

SUMMARY
Ants are an emerging model system for neuroepigenetics, as embryos with virtually identical genomes develop into different adult castes that display diverse physiology, morphology, and behavior. Although a number of ant genomes have been sequenced to date, their draft quality is an obstacle to sophisticated analyses of epigenetic gene regulation. We reassembled de novo high-quality genomes for two ant species, Camponotus floridanus and Harpegnathos saltator. Using long reads enabled us to span large repetitive regions and improve genome contiguity, leading to comprehensive and accurate protein-coding annotations that facilitated the identification of a Gp-9-like gene as differentially expressed in Harpegnathos castes. The new assemblies also enabled us to annotate long non-coding RNAs in ants, revealing caste-, brain-, and developmental-stage-specific long non-coding RNAs (lncRNAs) in Harpegnathos. These upgraded genomes, along with the new gene annotations, will aid future efforts to identify epigenetic mechanisms of phenotypic and behavioral plasticity in ants.
In Brief Using long-read sequencing, Shields et al. upgrade the genome assemblies for two ant species. Their results reveal a protein-coding gene preferentially expressed in worker ants and genes for long non-coding RNAs, several of which were expressed in the brain, in some cases at different levels in workers and reproductives.
INTRODUCTION
The ponerine ant Harpegnathos saltator is emerging as a model system to study the epigenetic regulation of brain function and behavior (Bonasio, 2012; Yan et al., 2014). Adult Harpegnathos workers can convert to acting queens, called gamergates, that are allowed to mate and lay fertilized eggs. We have previously shown that the worker-gamergate transition is accompanied by changes in brain gene expression (Gospocic et al., 2017), but the epigenetic mechanisms responsible for these changes remain unknown.
Previous work in Harpegnathos and in the more conventional Florida carpenter ant Camponotus floridanus has suggested that epigenetic pathways, including those that control histone modifications and DNA methylation, might be responsible for differential deployment of caste-specific traits (Bonasio et al., 2010, 2012; Simola et al., 2013a). Pharmacological and molecular manipulation of histone acetylation affects caste-specific behavior in Camponotus ants (Simola et al., 2016), suggesting a direct role for epigenetics in their social behavior. Although the molecular mechanisms by which environmental and developmental cues are converted into epigenetic information on chromatin remain subject of intense investigation (Allis and Jenuwein, 2016), it has become clear that non-coding RNAs play an important role in mediating this flow of information (Holoch and Moazed, 2015). In particular, long non-coding RNAs (lncRNAs)—transcripts longer than 200 bp that are not translated into proteins—have been proposed to participate in the epigenetic regulation of gene expression (Bonasio and Shiekhattar, 2014; Rinn and Chang, 2012).
Many proteins that regulate chromatin function bind RNA (Guttman et al., 2011; He et al., 2016; Hendrickson et al., 2016), and it is believed that these interactions might explain the epigenetic function of certain lncRNAs. Among epigenetic factors that bind to and are regulated by lncRNAs are SCML2 and EZH2 (Bonasio et al., 2014; Zhao et al., 2010), subunits of Polycomb repressive complex 1 and 2, which maintain lineage specifications and cell identity during development via epigenetic gene repression (Schuettengruber et al., 2017); WDR5 (Yang et al., 2014), a subunit of the MLL complex, which belongs to the Trithorax group of epigenetic activators (Schuettengruber et al., 2017); various DNA methyltransferases (Wang et al., 2015); and CTCF (Saldaña-Meyer et al., 2014; Sun et al., 2013), better known as the ‘‘master weaver of the genome’’ because of its role in organizing the genome in 3D loops (Phillips and Corces, 2009). In fact, lncRNAs have been directly implicated in maintaining looping interactions between promoters and enhancers (Lai et al., 2013) and as organizers of 3D genome architecture (Amaral et al., 2018; Engreitz et al., 2016a; Joung et al., 2017).
lncRNAs have been annotated extensively in human (Cabili et al., 2011; Derrien et al., 2012), mouse (Guttman et al., 2009; Pervouchine et al., 2015), model organisms such as zebrafish, Drosophila melanogaster and Caenorhabditis elegans (Gerstein et al., 2014; Nam and Bartel, 2012; Pauli et al., 2012; Ulitsky et al., 2011; Young et al., 2012), and the bees Apis mellifera and Apis cerana (Jayakodi et al., 2015); but, to our knowledge, no comprehensive annotation of lncRNAs in ants has been reported. This may be in part because ant genomes, including those of Camponotus and Harpegnathos (Bonasio et al., 2010), are still in draft, highly fragmented form due to the prevalent use of whole-genome shotgun sequencing to assemble them. In addition to making lncRNA annotation practically impossible, the fragmented nature of these genome assemblies also hamper the sophisticated genome-wide analyses required for epigenetic research, thus limiting the reach of these species as model organisms.
We upgraded the genomes of Harpegnathos and Camponotus to megabase level with a combination of de novo assembly of Pacific Biosciences (PacBio) long reads, scaffolding with mate pairs and long reads, and polishing with short reads. The contiguity of both genomes was greatly improved while maintaining the high accuracy of the short-read-only assemblies. We used these new assemblies to annotate protein-coding genes and lncRNAs, leading to the discovery of lncRNAs differentially expressed between Harpegnathos castes, developmental stages, and tissues. These improvements to the Harpegnathos and Camponotus genomes will lead to greater understanding of the genetic and epigenetic factors that underlie the behavior of these social insects.
RESULTS
Long-Read Sequencing Improves Contiguity
We sequenced genomic DNA isolated from Harpegnathos and Camponotus workers using PacBio single-molecule real-time technology, obtaining a sequence coverage of 703 for Harpegnathos and 533 for Camponotus, compatible with PacBio-only genome assembly (Koren et al., 2017). PacBio reads are much longer than those used for whole-genome shotgun draft assemblies, including the previously reported assemblies for these two ant species (Bonasio et al., 2010), and are thus expected to yield longer contigs and scaffolds with fewer gaps (scheme, Figure 1A).
Figure 1. PacBio Sequencing Improves Contiguity of Two Ant Genomes.
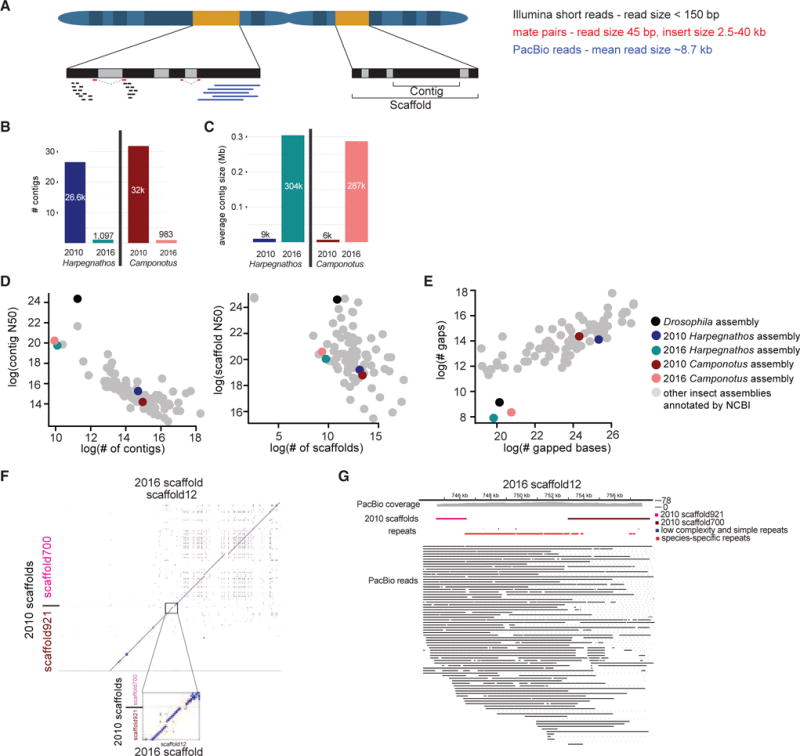
(A) Scheme showing types of reads used in assembly.
