R2 position that interacts with the amino acid residues of the β2AR site. In the case of the R3 position, our analysis indicates that bulky and hydrophobic groups, such as tert-butyl benzene chemical scaffolds, are favored. This finding therefore suggests that this region of the molecule may occupy a hydrophobic pocket, deep within a putative β2AR allosteric binding site to establish contacts with core hydrophobic residues. Solution of an X-ray crystallographic structure of the β2AR in complex with Cmpd-6 will provide further insights into the binding modes of the compound.
Because allosteric ligands are able to freeze or lock receptors into specific conformations by virtue of their cooperative interactions with orthosteric ligands, they can facilitate the study of receptors by biophysical techniques. Recently, atomic-level structural features of several GPCRs occupied by their allosteric ligands have been elucidated through X-ray crystallographic analyses (Kruse et al., 2013; Zhang et al., 2015; Jazayeri et al., 2016; Oswald et al., 2016; Zheng et al., 2016; Liu et al., 2017; Wacker et al., 2017). Such biophysical studies have revealed mechanisms by which allosteric ligands modulate the binding and action of orthosteric ligands. To date, most of the solved GPCR crystal structures together with allosteric ligands have been achieved with allosteric antagonists (or NAMs). An X-ray crystallographic study of the M2 muscarinic acetylcholine receptor (mAChR) occupied by its high-affinity orthosteric agonist and PAM, in a complex together with a transducer mimic nanobody, is the only case previously reported with a PAM (Kruse et al., 2013). Therefore, it will be of great interest to obtain atomic-level information with the Cmpd-6–occupied β2AR to determine how the compound positively cooperates with orthosteric agonists. Such studies will expand our understanding on the mechanistic details, by which PAMs exert their effects on GPCRs.
Cmpd-6 and its analogs are the first small-molecule PAMs for the β2AR. To date, several PAM molecules have been isolated for some other GPCRs (Christopoulos, 2014; Gentry et al., 2015). These include, for example, small molecules for different subtypes of mAChRs, namely benzyl quinolone carboxylic acid for the M1 mAChR (Canals et al., 2012) and LY2033298 for the M2 and M4 mAChRs (Chan et al., 2008; Kruse et al., 2013); 4-(3-benzyloxyphenyl)-2-ethylsulfinyl-6-(trifluoromethyl)pyrimidine for the glucagon-like peptide-1 receptor (Wootten et al., 2012); and VCP171 [2-amino-4-(3-(trifluoromethyl)phenyl)(thiophen-3-yl)(phenyl)methanone] for the adenosine A1 receptor (Aurelio et al., 2009). Among others, cinacalcet, which is a PAM for the calcium-sensing receptor, is currently used as a therapeutic drug for hyperparathyroidism (Lindberg et al., 2005). Likewise, Cmpd-6 or its analogs may have potential to serve as lead molecules for the development of new therapeutic drugs for β2AR-related diseases. Cmpd-6 has strong specificity for the β2AR (found predominantly in smooth muscle) over the most closely related, cardiac-specific subtype β1AR. Its activity also has a clear ceiling level, which can reduce risks from target-based overdoses. As an allosteric modulator, when used in a therapeutic context, Cmpd-6 will only exert its modulating activity when an endogenous agonist of the β2AR, e.g., epinephrine, is available. Altogether, PAMs such as Cmpd-6, which can fine-tune the activity of the β2AR, hold great potential for the development of better therapeutic treatments for diseases like asthma, for which the clinical use of current βAR agonists is limited by adverse side effects (National Asthma Education and Prevention Program, 2007).
The present study yielding PAMs, together with our previous work isolating a NAM (Ahn et al., 2017), strongly demonstrates that our current DEL screening approach with purified GPCRs can be used to accomplish target conformation-specific selection through in vitro manipulation of the receptors. Accordingly, we successfully isolated a NAM using the unoccupied β2AR in an inactive conformation, but obtained PAMs using the high-affinity agonist-occupied receptor in active conformations. In the future, complexes of the receptors with transducers G protein and β-arrestin, which are also allosteric molecules, could be used to isolate allosteric molecules that might have even more unique biased functional profiles.
In conclusion, in this study we introduce the discovery of the first small-molecule PAMs for the β2AR through an in vitro affinity-based iterative selection of highly diverse DELs against the agonist-occupied receptor in HDL particles. Characterization of the strongest PAM among these molecules reveals its positive cooperativity with orthosteric agonists in a wide range of receptor functions and its high selectivity for the β2AR. A number of pharmacological features of this compound suggest potential advantages of such a PAM over orthosteric agonists as a therapeutic drug. Finally, our current findings, together with our previous isolation of the first β2AR NAM (Ahn et al., 2017), establish a proof-of-concept strategy to isolate allosteric molecules with tailored functional profiles.
