

Abstract
Peptides are small biological molecules that are attractive in drug delivery and materials engineering for applications including therapeutics, molecular building blocks and cell-targeting ligands. Peptides are small but can possess complexity and functionality as larger proteins. Due to their intrinsic properties, peptides are able to overcome the physiological and transport barriers presented by diseases. In this review, we discuss the progress of identifying and using peptides to shuttle across biological barriers and facilitate transport of drugs and drug delivery systems for improved therapy.
Here, the focus of this review is on rationally designed, phage display peptides, and even endogenous peptides as carriers to penetrate biological barriers, specifically the blood-brain barrier(BBB), the gastrointestinal tract (GI), and the solid tumor microenvironment (T). We will discuss recent advances of peptides as drug carriers in these biological environments. From these findings, challenges and potential opportunities to iterate and improve peptide-based approaches will be discussed to translate their promise towards the clinic to deliver drugs for therapeutic efficacy.
Keywords: Peptides, drug delivery, carrier, blood-brain barrier, GI, tumor
Introduction
Peptides are a class of biological molecules that are typically less than 50 amino acids and possess unique properties that are attractive for use in biomedicine (Sato et al. 2006). Due to their small size, they can behave like small molecule drugs and have similar transport properties; simultaneously, they possess same functionalities as proteins due to the similar composition. These “biologically active” small bio-molecules have a wide range of functions, such as therapeutics, building blocks to higher order self-assembled structures and proteins, and targeting ligands against biomarkers expressed on cells and tissues during homeostasis and disease (Komin et al. 2017). Importantly, peptides can bind and penetrate different barriers present in cells and tissues due to their physical, chemical, and biological properties, therefore those functional peptides can serve as effective drug carriers to overcome the extracellular and intracellular barriers present in various disease states. Here in this review, we provide a summary of recent advances of peptides as carriers to shuttle drug or drug delivery systems across biological barriers, including the blood-brain barrier, the gastrointestinal tract, and the tumor microenvironment. An overview of peptides that have been identified to penetrate these three types of barriers are listed in Tables 1 and 2. From the progress, we also address challenges in translating the promise of these biological-based carriers for future use.
Table 1.
Identification methods, shuttling mechanisms of physical barriers penetrating peptides
| Identification methods, shuttling mechanisms of physical barriers penetrating peptides | ||||
|---|---|---|---|---|
| Identification methods | Name of peptides | Barrier (BBB, GI, T) | Mechanism | References |
| Rational design | Angiopep | BBB | LRP receptor mediated transcytosis | Demeule et al. 2008 |
| Cell-penetrating peptides (CPPs) | Tat | GI, BBB, T | Transcytosis, heparin sulfate-dependent endocytosis | Liang and Yang 2005, Mahmood et al. 2016, Yan et al. 2015, Boohaker et al. 2012, Green and Loewenstein 1988 |
| R6, R8, R10 | GI, | Energy-dependent endocytosis | Kamei et al. 2008b | |
| L-penetratin | GI, BBB | Macropinocytosis | Kamei et al. 2008a, Tremmel R et al.2016 | |
| L-pVEC | GI | Macropinocytosis | Kamei et al. 2008a | |
| L-RRL helix | GI | Macropinocytosis | Kamei et al. 2008a | |
| Sec | GI | Exocytosis | Zhu et al. 2015 | |
| SynB1 | BBB | Adsorptive endocytosis | Rousselle et al. 2002 | |
| ACPP | T | Unclear if transcellular or exocytosis | Jiang et al. 2004 | |
| Phage-displayed peptides | 13C (CTANSSAQC) | GI | Low affinity receptor binding | Kenngott et al. 2016 |
| DNPGNET | GI | Macropinocytosis | Yamaguchi et al. 2017 | |
| CSK (CSKSSDYQC) | GI | Receptor-mediated uptake | Kang et al. 2008, Kenngott et al. 2016 | |
| P8 (LETTCASLCYPS) | GI | Receptor-mediated | Higgins et al. 2004 | |
| P25 (VPPHPMTYSCQY) | GI | Receptor-mediated | Higgins et al. 2004 | |
| CTGKSC | GI | Receptor-mediated transcytosis | Fievez et al. 2010 | |
| PAVLG | GI | Receptor-mediated transcytosis | Fievez et al. 2010 | |
| LRVG | GI | Receptor-mediated transcytosis | Fievez et al. 2010 | |
| CKS9 (CKSTHPLSC) | GI | Binding affinity to M cells | Yoo et al. 2010 | |
| RGD | T | Integrin binding | Arap 1998, Koivunen et al. 1995 | |
| RGD-4C | T | binding to αvβ3 and αvβ5 integrin | Koivunen et al. 1995 | |
| NGR | T | aminopeptidase N recognition, αv-integrin binding | Arap 1998, Koivunen et al. 1995, Pasqualini et al. 2000, Curnis et al. 2008 | |
| Lyp-1 | T | CendR pathway | Laakkonen et al. 2002, Roth et al. 2012 | |
| iRGD | T | CendR pathway | Sugahara et al. 2009, Teesalu et al. 2009 | |
| tLyP-1 | T | CendR pathway | Roth et al. 2012 | |
| TT1 | T | Binding to p32 | Paasonen et al. 2016 | |
| F3 | T | NA | Porkka et al. 2002 | |
| iNGR | T | CendR motif specificity to endothelial CD13 | Alberici et al. 2013 | |
Table 2.
Identification and application of BBB shuttle peptides identified from phage display library
| Peptide | Sequence (phage displayed library) | BBB model (in vitro and in vivo) | Transport mechanism | Sanger/(NGS) | Advanced application– (Nano formulations) | References |
|---|---|---|---|---|---|---|
| SGV |
SGVYKVAYDWQH (Ph.D.™-12 Phage Display) |
Co-cultured Endothelial cells and pericytes | N.A. | Sanger (31 clones) | N.D. | (Díaz-Perlas et al. 2016; Sánchez-Navarro et al. 2017) |
| Peptide-22 |
Ac-[cMPRLRGC]c-NH2 (15-mer cyclic phage library) |
CHO cells (h LDLR +) | LDLR-RMT | N.A. | PEG-PLA-peptide22-PTX; PEG-LIP-Peptide22-RGD-DOX. | (Malcor et al. 2012; Zhang et al. 2013a; Chen et al. 2017) |
| B6, B8 | B6: GHKAKGPRK B8: KWKTPKVRV (custom phage-display 9 mer library) |
Purified extracellular domain of hTfR | TfR-RMT | Sanger (43 clones) | N.D. | (Xia et al. 2000) |
| T7 or HAI |
HAIYPRH (Ph.D.™-7 Phage display Peptide Library) |
CEF (hTfR +) | TfR-RMT | Sanger (9 clones from 10th round) | PEG-LIP-T7-TAT-DOX; PEG-LIP-T7-ZL006; PEG-PLGA-T7-BCNU; PEG-PLGA-T7-PTX-BCNU; PEG-LIP-T7-DOX-VCR. | Lee et al. 2001; Zong et al. 2014; Wang et al. 2015b; Bi et al. 2016; Cui et al. 2016; Zhang et al. 2017) |
| THR |
THRPPMWSPVWP (Ph.D.™-12 Phage Display) |
CEF (hTfR +) | TfR-RMT | Sanger (9 clones from 7th round) | 68Ga-THR fallypride-THR | (Lee et al. 2001; Wängler et al. 2011) |
| G23 |
HLNILSTLWKYRC (Ph.D.™-12 Phage Display) |
Immobilized ganglioside GM1 | GM1/GT1b-RMT | Sanger (N.A.) | G23fluor polymersomes | (Georgieva et al. 2012) |
| PepC7 |
CTSTSAPYC (Ph.D.™-C7C Phage Display) |
ICR mice | N.D. | Sanger (12 clones) | N.D. | (Li et al. 2012) |
| CRT |
CRTIGPSVC (Ph.D.™-C7C Phage Display) |
BALB/c mice (i.v. tail vein) | TfR-RMT | Sanger | PEG-PLGA-CRT-PTX | (Staquicini et al. 2011; Kang et al. 2015) |
| GLA GYR |
GLAHSFSDFARDFVA GYRPVHNIRGHWAPG (Filamentous f3-15mer) |
C57Bl/6 mice (In situ Perfusion) | N.D. | Sanger (17 clones) | P3-GLA-LIP | (Van Rooy et al. 2010, 2012) |
| TGN |
TGNYKALHPHNG
(Ph.D.™-12 Phage Display) |
ICR mice | N.D. | Sanger (20 clones) | PEG-PLA-TGN; PEG-PLGA-TGN; PEG-PLA-LIP-TGN-PTX | (Li et al. 2011, 2013, Gao et al. 2012, 2014, Zhang et al. 2013b, 2014; Ma et al. 2014) |
| c-SxTSSTx-c |
C-SYTSSTM-C (Ph.D.™-C7C PhageP |
Sprague-Dawley (SD) rats | N.D. | Sanger (500 clones) | N.D | (Smith et al. 2012) |
| CAGALCY |
CAGALCY (T7-select-415-CX10 C) |
6 Balb/C mice | N.D. | Sanger (200 clones) | CAGALCY-AgNP | (Fan et al. 2007; Toome et al. 2017) |
| #2077 |
RLSSVDSDLSGC (T7-select 10-3b-12 mer) |
Wistar rats | N.D. | NGS | N.D. | (Urich et al. 2015) |
Peptides as carriers to deliver therapeutics into the brain
Challenges of delivery across BBB and mechanisms of transport across BBB
The blood-brain barrier (BBB) acts as a physiological and transport barrier to successful delivery of therapeutic agents into the brain, with approximately 98% of all small molecule drugs and nearly 100% of all macromolecules unable to shuttle across the BBB for treatment of brain diseases. The three special characteristics of BBB, which are the tight-tight junction complex between endothelial cells, efflux transporters, and the vesicle compositions of the brain capillary endothelium, make drug delivery undergo very limited paracellular and cellular transport across the BBB and into the brain parenchyma. To address this challenging problem, tremendous efforts have been made to improve drug delivery to the brain, especially by the transcellular mechanism. Nowadays, peptides and proteins have been identified and engineered to penetrate the BBB through a variety of transport mechanisms (Schwarze 1999, Pardridge, 2006). Peptides, in particular, have received interest as delivery vectors to shuttle therapeutic cargo across the BBB. The concept of BBB shuttle peptides was initially conceived by Dr. William M. Pardridge, with a focus on receptor-mediated (active) peptide transport, to improve drug delivery to the brain (Pardridge, 2006). Here, we summarize findings on peptide mediated shuttling across BBB by two main mechanisms, active and passive transport.