(B and C) Comparison of contig number (B) and average size (C) in 2016 and 2010 assemblies.
(D) Comparison of Harpegnathos and Camponotus genome assemblies to other insect genomes using contig number and N50 (left) and scaffold number and N50 (right).
(E) Number of gaps and gapped bases in insect assemblies.
(F) Two 2010 scaffolds, scaffold921 and scaffold700, are depicted along the y axis, with the 2016 scaffold, scaffold12, along the x axis. Dots indicate regions where there is significant sequence similarity. The boundary region between the 2010 scaffolds is shown in the inset.
(G) A genome browser view of region from (F) shows coverage by several PacBio reads that span the stretch of repetitive sequence across the gap between the two 2010 scaffolds.
See also Table S1 and Figures S1 and S2.
We used these long reads to assemble the two genomes de novo using a multistep process (Figure S1A), starting with the dedicated long read assembler Canu (Koren et al., 2017). Although this initial step produced assemblies that surpassed the contiguity of the current draft genomes (Figure S1A; Table S1), we leveraged long reads and previously generated sequencing data to maximize the quality of the newly assembled genomes.
The new PacBio sequencing-derived assemblies (‘‘2016 assemblies’’) compared favorably to the short-read assemblies currently available for both ant species (‘‘2010 assemblies’’). Despite capturing a larger amount of genomic sequence (Table S1), the number of contigs was dramatically decreased in the 2016 assemblies (Figure 1B) and their average size was more than 30-fold larger than in the 2010 assemblies (Figure 1C), reflecting greatly increased assembly contiguity. Scaffolding was also improved in the 2016 assemblies, which consisted of fewer, larger scaffolds (Figure S1B) and contained fewer gaps than the 2010 assemblies (Table S1). Improvements were also evident in the conventional metrics of assembly quality such as contig and scaffold N50s (Table S1). Overall, the contig N50 size increased by 22-fold (to 885 kb) and 65-fold (to 1.2 Mb) for Harpegnathos and Camponotus, respectively, and in both assemblies, the scaffold N50 size surpassed 1 Mb (Table S1).
The contig N50 sizes of our improved Harpegnathos and Camponotus assemblies top almost all other insect genomes available in the NCBI database, with the exception of two genomes also assembled using PacBio long-read sequencing, Drosophila serrata (Allen et al., 2017) and Aedes albopictus (Miller et al., 2018), as well as the classic model organism Drosophila melanogaster (Figure 1D, left). The number and size of scaffolds also compared favorably with other available genomes (Figure 1D, right), and the numbers of gaps in our new assemblies (240 and 326 in Harpegnathos and Camponotus, respectively) are lower than for any other insect genome in this set, including Drosophila melanogaster (Figure 1E).
PacBio reads can span long repetitive sequence that cannot be assembled properly using short reads (Roberts et al., 2013). We found several cases where distinct scaffolds from the 2010 assemblies mapped to a single scaffold (or contig) in the 2016 assemblies, separated by repetitive sequences. For example, scaffolds 921 and 700 from 2010 were joined into a larger scaffold in the improved 2016 assemblies (Figure 1F), separated by ~6.5 kb of repeats spanned by multiple PacBio reads (Figure 1G). Indeed, much of the newly assembled DNA sequence consisted of repeats (Figure S2).
Thus, long PacBio reads allowed us to assemble across longer repeats than previously possible, greatly improving the contiguity of the Harpegnathos and Camponotus genomes.
The New Assemblies Are Highly Accurate
We countered the high error rate of PacBio sequencing with deep sequence coverage (>50×) and by polishing our assemblies with the short reads from the original draft genomes (Bonasio et al., 2010).
RNA sequencing (RNA-seq) from various developmental stages in both species mapped better to the 2016 assemblies compared to the 2010 draft versions in all cases (Figure 2A), with a lower mismatch rate per base (Figure 2B), demonstrating that our strategy to correct PacBio sequencing errors successfully generated highly accurate genome sequences. The improved mapping rate suggests that the new assemblies capture transcribed but previously unassembled sequence.
Figure 2. Improved Accuracy of New Assemblies.
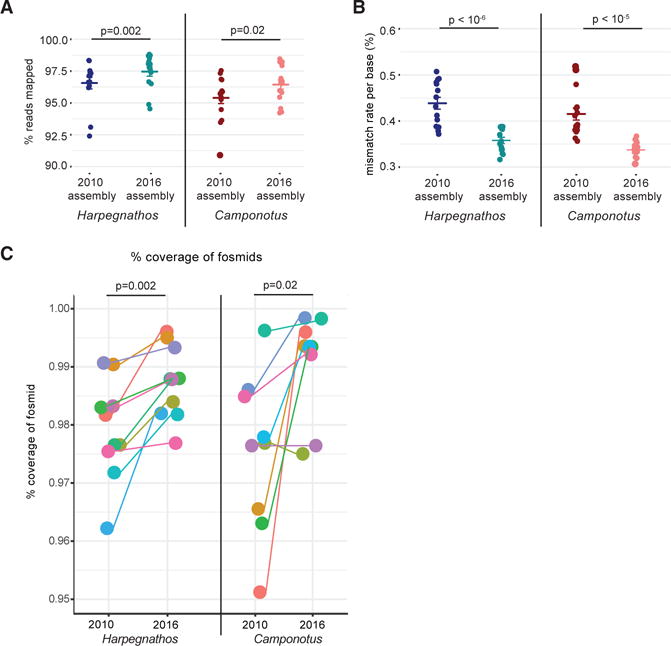
(A and B) Mapping (A) and sequence mismatch (B) rates for RNA-seq reads from various developmental stages of Harpegnathos (n = 14) and Camponotus (n = 15) to old and new assemblies. Horizontal bars indicate the means. p values are from two-sided, paired Student’s t test. Error bars indicate SEM.
(C) 2010 and 2016 assembly accuracy measured by percentage of fosmid Sanger sequence covered on a single scaffold. Each dot represents a fosmid. p value is from a two-sided Student’s t test.
See also Table S2.
Furthermore, alignment of Sanger sequences of 10 (Harpegnathos) and 9 (Camponotus) ~40-kb fosmid clones (Bonasio et al., 2010) showed similar or higher coverage in the new assemblies compared to the draft 2010 versions (Figure 2C; Table S2).
Neither the RNA-seq nor the fosmids were used in assembly construction, providing an orthogonal method of measuring genome completeness and accuracy.
Improvements in Protein-Coding Annotations
We annotated protein-coding genes using a combination of ab initio transcriptome reconstruction, homology-based searches, and de novo identification of gene structure (Figure S3A). We used MAKER2 (Holt and Yandell, 2011) to combine these sources of evidence and retained models consistently represented across evidence (Figure S3B) and/or with a protein domain, annotating 20,317 and 18,620 protein-coding genes for Harpegnathos and Camponotus, respectively (Figure S3C). The filtered protein-coding annotations recovered slightly higher percentages of a core set of evolutionarily conserved arthropod genes (Simão et al., 2015) compared to the 2010 annotations (Table S3).