Acknowledgments
We are grateful to Xinrong Jiang (Duke University), Darrell Capel (Duke University), David Hjort Pii (Nuevolution), and Cristina Delgado (Nuevolution) for technical assistance, and Quivetta Lennon and Joanne Bisson for secretarial assistance. We thank Dr. Irving Wainer (Laboratory of Clinical Investigation, National Institute on Aging Intramural Research Program, Baltimore, MD) and Dr. Andrew Kruse (Harvard Medical School, Boston, MA) for providing [3H](R,R′)-4-methoxyfenoterol and Nb6B9 plasmid, respectively. We are also grateful to Dr. Ryan Strachan and Dr. Justin English for providing the β2AR-Gsα fusion construct, which they generated in Dr. Bryan Roth’s laboratory at University of North Carolina (Chapel Hill, NC).
Abbreviations
- 3H-FEN
[3H](R,R′)-4-methoxyfenoterol
- β2AR
β2-adrenoceptor
- BSA
bovine serum albumin
- CLEN
clenbuterol
- Cmpd
compound
- CYP
cyanopindolol
- DEL
DNA-encoded small-molecule library
- DMSO
dimethylsulfoxide
- EPI
epinephrine
- FEN
fenoterol
- GPCR
G protein–coupled receptor
- HDL
high-density lipoprotein
- Nb80
nanobody-80
- ISO
isoproterenol
- ITC
isothermal titration caloriometry
- mAChR
muscarinic acetylcholine receptor
- MNG
maltose neopentyl glycol
- NAM
negative allosteric modulator
- NGS
next-generation sequencing
- PAM
positive allosteric modulator
- PCR
polymerase chain reaction
- qPCR
quantitative PCR
- SAR
structure–activity relationship
- ssDNA
sheared salmon sperm DNA
- ΤΜ6
transmembrane helix 6
- V2R
vasopressin 2 receptor
Authorship Contributions
Participated in research design: Ahn, Pani, Kahsai, Olsen, Wingler, Franch, Chen, Lefkowitz.
Conducted experiments: Ahn, Pani, Kahsai, Wingler, Rambarat, Simhal, Xu, Sun, Shim.
Contributed new reagents or analytic tools: Pani, Kahsai, Olsen, Husemoen, Vestergaard, Jin, Zhao, Staus, Huang, Franch, Chen.
Performed data analysis: Ahn, Pani, Kahsai, Olsen, Husemoen, Wingler, Rambarat, Simhal, Xu, Sun, Shim, Franch, Lefkowitz.
Wrote or contributed to the writing of the manuscript: Ahn, Pani, Kahsai, Rambarat, Simhal, Franch, Chen, Lefkowitz.
Footnotes
This work was supported, in part, by National Institutes of Health National Heart, Lung, and Blood Institute [Grant HL16037 (to R.J.L.) and Grants T32HL007101 and HL16037-45S1 (to A.W.K.)]; National Science Foundation of China [Grant 21272029 (to X.C.)]; Priority Academic Program Development of Jiangsu Higher Education Institution (to X.C.); and Medical Research Fellowship from Howard Hughes Medical Institute (to P.K.R.). R.J.L. is an Investigator with the Howard Hughes Medical Institute.
The authors declare no conflict of interest.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Ahn S, Kahsai AW, Pani B, Wang QT, Zhao S, Wall AL, Strachan RT, Staus DP, Wingler LM, Sun LD, et al. (2017) Allosteric “beta-blocker” isolated from a DNA-encoded small molecule library. Proc Natl Acad Sci USA 114:1708–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurelio L, Valant C, Flynn BL, Sexton PM, Christopoulos A, Scammells PJ. (2009) Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J Med Chem 52:4543–4547. [DOI] [PubMed] [Google Scholar]
- Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, Lee KJ. (2008) The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci USA 105:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassoni DL, Raab WJ, Achacoso PL, Loh CY, Wehrman TS. (2012) Measurements of β-arrestin recruitment to activated seven transmembrane receptors using enzyme complementation. Methods Mol Biol 897:181–203. [DOI] [PubMed] [Google Scholar]
- Binkowski BF, Fan F, Wood KV. (2011) Luminescent biosensors for real-time monitoring of intracellular cAMP. Methods Mol Biol 756:263–271. [DOI] [PubMed] [Google Scholar]
- Canals M, Lane JR, Wen A, Scammells PJ, Sexton PM, Christopoulos A. (2012) A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J Biol Chem 287:650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC, Christopoulos A, Lazareno S, Birdsall NJ, Bymaster FP, et al. (2008) Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci USA 105:10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A. (2014) Advances in G protein-coupled receptor allostery: from function to structure. Mol Pharmacol 86:463–478. [DOI] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. (1980) A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem 255:7108–7117. [PubMed] [Google Scholar]
- Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, et al. (2005) Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother 49:4721–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini RM, Randolph C. (2016) Chemical space of DNA-encoded libraries. J Med Chem 59:6629–6644. [DOI] [PubMed] [Google Scholar]
- Gentry PR, Sexton PM, Christopoulos A. (2015) Novel allosteric modulators of G protein-coupled receptors. J Biol Chem 290:19478–19488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow RA, Jr, Dumelin CE, Keefe AD. (2017) DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat Rev Drug Discov 16:131–147. [DOI] [PubMed] [Google Scholar]
- Jazayeri A, Doré AS, Lamb D, Krishnamurthy H, Southall SM, Baig AH, Bortolato A, Koglin M, Robertson NJ, Errey JC, et al. (2016) Extra-helical binding site of a glucagon receptor antagonist. Nature 533:274–277. [DOI] [PubMed] [Google Scholar]
- Kahsai AW, Wisler JW, Lee J, Ahn S, Cahill Iii TJ, Dennison SM, Staus DP, Thomsen AR, Anasti KM, Pani B, et al. (2016) Conformationally selective RNA aptamers allosterically modulate the beta2-adrenoceptor. Nat Chem Biol 12:709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK. (1995) Amino and carboxyl terminal modifications to facilitate the production and purification of a G protein-coupled receptor. Anal Biochem 231:269–271. [DOI] [PubMed] [Google Scholar]
