

Abstract
Motivation
Enrichment-based technologies can provide measurements of DNA methylation at tens of millions of CpGs for thousands of samples. Existing tools for methylome-wide association studies cannot analyze datasets of this size and lack important features like principal component analysis, combined analysis with SNP data and outcome predictions that are based on all informative methylation sites.
Results
We present a Bioconductor R package called RaMWAS with a full set of tools for large-scale methylome-wide association studies. It is free, cross-platform, open source, memory efficient and fast.
Availability and implementation
Release version and vignettes with small case study at bioconductor.org/packages/ramwas Development version at github.com/andreyshabalin/ramwas.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Methylome-wide association studies (MWAS) can make unique contributions to our understanding of disease etiology and identify clinical biomarkers. MWAS is commonly performed using arrays that measure only a small fraction (e.g. 2–3%) of all 28 million common CpGs in the human genome. The obvious risk of such sparse methods is the potential to miss numerous association signals. Enrichment methods can provide much better coverage. For instance, we have shown that after protocol optimization, methyl-CG binding domain sequencing (MBD-seq) assays 94% of all CpGs at a cost comparable to that of arrays (Aberg et al., 2017; Chan et al., 2017). However, existing software packages are not able to process large-scale enrichment data, and often also lack important features, such as fragment size estimation, principal component analysis of the full dataset, combined analysis of methylation and SNP data, or outcome predictions based on multiple methylation sites. To address these limitations, we developed RaMWAS, an R package available through Bioconductor that is free, cross-platform, open source and allows users to take advantage of the R environment to customize analyses. Although RaMWAS is primarily designed for the analysis of methylation enrichment data, it can be used with other platforms (e.g. arrays) or data types (e.g. genotypes).
2 Materials and methods
2.1 Data storage, memory economy and parallelization
It is common for R programs to store data in computer memory. This, however, is not feasible for MWAS with enrichment data (e.g. 20 million CpGs across 1000 samples would already occupy 160 GB, which far exceeds the capacity of most computers). RaMWAS solves this by using a specially developed system of file-backed data processing that avoids loading all data into memory, and allows for fast non-sequential access to large matrices (see R package ‘filematrix’). We have made RaMWAS fast by employing efficient algorithms and by parallelizing most tasks across multiple CPU cores. Parallelization is facilitated by the use of file-backed data processing as each job gets the data directly from the file matrices. Next, we present RaMWAS components in the order of the pipeline.
2.2 Reading data and quality control (QC)
RaMWAS input data are BAM files with aligned reads. Once processed, the large BAMs are no longer needed as all necessary information is saved in compact RaMWAS format using only a fraction of the disk space (typically 1–2%). After exclusion of duplicate reads and reads with low alignment scores, RaMWAS calculates QC summary statistics. For example, it calculates the fraction of reads aligned to the X and Y chromosomes, which can be used to check the sex against recorded in the phenotype data (Fig. 1a). Another QC metric is the CpG density at which CpG scores peak (Fig. 1b). As several aspects of the laboratory protocol affect this peak location, it can be used as a laboratory technical covariate in downstream analyses. Low quality samples are usually detected and excluded based on such QC metrics as the number of aligned reads, average alignment score, the number of duplicate reads and the number of reads aligned away from any CpGs. The full list of indices is available in the BAM QC vignette at the Bioconductor website.
Fig. 1.
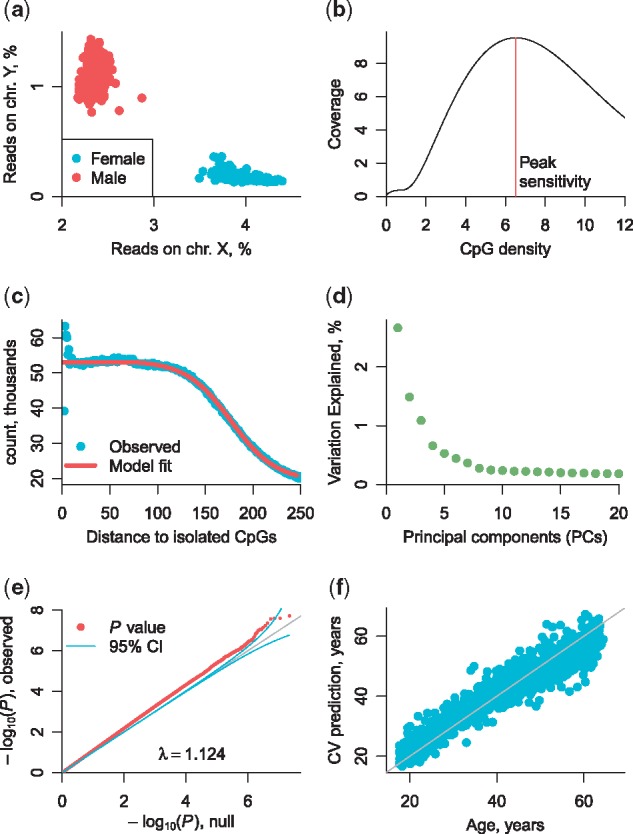
RaMWAS features. (a)––sex check via read count on chr. X and Y, (b)––enrichment diagnostic via average score by CpG density plot, (c)—fragment size distribution estimation, (d)—principal component analysis, (e)––QQ-plot for major depression disorder phenotype and (f)––methlylation risk score for age. The correlation between age and its MRS is 0.95. Details of the study are provided in the Supplementary Material
2.3 CpG score matrix
A natural way to quantify methylation with enrichment methods is to estimate the number of fragments covering a CpG. For enrichment approaches, single-end reads are most cost-effective (Chan et al., 2017). As fragment sizes are not observed with single-end libraries, RaMWAS estimates the fragment size distribution from reads around isolated CpGs (Fig. 1c) prior to calculating CpG scores using the approach proposed and validated by van den Oord et al. (2013). RaMWAS filters out CpGs with low average score across samples as they are non-methylated and unlikely to produce significant associations. The CpG scores are scaled to the same sample average to correct for varying number of total reads across samples.