The number of gene models encoding proteins conserved throughout evolution was more or less unchanged after the genome update (Figure 3A). Interestingly, a higher percentage of genes in the 2016 assemblies had no homology to known protein-coding genes in human, mouse, and a panel of insects, including several Hymenoptera (Figure 3A, red boxes). A majority of these gene models without homology to known proteins contained at least one recognizable protein families (PFAMs) domain (Figure 3B), suggesting that they might encode true protein-coding genes missed by annotation efforts in related organisms.
Figure 3. Annotation of Protein-Coding Genes.
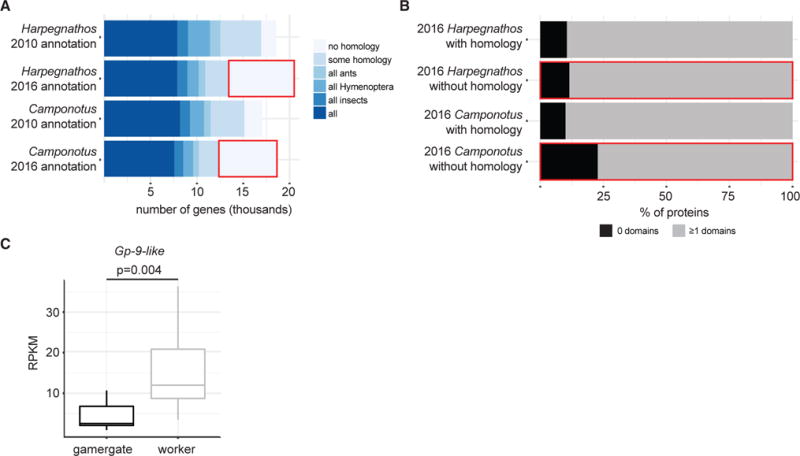
(A) Number of genes in 2010 and 2016 Harpegnathos and Camponotus annotations with a homolog in a panel of other ants, Hymenoptera, and animals.
(B) Fraction of genes with no detectable homology, as outlined in red in (A), that contains no (black) or ≥1 (gray) protein family (PFAM) domains.
(C) Expression of the previously unannotated Gp-9-like gene in Harpegnathos gamergates (n = 12) and workers (n = 11). p value is from a two-sided Student’s t test.
See also Table S3 and Figures S3 and S4.
We reasoned that the improved assemblies and protein-coding annotations might uncover biologically relevant genes missing in the older versions. Harpegnathos workers are characterized by their unique reproductive and brain plasticity that, in absence of a queen, allows some of them to transition to a queen-like phenotypic status called ‘‘gamergate’’ (Bonasio et al., 2010, 2012), which is accompanied by major changes in brain gene expression (Gospocic et al., 2017). Mapping this dataset to the new annotation, we found that a Gp-9-like gene had significantly higher expression in worker brains compared to gamergates (Figure 3C). This gene was not previously detected as differentially expressed, likely because its closest homolog in the old annotation contains many sequence disparities (Figure S4A), reducing the RNA-seq coverage mapped to this gene in both castes (Figure S4B). Mass spectrometry analyses identified two peptides mapping exactly to the newly predicted sequence (Figure S4A), confirming the accuracy of the updated gene model.
This Gp-9-like gene encodes one of several proteins with homology to a pheromone-binding protein well studied in the fire ant Solenopsis invicta because it marks a genomic element associated with the ability of the colony to accept one or more fertile queens (Wang et al., 2013). Other ant species, including Monomorium pharaonis (Warner et al., 2017), Vollenhovia emeryi (Miyakawa and Mikheyev, 2015), and Dinoponera quadriceps (Patalano et al., 2015), have several Gp-9-like homologs, some of which display worker-biased expression patterns (Figures S4C–S4E). Many other Hymenoptera also have Gp-9 or Gp-9-like homologs in their genomes (Figure S4F), and much of the Solenopsis invicta gene that associates with colony structure is conserved with these Gp-9-like gene models, especially within the odorant-binding domain (Figure S4G). Furthermore, this gene is likely under positive selection (significant by chi-square test, degrees of freedom [df] = 1, α = 0.001). These observations suggest that the role of this pheromone-binding protein in social organization is more conserved than previously appreciated.
One specific locus with better contiguity and improved protein-coding annotations is the Hox cluster, a group of developmental genes conserved throughout metazoa (Finnerty and Martindale, 1998). Homologs for two Drosophila Hox cluster genes, lab and abd-A, were surprisingly missing from the 2010 Harpegnathos annotation (Simola et al., 2013b); however, both genes were properly positioned in the Hox cluster of the new Harpegnathos assembly, in the same order as the corresponding Drosophila homologs (Figure 4A). The 2010 annotation did contain gene models overlapping the loci for Iab and abd-A, but they were incomplete (Figures 4B and 4C; data not shown), which had previously prevented their detection by homology searches. The contiguity of the Hox cluster is critical to its function, as genes in the cluster are expressed in a collinear fashion during development (Kmita and Duboule, 2003). Drosophila and the silkworm Bombyx mori have split Hox clusters (Negre et al., 2005; Yasukochi et al., 2004), but many other insects have an intact one (Brown et al., 2002; Devenport et al., 2000; Ferrier and Akam, 1996), including Apis mellifera (Dearden et al., 2006). In our previous assemblies, the Camponotus cluster was split among three different scaffolds, begging the question of whether this separation was due to the actual relocalization of genes during evolution or simply discontinuous assembly. The improved 2016 assemblies answered this question by showing that the entire Camponotus Hox clusters could be assembled into a single, larger scaffold (Figure 4A).
Figure 4. Reassembly of the Hox Clusters of Camponotus and Harpegnathos.
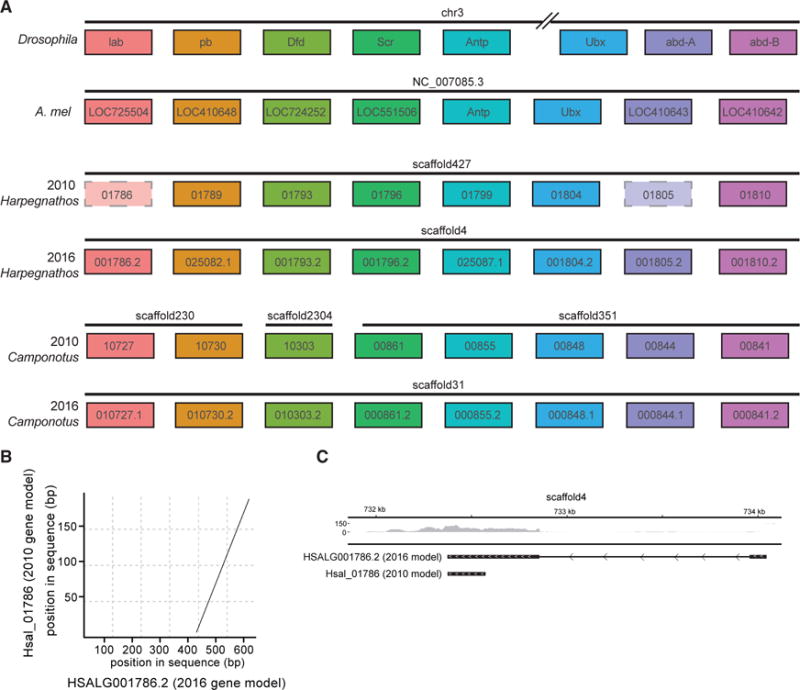
(A) Scheme of Hox gene organization in (from the top) Drosophila, Apis mellifera, Harpegnathos (old and new assembly), and Camponotus (old and new assembly).
(B) Example of a Hox gene in Harpegnathos updated in 2016 annotation. The 2010 gene model is depicted on the y axis, with the 2016 gene model on the x axis. Dots in the plot indicate regions of significant sequence similarity between 2010 and 2016 models.