Transporter- and receptor-mediated active transport of peptides
Active transport of peptides shuttle across the BBB is through specific, concentration independent and saturable pathways. For example, endogenous neuropeptides travel freely between capillary and brain tissue mainly by the transporter-mediated pathway. A group of transporter proteins expressed on the plasma membrane of the brain capillary endothelial cells, i.e. organic anion polypeptide transporter and organic cation polypeptide transporter, permit transport of anion and cation-based molecules (e.g. cationic and anionic drugs) (Qosa et al. 2016). The main active pathway involved in the transport of the majority of identified BBB penetrating peptides is receptor-mediated transcytosis (RMT) (Tuma and Hubbard 2003; Pardridge 2012). So far, insulin receptor, transferrin receptor (TfR), and low-density lipoprotein receptor (LDLR) are the main receptors expressed on BBB that mediate shuttling of peptides (and other macromolecules).
Diffusion driven cargo delivery-passive transport
Intracellular and paracellular diffusion can mediate passive transport of small hydrophobic peptides across the BBB. Typically hydrophobic peptides, which have low molecular weight (m.w. < 400 Da) and low density of hydrogen bonding (number of hydrogen bonds ≤ 7), are able to partition into the lipophilic membrane of the endothelium and subsequently shuttle across the endothelium of BBB (Pardridge 2012). Moreover, the molecule conformation (with or without secondary structure) and charge density of peptides also affect the ability of peptides to penetrate BBB.
BBB shuttle peptides as carriers of drug delivery across the BBB
Endogenous BBB shuttle peptide-mediated drug delivery
Endogenous neuropeptides can penetrate the BBB to regulate and maintain homeostasis of the brain functions; subsequently, these peptides can be exploited to facilitate drug delivery. Bradykinin (BK), is a well-characterized endogenous neuropeptide that downregulates the tight-tight junction complex to increase the permeability of BBB, its analog, RMP-7 was coupled with carboplatin to treat glioblastoma patients, which was proceeded to a Phase II clinical trial (Prados et al. 2003). While the clinical trial failed, identification and development of analogs of BK opened the use of other endogenous peptides to shuttle across the BBB. GSH, a three-amino acid endogenous peptide, was used to facilitate the exchange of substances between and bloodstream and brain; GSH transport was thought to be Na+ dependent, the mechanism of transport across the BBB is unclear (Kannan et al. 2000). GSH had been employed to couple with PEGylated liposomes to deliver anti-amyloid agents in a mouse model of Alzheimer’s disease (AD) (Rotman et al. 2015). 2B3-101, a doxorubicin-encapsulated liposome conjugated with GSH demonstrated efficacy in preclinical studies of the brain tumor. This GSH-functionalized formulation was recently investigated in Phase I and Phase IIa clinical trials for safety and efficacy in glioma patients (Gaillard et al. 2014; Maussang et al. 2016). Additionally, opioid peptides also showed enhanced brain uptake in vivo (Lindqvist et al. 2016).
Rational design of BBB shuttle peptides
While endogenous peptides have natural targets for delivery, they are already present in healthy tissues and as a result, do not offer sufficient specificity for BBB targeting in brain diseases. Through rational design, synthesized peptides can mimic the ability of other molecules to transport across the BBB. From sequence alignment of existing protein transduction domains or peptides able to transport across the BBB, motifs or truncated sequences can be identified to generate rationally designed BBB shuttle peptides. The following section will focus on two such cell-penetrating peptides, Tat and Angiopep-2, which have demonstrated great promise as BBB shuttle peptides and have been conjugated to therapeutics in preclinical and clinical studies to improve drug delivery in the brain.
Cell-penetrating peptides (CPPs) as delivery vehicles–Tat
Briefly, cell penetrating peptides (CPPs) are defined as a family of peptides (~747 CPPs) that can be internalized into live cells. Traditionally, CPPs have similar physicochemical properties, i.e. small size, cationic, median length 14 residues and median charge +5 (Kauffman et al. 2015). There are multiple potential mechanisms of CPPs translocation into the intracellular space. There is evidence of both energy-independent and -dependent (i.e. endocytosis) pathways that mediate CPPs internalization (Tyagi et al. 2001; Lindsay 2002; Richard et al. 2003; Nakase et al. 2004; Fittipaldi and Giacca 2005). However, not all CPPs can penetrate BBB due to the limitation of uptake or efflux capability of CPPs. A few CPPs, including SynB1(Rousselle et al 2002), penetratin (Tremmel R et al.2016), and Tat, have shown to increase the permeability of the BBB.
Tat (47-57), an 11-mer peptide (YGRKKRRQRRR), is a truncated version of HIV-1 transactivator protein ‘tat’ (86 amino acids in full length) and has been shown to penetrate BBB by a concentration-dependent, receptor- and transporter-independent protein transduction pathway (Green and Loewenstein 1988; Schwarze 1999). Tat is the most commonly used CPP, either as the drug carrier itself or conjugated with other BBB shuttle peptides to deliver cargos (e.g. small molecule chemo drugs, siRNA, antibody or therapeutic peptides) in animal models of glioblastoma (GBM), AD, Parkinson’s disease (PD), and ischemic brain injury diseases; additionally, Tat has been conjugated to numerous formulations to improve delivery to the brain by traversing the BBB and penetrating into cancer cells (Wu et al. 2015; Nguyen et al. 2017; Shi et al. 2017; Wang et al. 2017).
Rational design BBB shuttling peptide–Angiopep-2
Angiopeps are a family of peptides derived from sequence alignment of aprotinin (protease inhibitor, a ligand of low-density lipoprotein receptor-related protein (LRP)) and Kunitz domain of human proteins (Demeule et al. 2008). Angiopep-2 (TFFYGGSRGKRNNFKTEEY) exhibited the greatest transcytosis ability amongst the Angiopeps, which was 7-fold higher than native aprotinin (aprotinin transcytosis across BBB was 8-fold higher than transferrin). In addition, Angiopep-2 had a very high accumulation in the brain in an animal model (Demeule et al. 2008). Building on these results, Angiopep-2 has been used in numerous delivery systems to improve BBB transport and brain targeting. In particular, Angiopep-2 has been employed to functionalize as carriers to deliver anticancer drugs for the treatment of brain cancer since LRPs are overexpressed by cancer cells. Angiopep-2 significantly enhanced BBB penetration and improved therapeutic efficacy of multiple formulations delivering siRNA, doxorubicin, paclitaxel, or antibody in GBM (Bertrand et al. 2011; Wang et al. 2015c; Li et al. 2016). Angiopep-2 has also been conjugated to paclitaxel (ANG1005) and anti-HER2 monoclonal antibody (ANG4043) to develop the formulations that were investigated in Phase I and II clinical trials for the treatment of breast cancer, lung cancer, and GBM (Régina et al. 2008; Bertrand et al. 2011; Sullivan et al. 2016). Currently, Phase II trials with ANG1005 in GBM patients is ongoing, and studies with ANG4043 demonstrated higher distribution in brain parenchyma and prolonged survival in an animal model of breast cancer (Regina et al. 2015). These findings highlight the attractiveness of Angiopep-2 and overall, rationally designed peptides as drug carriers.
BBB shuttle peptides screened from combinatorial peptide libraries
While rationally designed peptides build from existing peptides and proteins to identify potential motifs, the number of candidates and the design space are limited and may not necessarily provide the complexity to identify and optimize peptides with desired or evolved functionalities for enhanced BBB transport. Alternatively, combinatorial peptide libraries can be used in a high-throughput approach to screen for BBB shuttle peptides. Here, random peptides can be genetically engineered into display systems (i.e. the surface of bacteria, yeast, or phage) or chemically synthesized peptide library. The genetically engineered phage display library or the chemically synthesized peptide library can be “biopanned” against an in vitro or in vivo model of the BBB to screen for peptides that can shuttle across the BBB. This biopanning process is iterated through multiple rounds of screening to identify peptide candidates that demonstrate enhanced penetration through the BBB (Fig. 1). The permutation of amino acids provide complexity, from which peptides “evolved” to shuttle across the BBB can be selected. There have been multiple phage display systems used for BBB penetrating peptides screening in cell line and animal models. Lysogenic filamentous M13 phages, viruses that infect F-pilus containing E. coli, have been engineered to generate random phage libraries and “biopanned” against BBB models to identify BBB shuttle peptides (Lee et al. 2001; Li et al. 2011). In addition, lytic T7 phage library has been employed to identify BBB shuttle peptides (Fan et al. 2007; Urich et al. 2015).