- Kontijevskis A. (2017) Mapping of drug-like chemical universe with reduced complexity molecular frameworks. J Chem Inf Model 57:680–699. [DOI] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hübner H, Pardon E, Valant C, Sexton PM, et al. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504:101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ. (2011) Determining allosteric modulator mechanism of action: integration of radioligand binding and functional assay data. Methods Mol Biol 746:195–209. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. (2007) Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) 190:9–19. [DOI] [PubMed] [Google Scholar]
- Lindberg JS, Culleton B, Wong G, Borah MF, Clark RV, Shapiro WB, Roger SD, Husserl FE, Klassen PS, Guo MD, et al. (2005) Cinacalcet HCl, an oral calcimimetic agent for the treatment of secondary hyperparathyroidism in hemodialysis and peritoneal dialysis: a randomized, double-blind, multicenter study. J Am Soc Nephrol 16:800–807. [DOI] [PubMed] [Google Scholar]
- Liu X, Ahn S, Kahsai AW, Meng KC, Latorraca NR, Pani B, Venkatakrishnan AJ, Masoudi A, Weis WI, Dror RO, et al. (2017) Mechanism of intracellular allosteric β2AR antagonist revealed by X-ray crystal structure. Nature 548:480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, et al. (2015) Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Asthma Education and Prevention Program (2007) Expert Panel Report 3 (EPR-3): guidelines for the diagnosis and management of asthma-summary report 2007. J Allergy Clin Immunol 120:S94–S138. [DOI] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, et al. (2011) Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal 4:ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C, Rappas M, Kean J, Doré AS, Errey JC, Bennett K, Deflorian F, Christopher JA, Jazayeri A, Mason JS, et al. (2016) Intracellular allosteric antagonism of the CCR9 receptor. Nature 540:462–465. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, Dewire SM, Violin JD, Lefkowitz RJ. (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol 80:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ. (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov 9:373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, et al. (2011) Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 469:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK. (2013) Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502:575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. (2006) Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem 281:1261–1273. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, et al. (2013) Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Strachan RT, Manglik A, Pani B, Kahsai AW, Kim TH, Wingler LM, Ahn S, Chatterjee A, Masoudi A, et al. (2016) Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535:448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Wingler LM, Strachan RT, Rasmussen SG, Pardon E, Ahn S, Steyaert J, Kobilka BK, Lefkowitz RJ. (2014) Regulation of β2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol Pharmacol 85:472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan RT, Sun JP, Rominger DH, Violin JD, Ahn S, Rojas Bie Thomsen A, Zhu X, Kleist A, Costa T, Lefkowitz RJ. (2014) Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J Biol Chem 289:14211–14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. (2003) The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem 278:6258–6267. [DOI] [PubMed] [Google Scholar]
- Toll L, Pajak K, Plazinska A, Jozwiak K, Jimenez L, Kozocas JA, Tanga MJ, Bupp JE, Wainer IW. (2012) Thermodynamics and docking of agonists to the β(2)-adrenoceptor determined using [(3)H](R,R′)-4-methoxyfenoterol as the marker ligand. Mol Pharmacol 81:846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Stevens RC, Roth BL. (2017) How ligands illuminate GPCR molecular pharmacology. Cell 170:414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JB, Ahn S, Kahsai AW, Liu R, Ren J, Hu K, Sun XQ, Chen X. (2013) Synthesis of beta(2)-AR agonist BI-167107. Chin J Org Chem 33:634–639. [Google Scholar]
- Whalen EJ, Rajagopal S, Lefkowitz RJ. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med 17:126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA 104:7682–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, Sexton PM. (2013) Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov 12:630–644. [DOI] [PubMed] [Google Scholar]
- Wootten D, Savage EE, Valant C, May LT, Sloop KW, Ficorilli J, Showalter AD, Willard FS, Christopoulos A, Sexton PM. (2012) Allosteric modulation of endogenous metabolites as an avenue for drug discovery. Mol Pharmacol 82:281–290. [DOI] [PubMed] [Google Scholar]
- Yao XJ, Vélez Ruiz G, Whorton MR, Rasmussen SG, DeVree BT, Deupi X, Sunahara RK, Kobilka B. (2009) The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc Natl Acad Sci USA 106:9501–9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, et al. (2015) Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 520:317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Qin L, Zacarías NV, de Vries H, Han GW, Gustavsson M, Dabros M, Zhao C, Cherney RJ, Carter P, et al. (2016) Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540:458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]