(C) RNA-seq from various developmental stages in Harpegnathos shows extension of the gene model past the 2010 boundaries. The 2010 and 2016 gene models are shown under the RNA-seq coverage track. Scale on RNA-seq track indicates reads per million.
Together, our analyses show that reannotation of the improved assemblies for Harpegnathos and Camponotus yielded more complete gene sets, better models of already annotated genes, and better contiguity of a tightly regulated gene cluster.
Annotation of lncRNAs
To annotate lncRNAs, we assembled a reference-based transcriptome from RNA-seq of various developmental stages and retained high-confidence transcripts longer than 200 bp not overlapping with existing protein-coding gene models (Figure S5A). Approximately 24% of de-novo-assembled Harpegnathos and Camponotus transcripts met this requirement (Figure S5B).
We filtered our non-coding annotations using PhyloCSF (Lin et al., 2011). Most protein-coding genes in both Harpegnathos and Camponotus had positive PhyloCSF scores, indicative of a coding model, whereas our newly annotated putative non-coding transcripts were skewed toward negative, non-coding scores (Figure 5A). After filtering by PhyloCSF score, 628 (28.2%) and 683 (30.1%) of the putative non-coding genes in Harpegnathos and Camponotus, respectively, were retained. We then removed lncRNA gene models with splice junctions to adjacent protein-coding genes, as they might constitute 5′ or 3′ UTRs missed by our protein-coding annotation pipeline (You et al., 2017) (Figure 5B). We also removed lncRNA models containing open reading frames to which we could assign PFAM domains or peptides from mass spectrometry (Figure 5B). We did not consider these models for protein-coding annotations.
Figure 5. Annotation of Long Non-Coding RNAs.
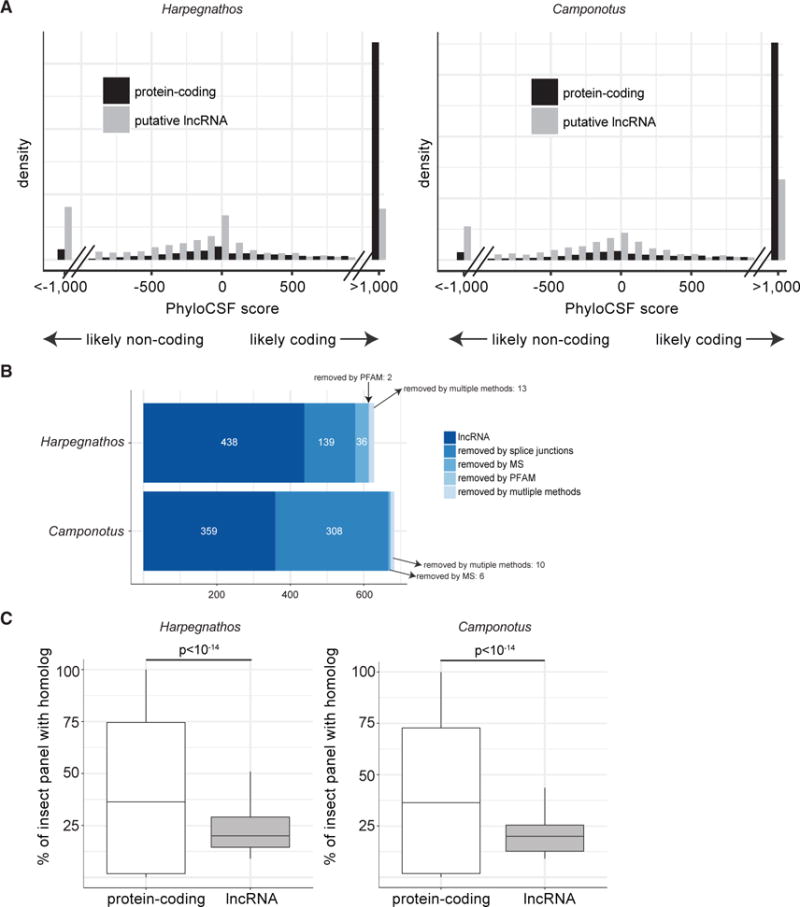
(A) PhyloCSF scores for transcripts with no overlap to coding sequences (gray) and known protein-coding genes (black). The x axis indicates the PhyloCSF scores in decibans, which represents the likelihood ratio of a coding model versus a non-coding model. Negative values indicate that a gene model is more likely to be non-coding than coding.
(B) Filtering of lncRNA using stranded, spliced RNA-seq reads and mass spectrometry.
(C) Boxplot for the number of homologs (BLASTN e-value <10−3) found in other insect genomes for lncRNAs compared to protein-coding gene models.
See also Figures S5 and S6.
At the end of all filtering steps, we annotated 438 and 359 high-confidence lncRNA gene models for Harpegnathos and Camponotus, respectively (Figures 5B and S5A), which we subdivided according to their spatial relationship to neighboring protein-coding gene models into intervening, promoter-associated, and intronic (Figures S5C and S5D), all of which showed a lack of coding potential, even when considered separately (Figure S6A). We could not detect a substantial number of antisense lncRNAs overlapping exons of protein-coding genes.
lncRNAs in other organism are less conserved, are shorter, have fewer exons, and, overall, are expressed at lower levels than protein-coding genes (Quinn and Chang, 2016). We detected most of these features in our ant lncRNAs; they were less conserved than protein-coding genes (Figure 5C), regardless of their genomic localization (Figure S6B); they had a smaller number of exons (Figure S6C); and they were expressed at lower levels than protein-coding genes (Figure S6D). However, the length distribution of the ant lncRNAs was similar to that of protein-coding genes (Figure S6E), which was a departure from what was observed in mammals, Drosophila, and C. elegans (Cabili et al., 2011; Nam and Bartel, 2012; Young et al., 2012). lncRNAs in other genomes tend to overlap with transposable elements at a higher rate than protein-coding genes, suggesting a role for these sequences in their function and diversification (Kapusta et al., 2013; Kelley and Rinn, 2012). We observe this in ant lncRNAs as well (Figure S6F).
Expression Patterns of lncRNAs
If our lncRNA gene models comprise functional loci with potential for epigenetic regulation we should be able to observe their differential expression in a number of relevant comparisons, such as through developmental stages, in different tissues, and perhaps even the same tissue from different castes.
We determined whether lncRNA transcription was differentially regulated during life transitions in Harpegnathos. We analyzed whole-body RNA-seq datasets from embryos, larvae, pupae, and adult workers. We clustered relative changes in the expression levels of lncRNAs across these samples into groups with distinct kinetics (Figure 6A), which allowed us to identify early development lncRNAs (Figure 6A, clusters 1–4), adult lncRNAs (clusters 8–10), and a set of lncRNAs predominantly expressed in the pupal stage (clusters 6 and 7), a critical phase in the life of holometabolous insects characterized by pronounced cell proliferation, morphogenesis, and neuronal remodeling.
Figure 6. Differential Expression of LncRNAs in Harpegnathos Developmental Stages, Tissues, and Brain Regions.