Fig.1.

Strategy to identify BBB shuttle peptides from combinatorial peptide libraries (genetically engineered phage display and chemically synthesized libraries)
The chemically synthesized peptide library is a family or a group of families of peptides that have been designed and synthesized following specific algorithms and chemical synthesis. Typically, mix-and-split method and solid-phase peptide synthesis (SPPS) by F-moc chemistry are used to produce the chemically synthesized peptide library. To prevent degradation, most chemical libraries are synthesized with D-amino acids, along with termini modification, such as N-terminus acetylation and C-terminus amidation. There are several differences between chemically synthesized peptide library and phage display peptide library. First, the chemically synthesized peptide libraries are created by chemical synthesis and purification, whereas phage display peptide libraries are constructed by molecular cloning (i.e. degenerate codons that translate into a library of random peptides are inserted into phage genomic vector). Second, chemical libraries typically consist of a combination of fixed and partially randomized residues. Phage display libraries can be constructed to possess more randomness and thus, larger diversity, than chemical libraries. Finally, a chemically synthesized peptide library can consist of all D-amino acids, whereas phage display libraries mostly use natural L-amino acids. There are limited studies using chemically synthesized peptide library to select BBB shuttle peptides (Teixidó et al. 2005; Guixer et al. 2016); most peptides are identified from rational design or phage display and then subsequently chemically synthesized for future use.
BBB shuttling peptides identified from phage display and their use in nanomedicine
From in vitro panning using M13 phage library, Malcor et al. identified Peptide 22 (P22) and demonstrated that it had strong affinity to LDLR and without competition with endogenous low-density lipoprotein (LDL), which suggests that the identified peptide binds to LDLR at a different location than LDL (Malcor et al. 2012). From this finding, Zhang et al. developed P22-functionalized PEG-PLGA-paclitaxel (PTX) nanoparticles (NPs), which improved BBB penetration and PTX accumulation in GBM compared to controls (Zhang et al. 2013a). In another study, Peptide 22 with cRGD peptide were co-conjugated to liposomes and exhibited enhanced delivery of doxorubicin by overcoming BBB and blood-brain tumor barrier (BBTB) (Chen et al. 2017). Since human transferrin receptor (hTfR) is involved in transcytosis of the BBB, it has been screened and identified T7 or HAI peptide against hTfR (Lee et al. 2001). T7 peptide has been frequently used and conjugated onto different NPs to enhance drug delivery in ischemia, glioblastoma, and ovarian cancer. T7 peptide-functionalized PEG-DSPE liposomes loaded with ZL006 (a drug for ischemic stroke) demonstrated therapeutic efficacy in an animal model (Wang et al. 2015d). Moreover, T7 peptide has been conjugated onto PEG-PLGA NPs (Bi et al. 2016; Cui et al. 2016), DSPE-PEG2000-DA7R NPs (Zhang et al. 2017), and DSPE-PEG2000 liposomes (Zong et al. 2014) to improve biodistribution, brain targeting and efficacy of chemotherapeutics in various animal models of GBM.
In addition to in vitro panning, phage libraries can be screened in vivo (i.e. animal models) to identify phage-displayed peptides that can withstand systemic circulation and the immune system to accumulate at the site of interest. For example, GLA and GYP peptides were identified from a phage 15-mer library administered by cardiac perfusion and were subsequently validated in hCMEC/D3 BBB cell model; GLA and GYP phage clones showed better BBB targeting compared to the wild-type (WT) phage (Van Rooy et al. 2010). However, GLA and GYP peptide functionalized liposomes did not demonstrate stronger binding affinity compared to non-functionalized liposomes. Only when GLA peptide was re-engineered in the structural context of the phage p3 coat protein showed the p3-GLA-conjugated liposomes bind more extensively to hCMEC/D3 cells (Van Rooy et al. 2012). In addition, from in vivo panning, iron mimic peptide CRT was discovered to shuttle across intact BBB via TfR mediated transcytosis (Staquicini et al. 2011). CRT conjugated PEG-PLGA NPs penetrated through a BBB cell model and GBM spheroids than negative controls (Kang et al. 2015). TGN peptide (Li et al. 2011) has been extensively used in multiple formulations of functionalized NPs to deliver small molecules, therapeutic peptides, DNA or imaging agents in the models of AD and GBM (Li et al. 2013; Zhang et al. 2013b, 2014). Using T7 phage library for BBB biopanning in vivo, CAGALCY was identified as the dominant motif. Structure-activity relationship analysis of CAGALCY peptide demonstrated the free carboxylic acid side chain and N-tyrosine as the key pharmacophore of the peptide (Fan et al. 2007). CAGALCY coupled to silver NPs improved their brain homing effect in vivo (Toome et al. 2017). Finally, Urich et al identified RLSSVDSDLSGC by next-generation sequencing (NGS) of T7 display phage library screened against in-vivo BBB model (rat). Here, RLSSVDSDLSGC exhibited 1000-fold higher binding than control wild-type T7 phage in vivo and 10-fold higher distribution in cerebral spinal fluid than the scramble control (Urich et al. 2015).
In summary, T7/HAI peptide was most frequently utilized by multiple groups to transport multifunctional NPs across the BBB and subsequently improve brain tissue penetration and targeting of the therapeutics and imaging agents (Table 2). From these NPs, PEGylated liposomes and PEG-PLGA NPs were most common frameworks with BBB shuttle peptides, along with other functional peptides (e.g. CPPs or tissue targeting peptide–NAP, QSH) to improve the brain delivery of therapeutics in GBM, AD or ischemic disease models (Table 2). Table 2 provides a summary of various phage-displayed BBB shuttle peptides, their method of panning, and applications of these identified peptides as carriers to enhance drug delivery.
BBB shuttle peptide screened from chemically synthesized peptide library
While most efforts with combinatorial screening involve phage display libraries, using chemically synthetic libraries involves screening without being displayed or having the structural context of the phage; peptides selected from chemical libraries circumvent the potential problem of structure-function activity of peptides “taken off” of the phage protein coat. Giralt et al. adapted generic algorithms to design two generations of peptide libraries comprising of a set of physicochemical properties. Peptide 3 (I-MeD-F-P-MeA-MeE-MeF) and peptide 22 (R-W-I-R) were identified and confirmed to efficiently penetrate BBB via diffusion (Teixidó et al. 2005). Later, they also designed and synthesized a peptide library consisting of a 5-mer random library using seven different D-amino acids. Here, several peptide families were hypothesized to shuttle BBB by passive transport or any other mechanisms (Guixer et al. 2016). While initial findings are promising, further studies are warranted to optimize the generation of these chemically synthesized libraries, their functionalities, and understanding their mechanism of transport.
Peptides as carriers to deliver therapeutics across the gastrointestinal (GI) tract
Challenges of delivery across GI tract and mechanisms of transport
Oral delivery is the most frequent route of drug administration, although delivering macromolecules such as peptides and proteins to the gastrointestinal tract (GI) remains challenging (Morishita and Peppas 2006). Despite the large number of proteins and peptides recently discovered, poor oral bioavailability is still a major problem faced mainly due to their physicochemical properties, short half-lives, rapid clearance, and different biological barriers in the gastrointestinal tract. Larger molecular weight compounds (compared to small m.w. drug molecules) and high hydrophilicity (LogP values less than zero (Morishita and Peppas 2006)) contribute to poor drug absorption into systemic circulation, resulting in low bioavailability (most cases below 1% (Zupančič and Bernkop-Schnurch 2017)) and consequently limited therapeutic success.
Drugs and drug delivery systems encounter several physical, chemical and biologic barriers after oral administration en route to their pharmacological target site of action: acidic and enzymatic degradation in the stomach and intestine, mucosal barriers, and limited permeation across the intestinal epithelium (Bernkop-Schnurch 1998; Kompella and Lee 2001; Morishita and Peppas 2006). Exposure to low pH and enzymes may affect the secondary and tertiary structures of peptides and proteins, leading to hydrolysis of peptide bonds (Lee and Yamamoto 1989; Woodley 1994) and loss of function and aggregation. Additionally, the mucus layer on the GI epithelium hinders the diffusion of drugs and macromolecules (Larhed et al. 1997, 1998; Boegh et al. 2015).