2.4 Principal component analysis
RaMWAS can perform principal components analysis (PCA) on the whole matrix of CpG scores. The PCA can account for measured covariates to capture only the major sources of remaining variation. A scree plot (Fig. 1d) is used to select the number of PCs to include in the MWAS. The PCA can also be used for QC, as large individual PC scores can indicate (multidimensional) outlying samples.
2.5 MWAS
Association tests are performed using linear regression while accounting for selected covariates and top PCs. RaMWAS generates QQ-plots with confidence intervals and inflation factor lambda (Fig. 1e). It also reports the test statistics, p-values and q-values for all CpGs. Top findings can be annotated with genomic tracks from the online database BioMart.
2.6 Methylation risk scores (MRS)
For the purpose of making predictions from the methylation data, it is more convenient and powerful to combine information from multiple CpGs into a single risk score. RaMWAS builds such predictor by applying elastic net to the top CpGs from MWAS, as Horvath (2013) did for predicting biological age. To avoid overfitting and correctly estimate the predictive power, we use k-fold cross-validation. Specifically, for each training set, RaMWAS performs MWAS, selects top sites, trains the elastic net and makes predictions for the test samples. The set of predictions is recorded as the MRS (Fig. 1f).
2.7 CpG-SNP analysis
Point mutated CpGs, called CpG-SNPs, are particularly interesting sites because in addition to the sequence variation they may show differences in methylation. RaMWAS can perform CpG-SNP analyses if SNP data from the same subjects/samples is also available. These tests are performed using a regression model that assesses whether the case-control differences are proportional to the number of CpGs (van den Oord et al., 2015).
2.8 Performance, memory use and comparison with existing software
We tested RaMWAS on a dataset with 1132 samples and over 21 million CpGs that passed QC. Both the PCA and MWAS ran in under 30 min on a regular desktop computer with 16 GB of RAM.
We also compared RaMWAS with the main alternative software QSEA (Lienhard et al., 2016) which, although not exclusively designed for large-scale MWAS, is the main alternative. The full details of the comparison are provided in the Supplementary Material. QSEA required vast amounts of RAM; to run it we had to limit the data from 1132 to 200 samples and further needed to restrict the analysis to a few select chromosomes. Clearly, this is a serious limitation as MWAS sample sizes are typically larger, and for the best analysis it is critical to analyze all chromosomes simultaneously. Both RaMWAS and QSEA detected the well-known smoking AHRR association [Andersen et al. (2015), Supplementary Fig. S1]. However, the QSEA P-values had a highly deflated distribution (Supplementary Fig. S3–4, inflation factor λ = 0.237). RaMWAS showed excellent control of type I error (Supplementary Fig. S5, λ = 1.004). QSEA supports estimation of absolute methylation levels at single-base resolution from enrichment data. However, results obtained with these transformed scores did not show any advantage in terms of detecting associations and still suffered severe deflation (Supplementary Fig. S2).
In summary, in terms of performance, functionality and accuracy, RaMWAS outperformed QSEA and is currently the most viable option for performing large-scale MWAS.
Funding
This research was supported by the National Institute of Mental Health (R03MH102723, R01MH104576 to KA, R01MH099110, 1R01MH104576, RC2MH089996 to EO). MH received salary support from the National Institute on Drug Abuse (2R25DA026119).
Conflict of Interest: none declared.
Supplementary Material
References
- Aberg K.A. et al. (2017) A MBD-seq protocol for large-scale methylome-wide studies with (very) low amounts of DNA. Epigenetics, 12, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen A.M. et al. (2015) Current and future prospects for epigenetic biomarkers of substance use disorders. Genes, 6, 991–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan R.F. et al. (2017) Enrichment methods provide a feasible approach to comprehensive and adequately powered investigations of the brain methylome. Nucleic Acids Res., 45, e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S. (2013) DNA methylation age of human tissues and cell types. Genome Biol., 14, R115.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhard M. et al. (2016) QSEA—modelling of genome-wide DNA methylation from sequencing enrichment experiments. Nucleic Acids Res., 45, e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Oord E.J. et al. (2013) Estimation of CpG coverage in whole methylome next-generation sequencing studies. BMC Bioinformatics, 14, 50.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Oord E.J. et al. (2015) A whole methylome CpG-SNP association study of psychosis in blood and brain tissue. Schizophrenia Bull., 42, 1018–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.