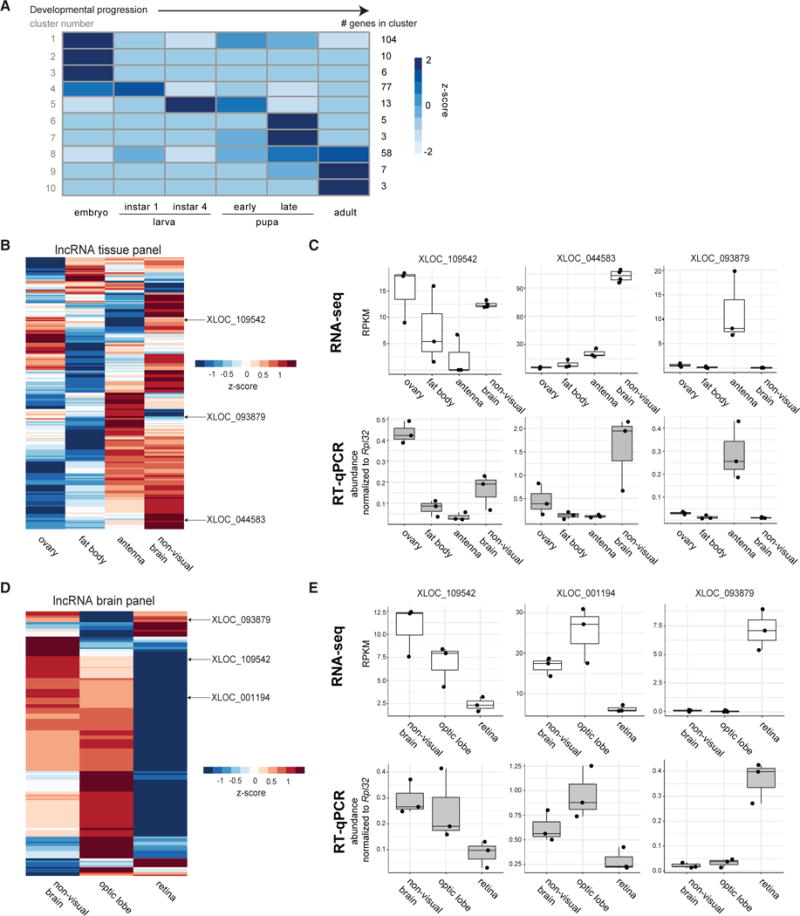
(A) K-means clustering of changes in lncRNA expression across the indicated developmental stages (all n = 2). The cluster number is displayed to the left of the heatmap, while the number of lncRNAs in each cluster is shown to the right.
(B) Heatmap of lncRNA expression patterns from RNA-seq in ovary, fat body, antenna, and non-visual brain (all n = 3). Heatmap shows Z scores of log(RPKMs) (read per kilobase per million) by row. Arrows point to lncRNAs that have expression specific to one or more tissues.
(C) RNA-seq and qRT-PCR for the three lncRNAs highlighted in (B).
(D) Heatmap of lncRNA expression patterns from RNA-seq in non-visual brain, optic lobe, and retina (all n = 3). Heatmap shows Z scores of log(RPKMs) by row. Arrows point to lncRNAs that have expression specific to one or more regions.
(E) RNA-seq and qRT-PCR for the three lncRNAs highlighted in (D).
See also Figure S7.
We also identified lncRNAs with tissue-specific expression in Harpegnathos adults by comparing the transcriptomes of antenna, non-visual brain (the central part of the brain after removal of the optic lobes; Gospocic et al., 2017), fat body, and ovary. Many lncRNAs were expressed specifically in one tissue, especially the brain (Figure 6B), perhaps indicating a dedicated function. We validated the tissue-specific expression of three lncRNAs by RT-qPCR (Figure 6C): XLOC_109542, which showed higher expression in ovary and non-visual brain; XLOC_044583, with highest expression in the brain; and XLOC_093879, which was restricted to the antenna (and the retina; see below).
As lncRNAs have been previously shown to be expressed in different regions of the mouse brain (Mercer et al., 2008), we compared lncRNA expression levels in RNA-seq data from non-visual brain, optic lobe, and retina. We used region-specific controls corazonin (non-visual brain), Gabbr2 (optic lobe), and Arr2 (retina) to ensure that our dissections had been performed accurately (Figure S7A). We detected many lncRNAs with higher expression in one region of the brain (Figure 6D), and validated three by RT-qPCR (Figure 6E): XLOC_109542, which was expressed at higher levels in the non-visual brain; XLOC_ 001194, expressed at higher levels in the optic lobe; and XLOC_093879, restricted to the retina (and the antenna, see above).
We previously showed that the adult caste transition between worker and gamergates in Harpegnathos is accompanied by major changes in protein-coding gene expression (Gospocic et al., 2017). We reanalyzed that dataset in the context of our new lncRNA annotation and found 17 lncRNAs that were differentially expressed in worker and gamergate brains with a p value cutoff of 0.05 (Figure 7A). We also looked for lncRNAs that might be responsible for co-regulating a protein-coding gene. XLOC_094172 caught our attention because its expression strongly correlated with that of the neighboring protein-coding gene vps26 (Figures 7B, 7C, and S7B), a subunit of the retromer complex implicated in neurological disorders (Linhart et al., 2014; McMillan et al., 2017). This lncRNA and its co-regulated protein-coding gene are ~22 kb apart and on opposite strands (Figure 7D), excluding the possibility that they are spanned by the same primary transcript. Instead, we propose that this lncRNA controls expression of the protein-coding gene, as is the case for several cis-acting lncRNAs in other organisms (Engreitz et al., 2016b).
Figure 7. Differential LncRNA Expression and LncRNA/Protein-Coding Co-regulation in Harpegnathos Castes.
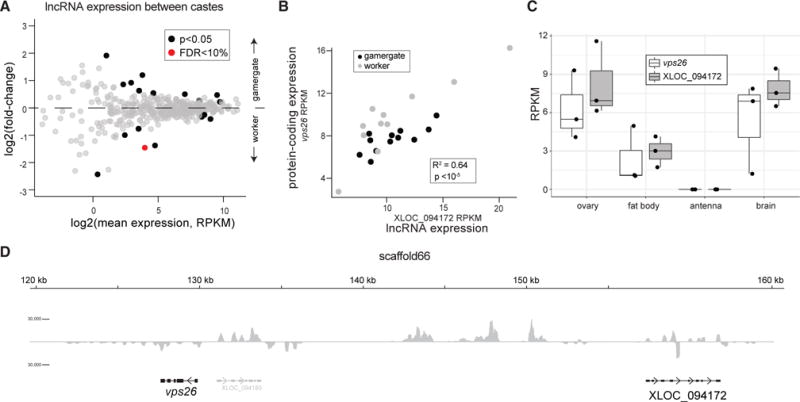
(A) MA plot of lncRNAs in RNA-seq data comparing worker and gamergates (Gospocic et al., 2017). Genes with unadjusted p < 0.05 are highlighted in black, genes with <10% false discovery rate (FDR) in red. Data are from ≥10 biological replicates per condition (individual ants; worker, n = 11; gamergate, n = 12).
(B) The expression levels of XLOC_094172 lncRNA (x axis) and the protein-coding gene vps26 (y axis) correlate in both gamergate and worker. Each dot represents one biological sample (worker, n = 11; gamergate, n = 12). p value from Pearson correlation is indicated.
(C) Expression patterns of XLOC_094172 and vps26 in worker brains by RNA-seq in non-visual brain, optic lobe, and retina (all n = 3).
(D) Positions of XLOC_094172 and vps26 on scaffold66, with RNA-seq coverage from combined workers (n = 11) and gamergates (n = 12). Scale on RNA-seq track indicates reads per million.
See also Figure S7.
We also confirmed the expression in the brain of lncRNAs with homologs in other insects (Jayakodi et al., 2015; Li et al., 2012) (Figure S7C). Most notably, the Harpegnathos homolog of CASK regulatory gene (CRG), a lncRNA involved in locomotor behavior in Drosophila (Li et al., 2012), was expressed in neurons throughout the brain, as demonstrated by RNA-seq analyses (Figure S7D; XLOC_081169) and by its co-localization with the pan-neuronal marker elav by fluorescence in situ hybridization (FISH) (Figures S7D and S7E).