The GI tract exhibits site-specific absorption depending on the physicochemical properties of small molecule and macromolecule drugs and the local differences such as pH, enzyme activity, mucosal thickness, transit time, and surface area (Kompella and Lee 2001). Several factors limit the absorption of proteins and peptides through the stomach, such as the harsh acidic environment, low surface area, and pepsin degradation (Kompella and Lee 2001). More than 90% of nutrients (carbohydrates, proteins, lipids, water, vitamins, and minerals) are absorbed by the small intestine, while the rest is absorbed in the stomach and large intestine (Renukuntla et al. 2013). The intestinal barrier contains a monolayer of epithelial cells, tight junctions, and a mucus barrier preventing direct access by microorganisms to the intestinal mucosa (Langguth et al. 1997; Goldberg and Gomez-Orellana 2003; Pelaseyed et al. 2014). The epithelial layer contains mostly enterocytes with tight junctions that serve as the primary physiological barrier. Additional intestinal cells include goblet cells, M cells, dendritic cells (DC), lysozyme secreting Paneth cells, intraepithelial lymphocytes and hormonesecreting endocrine cells (O’Neill et al. 2011; Renukuntla et al. 2013; Pelaseyed et al. 2014; Peterson and Artis 2014). M cells and DCs are primarily located in the epithelium of Peyer’s patches, and are able to deliver proteins and peptides from the lumen to the underlying lymphoid tissues, as well as induce immune responses. Due to the high endocytic ability of M cells and antigen-presenting characteristics of DCs, such cells can be exploited as a potential route for delivery of proteins and peptides (Renukuntla et al. 2013; Vela Ramirez et al. 2017). Therefore, they are potential targets for the development of new intestinal vaccines. In association with the apical membrane of enterocytes is the glycocalyx, a glycoprotein and polysaccharide layer with a thickness of 400-500 nm acting as a filtering barrier to certain viruses, bacteria, and particles into the underlying plasma membrane (Frey et al. 1996). Upon oral administration, molecules must traverse across the intestinal lipophilic membrane before entering the systemic circulation.
The organization of the intestinal mucosa allows molecules to cross the cellular barrier via paracellular or transcellular routes of absorption (Pauletti et al. 1996). The factors governing mechanisms of transport depend mainly on the physicochemical properties of molecules (Renukuntla et al. 2013). Due to their general hydrophilic nature, most peptides will not likely partition across the lipophilic membrane and passively diffuse (Camenisch et al. 1998). For paracellular transport, the pore size ranges from 10-50 Å (Morishita and Peppas 2006) and thus, precludes the absorption of most peptides via this route (Rubas et al. 1996). Other possible mechanisms of peptide translocation across the intestinal epithelium are endocytosis (Agarwal 2001) and active carrier mediated transport (Bastian et al. 1999). Although most peptides are poorly bioavailable by the oral route of administration, some were reported to resist proteolytic degradation and were absorbed in the GI tract at sufficient amounts to exert pharmacologic effects (Morishita et al. 1993; Tozaki et al. 1998).
Considerable effort has been made to understand the mechanisms governing gastrointestinal absorption of proteins and peptides. Indeed, extensive knowledge has been gained in the past few decades about macromolecular drug absorption and delivery strategies to overcome this selective barrier (Walter et al. 1996; Morishita and Peppas 2006; Järver et al. 2010; Khafagy and Morishita 2012; Wang et al. 2015b; Aguirre et al. 2016; Lundquist and Artursson 2016; Moroz et al. 2016; Nielsen et al. 2017).
Current strategies to improve mucosal permeability and stability of peptides in the GI tract include the use of permeation enhancers, enzyme inhibitors, polymers, mucoadhesive systems, liposomes, and other nanoparticle-based formulations (Scott Swenson and Curatolo 1992; Tozaki et al. 1998; Shah et al. 2002; Renukuntla et al. 2013; Yun et al. 2013; Hwang and Byun 2014). However from the translational and clinical perspective, their safety and efficacy remain to be proven, especially where long-term therapies should be required. For example, an enzyme inhibitor might create unbalanced digestion of proteins and peptides and a potential feedback regulation resulting in increased protease secretion (Shah et al. 2002; Morishita and Peppas 2006). Furthermore, oral delivery strategies directed only to overcome the mucus and enzymatic barrier will not address problems where systemic delivery is desired. Increasing oral bioavailability also requires membrane permeation across the gastrointestinal mucosa.
Application of peptide-based strategies to the delivery of therapeutic molecules
While advances using peptides as therapeutics are promising, peptides have more recently been developed as transport carriers to enhance targeted delivery and bioavailability of compounds with poor gastrointestinal absorption. Due to their small size and complexity, peptides are specific ligands against cell targets and help transport and internalize attached cargo molecules across the gastrointestinal mucosa.
Rationally designed CPPs as delivery vehicles
Rationally designed CPPs have been studied to improve gastrointestinal absorption of drugs. Indeed, various studies were performed to improve intestinal insulin absorption with different CPPs and delivery strategies. A study with conjugated insulin and Tat peptide revealed an increased absorption 6 to 8-fold of the insulin-CPP conjugate when compared to normal insulin in-vitro across a Caco-2 cell monolayer (Liang and Yang 2005). Another study evaluated intestinal insulin absorption in rats with coadministration of six, eight or ten arginine residues D-peptides (R6, R8, R10) and D- and L-forms of R6 through an ileal loop. Insulin absorption increased in a dose-dependent manner, and D-R8 presented the greatest increase in BA (relative bioavailability to the subcutaneous route) with a 35-fold increase compared to insulin solution. Also, D-R6 resulted in up to a 13.7-fold increased insulin absorption when compared with L-R6, suggesting that a higher metabolically stable form of oligoarginine induced higher insulin absorption across the ileum due to lower enzymatic degradation (Morishita et al. 2007). Later, the same research group investigated the mechanisms involved in the absorption improvement of insulin when coadministered with oligoarginine peptides. In a permeation study of D-R6, L-R6 oligoarginine in isolated rat intestinal epithelial membranes, this CPP internalization showed to be mediated by adsorption to proteoglycans in the cell membrane surface, followed by transduction via an energy-dependent pathway (Kamei et al. 2008b). Furthermore, in situ intermolecular binding was analyzed by surface plasmon resonance (SPR)-based binding assay using a rat intestinal loop. From 16 different peptide drugs tested, only gastrin, insulin and glucagon-like peptide-1 (GLP-1) bound to D-R8 and had improved intestinal absorption by the coadministration with the CPP. Conversely, the intestinal absorption of peptide drugs that did not bind to D-R8 was not affected in the presence of the CPP, confirming that intermolecular binding is one mechanism governing the intestinal absorption enhancing effect of drugs and macromolecules by the CPP (Kamei et al. 2009). In addition, electrostatic interactions between the positively charged arginine residues and the negatively charged proteoglycans onto the cell membrane surface play a role in CPP cell internalization (Renukuntla et al. 2013). Insulin intestinal absorption was further evaluated with the coadministration of R8 oligoarginine and hydroxypropyl-β-cyclodextrin (HP-β-CD) both in vitro in a Caco-2 cell monolayer and in vivo orally in diabetic rats. The in vitro transcytosis assay indicated that insulin-HP-β-CD- R8 improved insulin effective permeability with a 9-fold increase compared to native insulin, a 7-fold increase of insulin-R8, and a 2-fold increase in effective permeability of insulin-HP-β-CD formulation when compared to native insulin. In vivo studies demonstrated a 19% and 35% decrease in blood glucose levels 2 hours following oral administration of insulin-R8 and insulin-HP-β-CD-R8 formulations, respectively (Zhang et al. 2012). Another study of in situ loop administration of insulin in rat ileal segments with coadministration of various types of CPPs demonstrated a significant increase in insulin bioavailability when coadministered with D-R8, L-penetratin, L-pVEC, and L-RRL helix. L-penetratin showed the most significant enhancement effects for ileal insulin absorption among the CPPs used, with a 27.5-fold increase in bioavailability compared to insulin administration alone. Interestingly, the absorption enhancement of L- and D-forms of the evaluated CPPs varied among peptides. R8 D-form had a 30-fold higher bioavailability compared to the L-form, whereas L- penetratin, L-pVEC, and L-RRL helix, despite their limited stability. They also showed higher bioavailability compared to their D-form (i.e. isomer), indicating that chirality of a molecule might impact the CPPs intestinal insulin absorption enhancement effect (Kamei et al. 2008a). To better understand the enhancing mechanism exerted by penetratin, an in-depth study was performed to investigate the pharmacological effects of orally delivered insulin in combination with L- or D-penetratin in mice and rats. Interestingly, D-penetratin showed better efficacy in decreasing blood glucose levels and increasing insulin absorption, contradicting previous results. Nevertheless, the degradation kinetics of L-penetratin showed to be much faster in rat intestinal fluid than the D isomer, suggesting that under harsh enzymatic conditions such as the rat intestinal fluid, the D- isomer has higher stability and half-life to exert its function (Nielsen et al. 2014). Nevertheless, these findings demonstrate the relevance and impact of the design of in vitro and in vivo models in experiments results. Another rational peptide-based strategy to increase oral bioavailability of insulin involved the use of insulin-loaded PLGA nanoparticles surface-modified with cell penetrating peptides (R8, Tat, penetratin) and a secretion peptide (Sec), which is hypothesized to promote the exocytosis of the cargos. In vitro permeation studies across Caco-2 monolayers indicated a 2-fold increase in the apparent permeability coefficient of the nanoparticles co-modified with Sec and penetratin (Sec-Pen-NPs) compared to the nanoparticles modified only with penetratin (Pen-NPs). R8 and Tat peptide-functionalized nanoparticles did not show significant differences across the experiments. In addition, in vivo insulin relative bioavailability after ileal segments administration of Sec-Pen-NPs showed a 1.7-fold increase compared to Pen-NPs in rats (Zhu et al. 2015).