To confirm that our example lncRNAs are bona fide lncRNAs, we utilized an orthogonal method of measuring coding potential used in other lncRNA annotations (Jayakodi et al., 2015; Nam and Bartel, 2012; Ulitsky et al., 2011; Young et al., 2012), the Coding Potential Calculator (CPC) (Kong et al., 2007). CPC scores correlated strongly with PhyloCSF scores (p < 10−15 for both Harpegnathos and Camponotus) and scored as non-coding all lncRNAs shown in Figures 6, 7, and S7. The accuracy of our lncRNA gene models was further confirmed by the fact that all qRT-PCR reactions yielded products of the expected size (Figure S7F).
Thus, our improved genome assemblies allowed us to annotate lncRNAs, several of which displayed developmental-, brain-, or caste-specific expression patterns, which suggests that they might have important roles in development and brain function.
DISCUSSION
Social insects offer a unique perspective from which to study epigenetics (Bonasio, 2012; Yan et al., 2014). Striking morphological and behavioral differences between castes include phenotypes relevant to translational research, such as social behavior, aging, and development. These traits can be studied on an organism level within a natural social context, as full colonies can be maintained in the laboratory. However, to analyze these complex traits at a molecular level, proper genomic tools must be developed. We previously assembled ant genomes generating a workable draft using the best technology at the time, whole-genome shotgun using short Illumina reads (Bonasio et al., 2010); however, the fragmented nature of these draft genomes presented an obstacle to epigenomic studies.
Here, we used PacBio long reads to reassemble de novo the genomes of the two ant species currently in use as models in our laboratory, Camponotus floridanus and Harpegnathos saltator, and produced accurate assemblies with scaffold N50 sizes larger than 1 Mb and a number of gaps smaller than in all other insect genomes available on NCBI at the time of writing (Figure 1E). Although other insect assemblies have larger scaffold N50s than our new ant assemblies, which might be helpful for evaluating structural variations and interactions at great length scales, many cis regulatory and epigenomic mechanisms take place at short-to-medium range and their study is facilitated by longer gap-free regions of sequence (i.e., longer contigs). Thus, we prioritized contig length in our new assemblies, and chose to pursue greater PacBio sequencing coverage rather than techniques used to improve scaffold N50, such as optical mapping and proximity ligation.
Our greatly improved Harpegnathos and Camponotus assemblies deliver several critical benefits to further development of these ant species into molecular model organisms: (1) more comprehensive protein-coding annotations and more complete gene models (Figure 3; Table S3), (2) more continuity of co-regulated gene clusters (Figure 4), (3) high-quality lncRNA annotations (Figure 5), and (4) the ability to detect regulatory mechanisms functioning in cis at distances of 10–100 kb (Figure 7).
Although the annotation of protein-coding genes did not suffer excessively from the draft status of the 2010 assemblies, the new annotations contain potentially relevant genes that were previously missing. Most notably, a Gp-9-like gene previously unannotated in the Harpegnathos genome was found to be differentially expressed in worker brains compared to gamergates (Figure 3C). The importance of Gp-9 in ant biology is well established, as it was one of the first genetic markers discovered in ants for a colony-level phenotype. In the fire ant Solenopsis invicta, Gp-9 maps to a cluster of genes involved in a large genomic rearrangement that governs the choice between a polygyne (multiple queens) or monogyne (one queen) colony (Ross, 1997; Wang et al., 2013). We found that a Gp-9-like homolog is expressed at different levels in Harpegnathos castes as well as in three other ant species with different social structures (Figures S4C–S4E), suggesting a conserved role for this gene in colony organization and opening an avenue for future investigation on its molecular function.
Another advance granted by our improved genome assemblies was the ability to annotate lncRNAs. We developed a custom pipeline and discovered over 300 high-confidence lncRNAs in both Harpegnathos and Camponotus. The mechanism of action and biological impact of lncRNAs is the subject of intense investigation in various model systems and in several cases a dedicated role in brain function has been advocated, based in part on their expression patterns (Bonasio, 2012; Bonasio and Shiekhattar, 2014).
Most lncRNAs are believed to act in cis to regulate expression of neighboring genes (Bonasio and Shiekhattar, 2014; Engreitz et al., 2016b; Lee, 2012); therefore, an extended view of protein-coding genes in the vicinity of lncRNAs is critical to understand their regulatory role, and this information is provided by our updated genomes. Thanks to the increased continuity of the new assemblies, we were able to identify a lncRNAs-mRNA pair whose brain expression patterns were correlated, suggesting a potential regulatory relationship (Figures 7B–7D). Similar cases have been described in mammals; one lncRNA, HAR1F, is co-expressed in human Cajal-Retzius neurons with reelin, a protein-coding gene that regulates cortical development (Pollard et al., 2006). A murine lncRNA, Dali, regulates the expression of the nearby transcription factor Pou3f3, which is involved in nerve and growth development (Chalei et al., 2014). Our findings on brain- and caste-specific lncRNAs as well as lncRNAs co-expressed with protein-coding gene will allow us to prioritize candidates for future studies on the neuroepigenetic functions of lncRNAs in ants. This prospect is particularly intriguing in Harpegnathos, where we discovered major transcriptional changes that accompany the rewiring of the brain during adult caste transitions (Gospocic et al., 2017) and where we recently showed the feasibility of genetic manipulation of the germline (Yan et al., 2017).
EXPERIMENTAL PROCEDURES
Genome Assembly Strategy
The reads of insert extracted from raw PacBio data were error corrected, trimmed, and assembled by Canu v1.3 (Koren et al., 2017). Quiver was used to polish the assemblies, which were then scaffolded with extracted subreads from the PacBio data using PBJelly (English et al., 2012), and with mate pairs using SSpace-Standard (Boetzer et al., 2011). The assemblies were polished with paired-end Illumina short reads using Pilon (Walker et al., 2014).
Annotation of Protein-Coding Genes
Protein-coding genes were annotated on the Harpegnathos and Camponotus assemblies using iterations of the MAKER2 pipeline. The MAKER2 pipeline was run four times, each step updated with hidden Markov models trained on the previous step. On the fourth run, gene models produced directly from RNA-seq and homology were reported, and all gene models were filtered using the annotation edit distance and the presence of a PFAM domain, as detailed in Supplemental Experimental Procedures.
Annotation of lncRNA Genes
RNA-seq reads from various developmental stages of Harpegnathos and Camponotus were assembled using two reference-based transcriptome assemblers, Trinity and Stringtie. Transcripts common among the two methods that did not overlap with protein-coding genes were designated as putative lncRNAs. LncRNAs were further filtered using the PhyloCSF Omega Test (Lin et al., 2011), mass spectrometry, and presence of splice junctions to protein-coding genes.
qPCR
For qRT-PCR, 1 ng RNA was assayed per 10 μL reaction using the RNA-to-Ct single-step kit (Thermo Fisher). The RNA for Rpl32, encoding a ribosomal protein, was used as a normalization control.
Heatmaps and Clustering of lncRNA Expression Levels
Expression patterns of differentially expressed lncRNAs in the developmental stages of Harpegnathos were clustered based on the quantile-normalized log-fold expression changes between each pair of samples. K-means clustering with a preset number of clusters (10) and maximum number of iterations (50) was performed on this quantile-normalized matrix. Heatmaps were plotted using the pheatmap package for R, with color scaling by row.
In Situ Hybridization
500-bp DNA probes were designed against XLOC_081169 and included T7 (sense) and SP6 (anti-sense) RNA polymerase promoters. RNA in situ hybridization were performed according to published protocols (Morris et al., 2009; Søe et al., 2011), with minor modifications. Chromogenic ISH sections were imaged with a DS-Ri1 Digital Microscope Camera from Nikon. Fluorescent ISH sections were imaged with a Leica SPE laser scanning confocal microscope.