CPPs are attractive shuttling vectors for various therapeutic cargos, in particular the delivery of nucleic acids. Gene therapy involves the delivery of nucleic acids to target cells, including plasmid DNA (pDNA), CRISPR-Cas systems, and RNA interference (RNAi) systems. Vectors for gene therapy must demonstrate high uptake by the target cell and be able to encapsulate the desired DNA (Zuris et al. 2014). Therefore, CPPs are potential candidates to improve uptake for oral delivery of gene therapies. To be administered orally, gene delivery vectors need to resist the gastrointestinal milieu and efficiently transfect or traverse the mucosal epithelium (Goldberg and Gomez-Orellana 2003; O’Neill et al. 2011). The use of CPPs in oral gene delivery has been investigated using self-nanoemulsifying drug delivery systems (SEDDS). SEDDS loaded with conjugated lipidized HIV-1 Tat-protein and oleoyl chloride (TAT-OL) demonstrated a 1.5-fold improved mucus diffusion compared to unloaded nanoemulsions, and 2.3- and 2.6-folds increase in Caco-2 cells uptake after 2 and 4 hours, respectively. A 1.7-fold improved transfection efficiency for pDNA compared to Lipofectin on HEK-293-cells was also observed, indicating that these CPP-conjugated lipid and polymer-based systems are a potential alternative for mucosal gene delivery (Mahmood et al. 2016).
Phage display to identify peptides for targeted delivery
Although CPPs exhibit delivery efficacy to the GI, their non-specificity still presents a major issue. Strategies using CPPs are non-selective and uptake is feasible in any tissue. To avoid this problem and achieve peptide-mediated targeted delivery, efforts have centered on phage display technology to identify cell-specific peptide ligands. Phage display technique can identify novel selective peptides with specific functions, screened from highly diverse peptide libraries. It has been demonstrated that M13 bacteriophage can traverse the gastrointestinal barrier selectively. An in vivo study with oral administration of a 7-mer random peptide M13 phage library to rats identified a number of sequences with homologies with HIV gp120 that were able to cross the gastrointestinal mucosa (Duerr et al. 2004). Further sequence analysis of additional phage clones could potentially elucidate additional peptide sequences and motifs that can selectively cross the intestinal barrier. Likewise, a 7-mer M13 phage library screened in vivo orally in mice identified 77 different peptide sequences that translocated the intestinal mucosa. Analysis of amino acid frequencies per position, hydrophobicity, and BLASTP search for protein homology did not show specificity, motifs, physicochemical patterns or relevant proteins homology in any of the isolated sequences (Hamzeh-Mivehroud et al. 2008). A single round of phage selection performed in this experiment can partially explain the lack of correlation between the identified sequences, since typically multiple sequential rounds of panning are necessary to collapse peptide sequences with high affinity to the target ligand. Recently, high-throughput sequencing technologies such as next-generation sequencing (NGS) of phage libraries have been applied to significantly improve the selection of ligands by detecting low abundant clones and high copy numbers of clones without many rounds of selection (’T Hoen et al. 2012; Liu et al. 2015; Matochko and Derda 2015). A novel cyclic peptide showed increased uptake and transcytosis of peptides and proteins upon intestinal administration. The cyclic peptide, CTANSSAQC (13C) was identified from a 7-mer cyclic M13 phage display library screening in vivo, and could be rapidly transported across the intestinal mucosa and targeted distinct intestinal cells such as goblet cells and DCs; this suggests 13C is a potential candidate to improve mucosal delivery of macromolecules for oral immunization (Kenngott et al. 2016). Recently, intestinal absorption of cyclic peptides was further tested in a study that evaluated permeability of a cyclic 7-mer M13 phage library against a Caco-2 cell monolayer and mice intestinal epithelium in an in situ closed loop model. Among three potential hits, the peptide DNPGNET (DNP-phage) demonstrated greatest permeability across a Caco-2 cell monolayer and mouse intestinal epithelium, with 37.4-fold and 620-fold increased permeability at 30 min, respectively (compared to control phage). A macropinocytic mechanism of uptake was suggested, due to inhibition of DNP-phage uptake in cells at low temperature and in the presence of a macropinocytosis inhibitor (Yamaguchi et al. 2017).
As previously described, the intestinal epithelium anchors distinct specialized cells, such as goblet cells, M cells, and DCs. Goblet cells are mucus-secreting cells, thus creating a barrier against the absorption of drugs and macromolecules. Hence, few studies have been conducted targeting goblet cells to enhance oral delivery of macromolecules. An in vivo phage display experiment with a cyclic 7-mer random peptides library administered orally to rats was able to identify the CSKSSDYQC (CSK) peptide as a high affinity ligand to goblet cells, demonstrating that the CSK peptide could be transported across the intestinal mucosal barrier via goblet cells via specific intestinal mucosal binding (Kang et al. 2008). Indeed, it was demonstrated that goblet cell-targeting insulin-loaded nanoparticles prepared with modified chitosan and functionalized with CSK peptide showed significant increase in cell uptake in an in vitro Caco-2/HT-29-MTX co-culture model compared to unmodified nanoparticles. However, mucus produced by HT29-MTX cells partially decreased the uptake of both peptide-functionalized and unmodified nanoparticles. Additionally, in vivo studies in rats demonstrated a 1.5-fold increased relative bioavailability of CSK peptide modified nanoparticles compared with unmodified administered by intestinal loops (Jin et al. 2012). Similar results have been reported for exenatide-loaded chitosan-CSK functionalized nanoparticles in Caco-2/HT-29-MTX co-culture monolayer and orally administered mice, with a 1.7-fold increase in bioavailability of peptide-functionalized compared to unmodified nanoparticles (Li et al. 2015b). Despite the great relevance of these studies in enhancing insulin and exenatide oral absorption via goblet cell targeting ligands, further studies are necessary to evaluate the potential of goblet cell targeting delivery strategies.
Peptide-mediated targeted delivery to M cells has also been investigated for potential oral vaccine delivery. A 12-mer random peptide M13 phage library screened in vivo in rats by a closed ileal loop model identified lead ligands with high binding activity to M cells and Peyer’s patch tissue, peptides P8 (LETTCASLCYPS) and P25 (VPPHPMTYSCQY). In addition, in a mouse intestinal ileal loop model, P25-D peptide-coated nanoparticles co-localized with M cells in Peyer’s patches (Higgins et al. 2004). An in vitro screening of a cyclic 7-mer random peptide T7 phage library against a human follicle-associated epithelium (FAE) model containing both Caco-2 and M cells identified peptides with high affinity to M cells (CTGKSC, PAVLG and LRVG). The study demonstrated enhanced phage transport across M-like cells of CTGKSC and LRVG sequences 3- and 14-fold, respectively, when compared to the library. When conjugated to the surface of PLGA nanoparticles, 4- and 8-fold increased transport across co-cultures was observed, respectively when compared to non-conjugated particles (Fievez et al. 2010). Similarly, the CKSTHPLSC (CKS9) peptide sequence was selected by biopanning against an in vitro M cell co-culture model, and its ability to facilitate transport was validated by conjugation to chitosan nanoparticles (CKS9-CNs). The CKS9-CNs showed 1.5-fold enhanced transport across the M cell model in a transcytosis assay, and increased accumulation in Peyer’s patch regions in a closed ileal loop assay in rats when compared to non-conjugated particles (Yoo et al. 2010). An interesting strategy to deliver an oral anthrax vaccine involved using Lactobacillus acidophilus expressing the Bacillus anthracis protective antigen (PA) fused to a dendritic cell (DC) targeting peptide. Effective immune responses against B. anthracis were observed with a 75% survival rate compared to 25% survival rate of mice vaccinated with PA fused to a control peptide (Mohamadzadeh et al. 2009). These studies suggest that peptide mediated transport is a promising strategy to enhance oral efficacy of vaccines or vaccine-loaded nanoparticles across the intestinal mucosal barrier.
Peptides as carriers for tumor targeting and penetration
Challenges of delivery into tumors
The therapeutic efficacy of clinically available anticancer drugs is limited by their poor penetration into tumor tissues because of the use of passive drug delivery vehicles (Ruoslahti 2012). For systemic delivery, a therapeutic agent must navigate through three major transport pathways before reaching cells in the tumor bed: (1) vascular transport, (2) transvascular transport, and (3) interstitial transport (Jain et al. 2001). In solid tumors, many therapeutic drugs penetrate only 3-5 cell diameters from the blood vessels, leading to reduced efficacy and the development of drug resistance (Minchinton and Tannock 2006; T.W. Hambley 2009). This low penetration is paradoxically due in part to the Enhanced Permeability and Retention (EPR) effect, which results from the leaky tumor vasculature and dysfunctional lymphatic system (Jain 1999; Heldin et al. 2004). The EPR effect is based upon macromolecules able to escape the leaky vasculature and remain in the tumor bed due to abnormal and poor drainage of lymphatics within the tumor. However, the leaky tumor vasculature and dysfunctional lymphatic system results in enhanced interstitial fluid pressure in the tumor microenvironment; as a result, coupled with low vascular flow, convective transport through the tumor extracellular matrix (ECM) is poor and leaves diffusion as the primary mode of drug transport, which limits drug penetration (Jain, 1999). Additionally, fibrosis can further reduce diffusion of drugs and macromolecules through tumors (Olive et al. 2009).