Sequencing Data
The accession number for the RNA-sequencing data generated for this study is GEO: SuperSeries GSE102605. The accession number for the raw PacBio reads of the insert, as well as assembled genomes, is BioProject: PRJNA445978.
Supplementary Material
Highlights.
Long reads produce highly contiguous genome assemblies for two ant species
Formerly unannotated gene well studied in other ants has caste-biased expression
Upgraded genomes allow for annotation of long non-coding RNAs
Many long non-coding RNAs are expressed in the brain, some in caste-specific manner
Acknowledgments
The authors thank Janko Gospocic for providing ant samples; Karl Glastad for helpful comments on genome assembly; Katy Munson and the UW PacBio Sequencing service for performing PacBio sequencing; Shawn Little for helping with FISH; and Yoseph Barash, Ben Voight, and Paul Babb for comments on the manuscript. R.B. thanks Danny Reinberg (NYU) as well as Guojie Zhang, Cai Li, Zhensheng Chen, and Luohao Xu (BGI) for their intellectual support and efforts during an initial attempt at annotating lncRNAs. R.B. acknowledges financial support from the NIH (DP2MH107055), the Searle Scholars Program (15-SSP-102), the Linda Pechenik Montague Investigator Award, and the Charles E. Kaufman Foundation (KA2016-85223). E.J.S. acknowledges financial support from the NIH (T32HG000046). B.A.G. was supported by the NIH (GM110174 and HL122993).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.014.
AUTHOR CONTRIBUTIONS
A.K.W. performed mass spectrometry experiments and analysis in B.A.G.’s lab. L.S. performed brain and tissue dissections and carried out in situ hybridization experiments. All remaining experiments and analyses were performed by E.J.S. The manuscript was written by E.J.S. and R.B., with input from L.S.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Allen SL, Delaney EK, Kopp A, Chenoweth SF. Single-molecule sequencing of the Drosophila serrata genome. G3 (Bethesda) 2017;7:781–788. doi: 10.1534/g3.116.037598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- Amaral PP, Leonardi T, Han N, Viré E, Gascoigne DK, Arias-Carrasco R, Büscher M, Pandolfini L, Zhang A, Pluchino S, et al. Genomic positional conservation identifies topological anchor point RNAs linked to developmental loci. Genome Biol. 2018;19:32. doi: 10.1186/s13059-018-1405-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- Bonasio R. Emerging topics in epigenetics: ants, brains, and noncoding RNAs. Ann N Y Acad Sci. 2012;1260:14–23. doi: 10.1111/j.1749-6632.2011.06363.x. [DOI] [PubMed] [Google Scholar]
- Bonasio R, Shiekhattar R. Regulation of transcription by long noncoding RNAs. Annu Rev Genet. 2014;48:433–455. doi: 10.1146/annurev-genet-120213-092323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, Zhang G, Ye C, Mutti NS, Fang X, Qin N, Donahue G, Yang P, Li Q, Li C, et al. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science. 2010;329:1068–1071. doi: 10.1126/science.1192428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, Li Q, Lian J, Mutti NS, Jin L, Zhao H, Zhang P, Wen P, Xiang H, Ding Y, et al. Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr Biol. 2012;22:1755–1764. doi: 10.1016/j.cub.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, Lecona E, Narendra V, Voigt P, Parisi F, Kluger Y, Reinberg D. Interactions with RNA direct the Polycomb group protein SCML2 to chromatin where it represses target genes. eLife. 2014;3:e02637. doi: 10.7554/eLife.02637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SJ, Fellers JP, Shippy TD, Richardson EA, Maxwell M, Stuart JJ, Denell RE. Sequence of the Tribolium castaneum homeotic complex: the region corresponding to the Drosophila melanogaster antennapedia complex. Genetics. 2002;160:1067–1074. doi: 10.1093/genetics/160.3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalei V, Sansom SN, Kong L, Lee S, Montiel JF, Vance KW, Ponting CP. The long non-coding RNA Dali is an epigenetic regulator of neural differentiation. eLife. 2014;3:e04530. doi: 10.7554/eLife.04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearden PK, Wilson MJ, Sablan L, Osborne PW, Havler M, McNaughton E, Kimura K, Milshina NV, Hasselmann M, Gempe T, et al. Patterns of conservation and change in honey bee developmental genes. Genome Res. 2006;16:1376–1384. doi: 10.1101/gr.5108606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devenport MP, Blass C, Eggleston P. Characterization of the Hox gene cluster in the malaria vector mosquito, Anopheles gambiae. Evol Dev. 2000;2:326–339. doi: 10.1046/j.1525-142x.2000.00074.x. [DOI] [PubMed] [Google Scholar]
- English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, Gibbs RA. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE. 2012;7:e47768. doi: 10.1371/journal.pone.0047768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Ollikainen N, Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol. 2016a;17:756–770. doi: 10.1038/nrm.2016.126. [DOI] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016b;539:452–455. doi: 10.1038/nature20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier DE, Akam M. Organization of the Hox gene cluster in the grasshopper, Schistocerca gregaria. Proc Natl Acad Sci USA. 1996;93:13024–13029. doi: 10.1073/pnas.93.23.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerty JR, Martindale MQ. The evolution of the Hox cluster: insights from outgroups. Curr Opin Genet Dev. 1998;8:681–687. doi: 10.1016/s0959-437x(98)80037-3. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Rozowsky J, Yan KK, Wang D, Cheng C, Brown JB, Davis CA, Hillier L, Sisu C, Li JJ, et al. Comparative analysis of the transcriptome across distant species. Nature. 2014;512:445–448. doi: 10.1038/nature13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gospocic J, Shields EJ, Glastad KM, Lin Y, Penick CA, Yan H, Mikheyev AS, Linksvayer TA, Garcia BA, Berger SL, et al. The neuropeptide Corazonin controls social behavior and caste identity in ants. Cell. 2017;170:748–759. doi: 10.1016/j.cell.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Sidoli S, Warneford-Thomson R, Tatomer DC, Wilusz JE, Garcia BA, Bonasio R. High-Resolution Mapping of RNA-Binding Regions in the Nuclear Proteome of Embryonic Stem Cells. Mol Cell. 2016;64:416–430. doi: 10.1016/j.molcel.2016.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson D, Kelley DR, Tenen D, Bernstein B, Rinn JL. Widespread RNA binding by chromatin-associated proteins. Genome Biol. 2016;17:1–18. doi: 10.1186/s13059-016-0878-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16:71–84. doi: 10.1038/nrg3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011;12:491. doi: 10.1186/1471-2105-12-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakodi M, Jung JW, Park D, Ahn YJ, Lee SC, Shin SY, Shin C, Yang TJ, Kwon HW. Genome-wide characterization of long intergenic non-coding RNAs (lincRNAs) provides new insight into viral diseases in honey bees Apis cerana and Apis mellifera. BMC Genomics. 2015;16:680. doi: 10.1186/s12864-015-1868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Engreitz JM, Konermann S, Abudayyeh OO, Verdine VK, Aguet F, Gootenberg JS, Sanjana NE, Wright JB, Fulco CP, et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. 2017;548:343–346. doi: 10.1038/nature23451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, Yandell M, Feschotte C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013;9:e1003470. doi: 10.1371/journal.pgen.1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D, Rinn J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012;13:R107. doi: 10.1186/gb-2012-13-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmita M, Duboule D. Organizing axes in time and space; 25 years of colinear tinkering. Science. 2003;301:331–333. doi: 10.1126/science.1085753. [DOI] [PubMed] [Google Scholar]
- Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–9. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, Shiekhattar R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501. doi: 10.1038/nature11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–1439. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- Li M, Wen S, Guo X, Bai B, Gong Z, Liu X, Wang Y, Zhou Y, Chen X, Liu L, Chen R. The novel long non-coding RNA CRG regulates Drosophila locomotor behavior. Nucleic Acids Res. 2012;40:11714–11727. doi: 10.1093/nar/gks943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–i282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhart R, Wong SA, Cao J, Tran M, Huynh A, Ardrey C, Park JM, Hsu C, Taha S, Peterson R, et al. Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson’s disease mutant of leucine-rich repeat kinase 2 (LRRK2) Mol Neurodegener. 2014;9:23. doi: 10.1186/1750-1326-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan KJ, Korswagen HC, Cullen PJ. The emerging role of retromer in neuroprotection. Curr Opin Cell Biol. 2017;47:72–82. doi: 10.1016/j.ceb.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci USA. 2008;105:716–721. doi: 10.1073/pnas.0706729105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, Koren S, Dilley KA, Puri V, Brown DM, Harkins DM, Thibaud-Nissen F, Rosen B, Chen XG, Tu Z, et al. Analysis of the Aedes albopictus C6/36 genome provides insight into cell line utility for viral propagation. Gigascience. 2018;7:1–13. doi: 10.1093/gigascience/gix135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa MO, Mikheyev AS. QTL mapping of sex determination loci supports an ancient pathway in ants and honey bees. PLoS Genet. 2015;11:e1005656. doi: 10.1371/journal.pgen.1005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CA, Benson E, White-Cooper H. Determination of gene expression patterns using in situ hybridization to Drosophila testes. Nat Protoc. 2009;4:1807–1819. doi: 10.1038/nprot.2009.192. [DOI] [PubMed] [Google Scholar]
- Nam JW, Bartel DP. Long noncoding RNAs in C. elegans. Genome Res. 2012;22:2529–2540. doi: 10.1101/gr.140475.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre B, Casillas S, Suzanne M, Sánchez-Herrero E, Akam M, Nefedov M, Barbadilla A, de Jong P, Ruiz A. Conservation of regulatory sequences and gene expression patterns in the disintegrating Drosophila Hox gene complex. Genome Res. 2005;15:692–700. doi: 10.1101/gr.3468605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patalano S, Vlasova A, Wyatt C, Ewels P, Camara F, Ferreira PG, Asher CL, Jurkowski TP, Segonds-Pichon A, Bachman M, et al. Molecular signatures of plastic phenotypes in two eusocial insect species with simple societies. Proc Natl Acad Sci USA. 2015;112:13970–13975. doi: 10.1073/pnas.1515937112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, Levin JZ, Fan L, Sandelin A, Rinn JL, Regev A, Schier AF. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012;22:577–591. doi: 10.1101/gr.133009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervouchine DD, Djebali S, Breschi A, Davis CA, Barja PP, Dobin A, Tanzer A, Lagarde J, Zaleski C, See LH, et al. Enhanced transcriptome maps from multiple mouse tissues reveal evolutionary constraint in gene expression. Nat Commun. 2015;6:5903. doi: 10.1038/ncomms6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RJ, Carneiro MO, Schatz MC. The advantages of SMRT sequencing. Genome Biol. 2013;14:405. doi: 10.1186/gb-2013-14-7-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross KG. Multilocus evolution in fire ants: effects of selection, gene flow and recombination. Genetics. 1997;145:961–974. doi: 10.1093/genetics/145.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldaña-Meyer R, González-Buendía E, Guerrero G, Narendra V, Bonasio R, Recillas-Targa F, Reinberg D. CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev. 2014;28:723–734. doi: 10.1101/gad.236869.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell. 2017;171:34–57. doi: 10.1016/j.cell.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Simola DF, Ye C, Mutti NS, Dolezal K, Bonasio R, Liebig J, Reinberg D, Berger SL. A chromatin link to caste identity in the carpenter ant Camponotus floridanus. Genome Res. 2013a;23:486–496. doi: 10.1101/gr.148361.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simola DF, Wissler L, Donahue G, Waterhouse RM, Helmkampf M, Roux J, Nygaard S, Glastad KM, Hagen DE, Viljakainen L, et al. Social insect genomes exhibit dramatic evolution in gene composition and regulation while preserving regulatory features linked to sociality. Genome Res. 2013b;23:1235–1247. doi: 10.1101/gr.155408.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simola DF, Graham RJ, Brady CM, Enzmann BL, Desplan C, Ray A, Zwiebel LJ, Bonasio R, Reinberg D, Liebig J, Berger SL. Epigenetic (re)programming of caste-specific behavior in the ant Camponotus floridanus. Science. 2016;351:aac6633. doi: 10.1126/science.aac6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søe MJ, Møller T, Dufva M, Holmstrøm K. A sensitive alternative for microRNA in situ hybridizations using probes of 2′-O-methyl RNA + LNA. J Histochem Cytochem. 2011;59:661–672. doi: 10.1369/0022155411409411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Del Rosario BC, Szanto A, Ogawa Y, Jeon Y, Lee JT. Jpx RNA activates Xist by evicting CTCF. Cell. 2013;153:1537–1551. doi: 10.1016/j.cell.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–1550. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang YC, Shoemaker D, Keller L. A Y-like social chromosome causes alternative colony organization in fire ants. Nature. 2013;493:664–668. doi: 10.1038/nature11832. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhao Y, Bao X, Zhu X, Kwok YKY, Sun K, Chen X, Huang Y, Jauch R, Esteban MA, et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. 2015;25:335–350. doi: 10.1038/cr.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner MR, Mikheyev AS, Linksvayer TA. Genomic signature of kin selection in an ant with obligately sterile workers. Mol Biol Evol. 2017;34:1780–1787. doi: 10.1093/molbev/msx123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Simola DF, Bonasio R, Liebig J, Berger SL, Reinberg D. Eusocial insects as emerging models for behavioural epigenetics. Nat Rev Genet. 2014;15:677–688. doi: 10.1038/nrg3787. [DOI] [PubMed] [Google Scholar]
- Yan H, Opachaloemphan C, Mancini G, Yang H, Gallitto M, Mlejnek J, Leibholz A, Haight K, Ghaninia M, Huo L, et al. An engineered orco mutation produces aberrant social behavior and defective neural development in ants. Cell. 2017;170:736–747. doi: 10.1016/j.cell.2017.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YW, Flynn RA, Chen Y, Qu K, Wan B, Wang KC, Lei M, Chang HY. Essential role of lncRNA binding for WDR5 maintenance of active chromatin and embryonic stem cell pluripotency. eLife. 2014;3:e02046. doi: 10.7554/eLife.02046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukochi Y, Ashakumary LA, Wu C, Yoshido A, Nohata J, Mita K, Sahara K. Organization of the Hox gene cluster of the silkworm, Bombyx mori: a split of the Hox cluster in a non-Drosophila insect. Dev Genes Evol. 2004;214:606–614. doi: 10.1007/s00427-004-0441-1. [DOI] [PubMed] [Google Scholar]
- You BH, Yoon SH, Nam JW. High-confidence coding and noncoding transcriptome maps. Genome Res. 2017;27:1050–1062. doi: 10.1101/gr.214288.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RS, Marques AC, Tibbit C, Haerty W, Bassett AR, Liu JL, Ponting CP. Identification and properties of 1,119 candidate lincRNA loci in the Drosophila melanogaster genome. Genome Biol Evol. 2012;4:427–442. doi: 10.1093/gbe/evs020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40:939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.