On the other hand, coupling a targeting moiety onto the drug delivery vehicle that specifically binds to the target site in tumors could enhance the delivery of diagnostic and therapeutic agents into tumors (Ruoslahti 2012). Conjugation of a specific ligand onto a drug will improve tumor targeting and internalization, potentially resulting in greater therapeutic efficacy and reduced side effects in the body (Ruoslahti et al. 2010; Ruoslahti 2012; Yao et al. 2016). Targeting ligands can be an antibody, a peptide or a natural ligand of a receptor preferentially expressed in tumors (Ruoslahti 2012). In particular, peptides are an attractive class of targeting ligands able to better penetrate tissues to reach target cells. They are small in size, easy to synthesize and typically non-immunogenic. Peptides can be easily functionalized on nanoparticle carriers, and the multivalent presentation of a peptide on a nanoparticle carrier provides high avidity for the target (Ruoslahti 2012). Peptides can bind to numerous targets overexpressed and/or specific to tumors, including but not limited to integrins, fibrin deposits, and tumor antigens (Ruoslahti 2017). The targeted receptors are present either on tumor vessels, cancer cells, or both; the availability of these receptors dictate active transport and delivery of molecules including peptides in tumors (Ruoslahti et al. 2010).
In the tumor microenvironment, the first barrier for systemic drug delivery is the tumor vasculature. Compared to normal blood vessels, they are heterogeneous with variable diameters, tortuosity, and leakiness and have aberrant expression of various cell surface and extracellular matrix proteins (Ruoslahti 2002; Ruoslahti et al. 2010). During tumor progression, dysregulated angiogenesis leads to expression of these receptors (Hanahan and Folkman 1996; Alitalo and Carmeliet 2002). By targeting markers present on tumor vessels for drug delivery, such as tumor endothelial markers, delivery of drugs and nanocarriers is not dependent on the leakiness of the vasculature, and there is no need for tumor penetration for therapeutic delivery. Regardless, targets present on tumor vasculature can facilitate transport across the vessel wall to get into the tumor parenchyma (Ruoslahti et al. 2010). After extravasation from the tumor vasculature, carriers must be able to penetrate throughout the tumor interstitium to reach their desired target, such as cancer cells or cells supporting tumor progression (e.g. cancer-associated fibroblasts, specific immune cells). Due to the leaky vasculature that enables passage of plasma proteins, cells, and other vascular components into the tumor microenvironment and dysfunctional lymphatics in the tumor, there is an enhanced interstitial fluid pressure that can reach similar values of the mean vascular pressure (Jain 1988). The negligible pressure difference results in the loss of convective flow and restricts delivery of molecules to diffusion. As a result, even after extravasation from tumors, it remains a challenge to diffuse and bind to cancer cells or other target cells distal to the vasculature.
Tumor-targeting and tumor-penetrating peptides as carriers
However, there has been tremendous progress to use peptides not only as targeting ligands against blood and lymphatic vasculature for tumor targeting, but recent work suggests peptides can facilitate penetration throughout the tumor parenchyma for deep penetration of drugs and nanocarriers for enhanced therapeutic efficacy. The following sections will discuss the development of tumor-targeting and penetrating peptides through rational design and phage display.
Rationally designed CPPs as delivery vehicles
To traverse multiple barriers of the tumor microenvironment for targeted delivery, peptides have been rationally designed to mimic the binding ability of viruses. Short cationic cell-penetrating peptides (CPPs) derived from these viral proteins carry the membrane-translocating sequence, which can deliver attached payloads into mammalian cells without specific receptors (Green and Loewenstein 1988). In particular, Tat peptide, a cationic peptide that mimics Tat protein and has been used for delivery across the BBB and GI tract (see earlier sections), has also been exploited for delivery and uptake by tumor endothelial cells for vascular targeting. Tat-functionalized dendrimers (Yan et al. 2015) and Tat fused to apoptotic peptides and transcription factor inhibitors have been used to inhibit angiogenesis and subsequent tumor growth (Boohaker et al. 2012). As stated before, CPPs are taken up into nearly all types of cells. To achieve tumor-selective delivery and minimize non-specific uptake, a CPP derivative with cationic and anionic modules were linked by a tumor protease-sensitive linker (Jiang et al. 2004). An inhibitory domain made up of negatively charged residues were fused to the CPPs via peptide linker sensitive to matrix metalloproteinases. This resulting activatable CPPs (ACPPs) would have its positive charge shielded until MMPs secreted by the tumor microenvironment cleave the peptide linker and effectively exposes the cationic domain (CPP) to bind and enter the cancer cell for specific cell uptake (Jiang et al. 2004). While most rationally designed peptide carriers achieve successful cell uptake, they do not necessarily address the challenges of transvascular transport and transport through the tumor interstitium. Due to their highly positive net charge, these rationally designed peptides would bind to cells surrounding the perivasculature and/or negatively charged extracellular matrix of tumors and be unable to penetrate through the tumor microenvironment, thereby limiting their attractiveness for tumor-penetrating delivery (Teesalu et al. 2013). Also, while Tat peptide translocates into cells, it is unclear whether these positively-charged peptide carriers achieve transcellular transport and exocytosis for tumor-penetrating delivery.
Phage display to identify peptides targeting tumor vasculature
Prior to targeting cancer cells, targeting selective receptors overexpressed in the tumor vasculature will concentrate and localize drug delivery systems at the tumor site and may improve tumor accumulation (Ruoslahti et al. 2010). Peptide-mediated targeting to tumor vasculature has involved the development and use of rationally designed peptides and functionally-screened peptides. Screening phage-displayed peptide libraries in vitro and in vivo would allow for combinatorial screen of a large number of peptides to identify “evolved” peptide ligands with specificity to tumor vasculature, while at the same time serve as a tool to identify novel vascular markers that are accessible to targeted delivery (Pasqualini and Ruoslahti 1996).
An early study validated the method of in vivo phage screening by isolating peptides that home selectively to the tumors of a nude mice bearing human breast carcinoma xenograft (Arap 1998). They yielded two peptide motifs RGD and NGR, which were known to bind to integrin (Koivunen et al. 1995). Coupling of the peptides to anticancer drug doxorubicin enhanced the drug efficacy against human breast cancer xenografts in nude mice and also reduced its toxicity (Arap 1998). The affinity of NGR for integrins is about three orders of magnitude less than that of RGD peptides (Ruoslahti 1996). However, the homing ratio of phage displaying NGR motif was three times that of the RGD-4C phage, suggesting both the peptides bind to different receptors in the tumors (Arap 1998). Later, it was understood that the NGR peptides recognizes aminopeptidase N (Pasqualini et al. 2000), and v-integrins after a chemical alteration (Curnis et al. 2008).
Many integrins recognize RGD sequence as a binding site in their extracellular matrix ligands (Ruoslahti 1996). The sequences surrounding the RGD motif and the confirmation of the peptide plays a significant role in the binding specificity of the RGD peptide (Ruoslahti 1996). For example, RGD-4C peptide (CDCRGDCFC) binds selectively to the αvβ3 and αvβ5 integrins (Koivunen et al. 1995). In addition, the high expression of αv-integrins in the tumor vasculature can be accessed by peptides containing the RGD integrin recognition motif (Pierschbacher and Ruoslahti 1984; Eliceiri and Cheresh 2001; Ruoslahti 2002, 2003). RGD-based active targeting has been successfully and extensively used to deliver small molecule drugs, biologicals, imaging agents, viruses and nanoparticles to tumor vasculature. Studies exploring development of tumor therapeutics targeting tumor vasculature has been summarized in Table 3.
Table 3.
Development of tumor therapeutics targeting tumor vessels
| Peptide | Function | Action | Tumor model | Reference |
|---|---|---|---|---|
| RGD (CDCRGDCFC) | Phage displayed peptides | Binding to αv integrins | Malignant melanoma and breast carcinoma | (Pasqualini et al. 1997) |
| RGD-4C (CDCRGDCFC) | Doxorubicin coupled to RGD-4C | Binding to human αv integrins: Binding to both tumor vessels and tumor cells | Mice with human MDA-MB-435 breast carcinoma xenograft | (Arap 1998) |
| NGR (CNGRC) | Doxorubicin coupled to NGR | Binding to human blood vessel | Mice with human MDA-MB-435 breast carcinoma xenograft | (Arap 1998) |
| RGD | RGD-Adenovirus | Utilization of CAR to bind to cell, followed by RGD mediated cellular internalization | (Wickham 2000) | |
| F3 (KDEPQRRSA RLSAKPAP PKPEPKPKK APAKK) CGNKRTRGC (LyP-1) |
Peptide-Quantum dot (ZnS-capped CdSe qdots) | Binds to blood vessels and tumor cells Binds to lymphatic vessels and tumor cells |
MDA-MB-435 tumor xenograft | (Akerman et al. 2002) |
| ACDCRGDCFCG | Fusion with tumor necrosis factor- α (TNF) | Binding to αv integrin | C57BL/6 mice | (Curnis et al. 2004) |
| RGD | RGD-NP encapsulating doxorubicin | Binding to αvβ3 tumor vasculature | pancreatic and renal cell orthotopic models | (Murphy et al. 2008) |
| F3 peptide | Alexa-488 linked cisplatin loaded nanoparticle with F3 F3-FITC-NP |
Tumor endothelial cells | Subcutaneous injection of SKOV3, A2780-GFP, and DsRED HEY1, of either C57Bl6 or nu/nu mice | (Winer et al. 2010) |
These αv-integrins are highly expressed in tumor endothelium, and their level of expression increases in more malignant tumors (Ceramide et al. 2000). αv-integrins are often overexpressed in cancer cells along with tumor vessels (Ruoslahti et al. 2010). Therefore, peptides (and the cargo they carry) that recognize αv-integrins are able to achieve two-step targeting, concomitantly targeting tumor vasculature and cancer cells of the tumor for enhanced delivery and targeting (Ruoslahti et al. 2010).
Phage display to identify tumor-penetrating peptides
Although RGD-based targeting has been successfully used in numerous studies, crossing the vessel wall and penetrating into the tumor parenchyma against elevated interstitial fluid pressure has been a long-standing challenge (Jain 1999; Heldin et al. 2004). Using in vivo phage display screening procedure, Laakkonen et al. detected a peptide that specifically accumulate in tumor lymphatics (Laakkonen et al. 2002). It was later discovered that Lyp-1 (CGNKRTRGC) primarily accumulates in tumor-associated macrophages; endothelial cells of tumor blood vessels and tumor cells also bind LyP-1, but there is less accumulation than observed in tumor macrophages (Sha et al. 2015). However, the mechanism of LyP-1 penetration through the tumor parenchyma is still unclear (67). There is another class of targeting probes having the ability to penetrate through the tumor tissue, called tumor-penetrating peptides, in which the peptide binds both to the tumor endothelium and the tumor cells (Ruoslahti et al. 2010).
Teesalu et al. introduced a class of peptides having the ability of cell internalization as well as tissue penetration (Teesalu et al. 2009). These peptides have been screened using phage display. It was found that they share a R/KXXR/K motif with the C-terminal domain of VEGF-A165 and some semaphorins. The R/KXXR/K motif has a binding tendency towards Neuropilin-1 (NRP-1), when exposed at the C terminus of the polypeptide chain (Teesalu et al. 2009). Therefore, the binding and internalization mechanism has been termed as the C-end rule or CendR (Teesalu et al. 2009). The overexpression of NRP-1 in tumor cells made this approach to be applicable for drug delivery in particular to tumor tissue.
Soon, Sugahara et al. reported a tumor homing peptide, internalizing RGD (iRGD), which can penetrate deep into the tumor parenchyma (Sugahara et al. 2009). Whereas conventional RGD peptides only delivered the conjugated drug cargo to the blood vessels, iRGD coupling could bind to the tumor vessel and extravasate into the tumor parenchyma. The mechanism of tumor homing of iRGD peptide is a three-step process (Sugahara et al. 2009). First, the RGD motif facilitates binding to αv-integrins, which is overexpressed on tumor endothelium. Other cells in tumors also express these integrins, which is the probable cause for the spreading of the peptide within tumor tissue (Sugahara et al. 2009; Ruoslahti 2017). Second, a proteolytic cleavage then exposes the CendR motif (R/KXXR/K) binding motif. The protease has not been identified, but speculated to be a furin or furin-like enzyme because of their preferred recognition for CendR motif (Ruoslahti 2017). However, any protease having the ability to cut after a basic residue can activate iRGD. Third, the CendR motif binds to either NRP-1 or NRP-2, which mediates penetration into tissue and cells (Sugahara et al. 2009). NRP is present and expressed by healthy cells; however, overexpression of NRP and v-integrins enables tumor selectivity. The interaction activates the CendR endocytotic/exocytotic transport pathway (Sugahara et al. 2009; Teesalu et al. 2009).
The CendR pathway starts with internalization via an endocytic pathway similar to macropinocytosis. The CendR pathway is receptor-initiated and nutrient dependent; increased activity of CendR pathway has been observed by nutrient deprived cells and tissues (Pang et al. 2014). The endocytic vesicles that form during the CendR pathway triggering is large, and can accommodate a considerable volume of extracellular fluid. If that fluid contains a drug, or any payloads of nanoparticle size range will be swept into the pathway (Ruoslahti 2017). Therefore, the CendR pathway is able to even transport co-administered payloads with peptides as well as the covalently attached payloads (Sugahara et al. 2010). Transportation of co-administered payload deep into the tumor tissue overcomes the need to achieve active targeting or functionalize drug carriers with targeting ligands and the possibility of low number of accessible receptors in the tumor tissue. While the initial endocytic process is well understood, limited work has been done to understand the mechanism of cell-to-cell transport (Ruoslahti 2017). The spreading of iRGD payloads in tumor tissue suggests the transport of the payload from one cell to another. There are two ways in which this can be explained: exosome transport which suggests that the CendR payload is protected by a biological membrane like exosome, and transportation through micro- or nanotubes (Ruoslahti 2017). Transportation though these tube conduits are more effective than the exosome transport (Connor et al. 2015), which explains the effectiveness and speed of the transportation by the CendR pathway (Sugahara et al. 2010).
Retrospective examination of the peptide sequence LyP-1 shows that it also contains a CendR motif and uses the CendR pathway for its transportation through tumor tissue (Roth et al. 2012). The primary receptor for LyP-1 is p32, which is expressed at the cell surface of highly activated cells such as tumor endothelial cells, tumor macrophages, and tumor cells. During homeostasis, p32 is intracellular in normal cells; as a result, Lyp-1 peptides have unique cell-surface targeting and selectivity for tumor cells (Fogal et al. 2008). A truncated form of Lyp-1 (CGNKRTR; tLyP-1) is also a tumor-specific CendR peptide (Roth et al. 2012), which has the characteristics complementary to those of iRGD (Ruoslahti 2017). Generally, it has the tendency to bind to all tissues as NRP-1 is not specific to tumor vessels (Teesalu et al. 2009); however, NRP-1 may be overexpressed in tumors that may confer the specificity (Bagri et al. 2009; Roth et al. 2012). Recently, an improved LyP-1 mimicking peptide, TT1 (CKRGARSTC) has been identified based on its binding affinity towards p32 (Paasonen et al. 2016). Tumor specificity of TT1 has been validated from its ability to home p32-expressing breast tumors in mice (Paasonen et al. 2016).
Another tumor-homing peptide screened from phage library is F3 (KDEPQRRSARLSAKPAPPKPEPKPKKAPAKK), which contains the CendR sequence (Porkka et al. 2002). Whether the peptide act as a CendR peptide has not been determined, However, it shows cell internalization (Porkka et al. 2002). Alberici et al. designed a new tumor-penetrating peptide using the NGR tumor-homing motif sequence, and termed it as iNGR (CRNGRGPDC) (Alberici et al. 2013). It contains the CendR motif (RNGR) which has specificity towards the endothelial CD13, and the sequence is embedded in the iRGD framework to impart its penetration in tumor tissue (Alberici et al. 2013). iNGR induced greater tumor penetration for both coupled nanoparticles and co-administered compounds than NGR (Alberici et al. 2013). These findings show the promise of peptides not only binding to cell surface targets but actively stimulating downstream cellular processes to achieve true tumor penetration to ultimately reach and kill distant cancer cells. A large number of papers have used tumor-penetrating peptides to target payloads either coupled to or co-administered with the peptide, which are tabulated in Table 4.
Table 4.
Administration of tumor-penetrating peptides for diagnosis and therapeutic application
| Peptide | Function | Action | Tumor model | Reference |
|---|---|---|---|---|
| iRGD | Peptide-functionalized Fluorescein-labeled |
Binding to tumor vessel and then tissue penetration | Xenografts of prostate cancers PC-3, PPC1, pancreatic cancer MIA PaCa-2, breast cancer BT474 | (Sugahara et al. 2009) |
| CREKA LyP-1 |
Fluorescein (FAM)-labeled CREKA-abraxane FAM-LyP-1-Abraxane |
Binding to tumor blood vessels | MDA-MB-435 human cancer xenografts | (Karmali et al. 2009) |
| iRGD (CRGDK/RGPD/EC) | Co-administration of peptide and drug (doxorubicin, nab-paclitaxel and doxorubicin liposome, trastuzumab) |
Tumor penetrating peptide | Orthotopic 22Rv1 human prostate tumors Orthotopic BT474 human breast tumors |
(Sugahara et al. 2010) |
| LyP-1 (CGNKRTRGC) | Peptide-functionalized Peptide displayed on Bi2S3 nanoparticle | Tissue penetration | Breast Cancer | (Kinsella et al. 2011) |
| CGKRK: tumor homing α-helical amphipathic peptide D[KLAKLAK]2 : proapoptic peptide | Tandem System: tumor homing peptide–proapoptotic peptide– multivalent presentation on iron oxide nanoparticles (for imaging) | Tumor homing and proapoptic behavior | Mice bearing orthotopic GBM tumors | (Agemy 2011) |
| iRGD | iRGD on paclitaxel loaded PCL-b-PVP copolymer | Tumor-penetrating | H22 tumor-bearing mice | (Zhu et al. 2011) |
| TP–LyP-1 transportan (TP) | Tumor-penetrating nano-complex (TPN) that comprised small interfering RNA (siRNA) complexed with a tandem peptide | Both Tumor penetrating and membrane-translocating | ovarian cancer, including the inhibitor of DNA binding 4 (ID4) | (Ren et al. 2012) |
| iRGD | CS-PAPBA NPs functionalized by iRGD and loaded with doxorubicin | Tumor-penetrating | H22 tumor-bearing mice | (Wang et al. 2013) |
| iRGD | (DOX)-loaded, iRGD-modified, sterically-stabilized liposome (SSL) iRGD-SSL-DOX |
Tumor-penetrating | B16-F10 tumor-bearing nude mice | (Yu et al. 2013) |
| iRGD MT1-AF7p (HWKHLHNTKTFLC) peptide (presents high binding affinity to membrane type-1 matrix metalloproteinase |
Coadministration of iRGD with MT1-AF7p pep-tide paclitaxel-loaded PEG-PLA nanoparticles (MT1-NP-PTX) | iRGD-tumor penetrating MT1-AF7p – matrix binding |
C6 glioma containing BALB/c mice | (Gu et al. 2013) |
| F3 tLyP-1 |
F3-functionalized nanoparticles (F3-NP), co-administered with tLyP-1 | Tumor-homing and penetration | mice bearing intracranial C6 glioma | (Hu et al. 2013) |
| iRGD | genetically inserted the iRGD peptide in the fiber C terminus of ICOVIR15K, an oncolytic tumor-retargeted adenovirus | Tumor penetrating | PaCa-2 implanted subcutaneously in female BALB/C nu/nu mice | (Puig-Saus et al. 2014) |
| iRGD | iRGD modified sterically stabilized liposomes (SSLs) containing conjugated linoleic acid– paclitaxel (CLA-PTX) iRGD-SSL-CLA-PTX |
Tumor-Penetrating | C57BL6/N mice containing a subcutaneous B16-F10 melanoma | (Du et al. 2014) |
| iRGD | iRGD conjugated D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) mediated co-delivery of paclitaxel (PTX) and survivin shRNA (shSur) | Tumor-Penetrating | Male nude Balb/c mice bearing A549/T lung cancer | (Shen et al. 2014) |
| iRGD | Dox loaded Exosome derived from mouse immature dendrite cells fused to iRGD (Dox loaded exosome-iRGD) | Tumor-Penetrating | Human breast cancer cells MDA-MB-231 transplanted into female BALB/c nude mice | (Tian et al. 2014) |
| iRGD | Linking ATAP to an iRGD peptide ATAP-potent inducer of apoptosis ATAP-iRGD-M8 |
iRGD-tumor penetrating ATAP-mitochondrial targeting peptide |
Prostate cancer xenograft | (De et al. 2014) |
| iRGD | Co-administration of iRGD peptide with size-shrinkable and tumor-microenvironment responsive (DOX-AuNPs-GNPs) | Tumor-penetrating | 4T1 tumor-bearing mouse model | (Cun et al. 2015) |
| anti-EGFR-iRGD: anti-EGFR VHH (the variable domain from the heavy chain of the antibody) fused to iRGD | Antibody vehicle-Peptide-drug anti-EGFR-iRGD-doxorubicin |
Tumor-penetrating | H22 cells injected xenograft | (Sha et al. 2015) |
| iRGD | Dual-labeled iRGD-modified multifunctional porous silicon nanoparticles (PSi NPs) | Tumor-penetrating | PC3-MM2 mouse xenograft model | (Wang et al. 2015a) |
| iRGD | iRGD-modified and doxorubicin-loaded sterically stabilized liposomes (iRGD-SSL-DOX) | Tumor-penetrating | B16 melanoma model | (Dai et al. 2014) |
| iRGD | iRGD, Dex-SA-DOX-CDDP doxorubicin-loaded cisplatin crosslinked polysaccharide-based nanoparticles Coadministration with iRGD |
Tumor-Penetrating | autochthonous colon cancer model 4T1 murine mammary carcinoma |
(Li et al. 2015a) |
| iRGD | Co-administration with Doxorubicin and Sorafenib | Tumor-penetrating | mice bearing HepG2 or Huh-7 xenografts | (Schmithals et al. 2015) |
| iRGD | Co-administration of Gemcitabine with iRGD | Tumor-penetrating | A549 cell line | (Zhang et al. 2015) |
| iRGD | iRGD–ICG-LPs iRGD modified indocyanine green (ICG) Liposomes |
Tumor-penetrating | (Yan et al. 2016) | |
| LyP-1 | Bismuth sulfide (Bi2S3) nanoparticle labeled with LyP-1 For tumor diagnosis |
Targeting tumor vessels along with tumor cells | 4T1 breast cancer in mice | (Kinsella et al. 2011) |
Conclusions
Peptides are attractive biological molecules that are able to carry biological functions like larger molecules, such as viral proteins and antibodies, while at the same time, take advantage of its small size to exhibit properties, such as permeability, similar to small molecules. Peptides are relatively low cost and scalable in synthesis and do not encounter potential immunogenicity concerns faced by proteins. And recent work summarized in this review has demonstrated that peptides are promising carriers of drug cargo and drug delivery systems to improve their bioavailability across multiple barriers to treat diseases of the brain, gastrointestinal tract and of tumors.
There is potentially transformative impact in discovering peptides able to shuttle cargo across the BBB to treat brain diseases. However, there is more work needed to understand the mechanism of transport across the BBB, whether it is real-time trafficking of receptor-mediated, adsorptive-mediated transcytosis, or possibly other mechanisms of transport across the BBB (Fig. 2). Also, it is not well understood the efficiency of transyctosis versus receptor recycling; to improve the potency and viability of peptides as drug carriers, future shuttle peptides would need to improve upon the efficiency of crossing the BBB as opposed to being recycled back to the capillary endothelium. Also, with new peptides being discovered, it is important to understand and discover the binding partner; it is feasible new targets can be discovered as opposed to existing molecules known to be involved in transcellular transport (e.g. transferrin, insulin, apoE receptors). In general, a greater mechanistic understanding of target binding, uptake, transcellular transport and export is needed to iterate and improve upon existing peptide carriers.
Fig. 2.
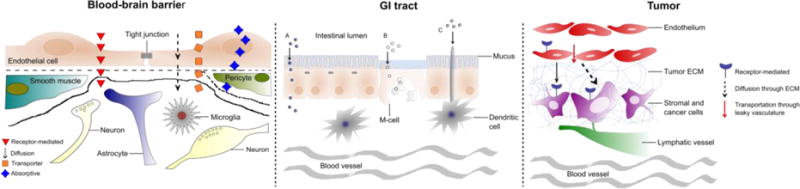
Cartoon of potential mechanisms of peptide-mediated transport across the blood-brain barrier (BBB), gastrointestinal tract (GI), and tumor microenvironment
The gastrointestinal tract confers a landscape of possibilities for the delivery of macromolecules, albeit several extracellular and cellular barriers can limit therapeutic success (Fig. 2). Oral bioavailability of peptides is still limited compared to intravenous route and thus higher doses must be administered in order to have the same pharmacological effect. Higher dosing regimens might result in toxicity, as well as prevent commercial viability. Future challenges include scaling up for manufacturing more complex delivery systems, and improving circulation half-lives of peptides. Ideally, a peptide delivery system would need to resist the extracellular milieu and efficiently internalize or traverse the mucosal epithelium, depending on the therapeutic strategy. Promising strategies rely on the development of technologies that increase selectivity and targeting efficacy of peptides in oral drug delivery.
Peptides have great potential in tumors to stimulate and improve drug perfusion throughout the tumor microenvironment to reach and treat difficult-to-reach cancer cells distant from the vasculature. In particular, tumor-penetrating peptides has been extensively studied in literature and proved to be a powerful method for the delivery of both covalently coupled and co-administered drug payloads into the tumor tissue. However, the mechanism of cell-to-cell transport through which the drug payload is distributed throughout the tissue is poorly understood. Although a large amount of work has been dedicated to investigate the transvascular and transcellular transport of peptides, transport through the tumor extracellular matrix (ECM) has been minimally explored (Fig. 2). The dense ECM in various types of solid tumors has been one of the major barriers for tumor therapeutics. Therefore, understanding the nature of the tumor specific ECM and potential binding sites present in different ECM proteins could lead to design higher penetrating peptide-based therapeutic carriers selective to tumor environment.
Regardless of the disease, peptides have shown the ability to overcome the biological barriers to improve drug delivery. However, these peptides can be further “evolved” to improve upon their existing functionality and achieve greater efficacy. Yet, there is no clinically approved peptide-drug or peptide-conjugated drug delivery system. There will be a great need to develop well-characterized, reproducible, and scalable peptide-based carriers. Underlying all peptide-based strategies for drug delivery into diseases, a greater understanding of mechanism of action is needed; how the peptide and/or peptide-functionalized drug delivery system interfaces with the local biological barrier and the immune system is critical to develop better design rules needed to improve transport, bioavailablity and ultimately, drug delivery for successful therapeutic interventions (Fig. 2).
Acknowledgments
The authors acknowledge and are thankful for support provided by the National Institutes of Health (R01-HL138251).
Footnotes
Compliance with ethical standards
This article does not contain any studies with human or animal subjects performed by any of the authors. This article follows ethical standards set by the International Standards for Editors and Authors. This article does not involve any studies conducted by the authors and informed consent was not needed.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- ’T Hoen PAC, Jirka SMG, Ten Broeke BR, et al. Phage display screening without repetitious selection rounds. Anal Biochem. 2012;421:622–631. doi: 10.1016/j.ab.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Agarwal V. Current status of the oral delivery of insulin. Pharm Technol. 2001;25:76–90. [Google Scholar]
- Agemy L. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc Nat Acad Sci. 2011;108:17450–17455. doi: 10.1073/pnas.1114518108. doi: 10.1073/pnas.1114518108/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1114518108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre TAS, Teijeiro-Osorio D, Rosa M, et al. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv Drug Deliv Rev. 2016;106:223–241. doi: 10.1016/j.addr.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Akerman ME, Chan WCW, Laakkonen P, et al. Nanocrystal targeting in vivo. Proc Natl Acad Sci U S A. 2002;99:12617–12621. doi: 10.1073/pnas.152463399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberici L, Roth L, Sugahara KN, et al. De Novo design of a tumor-penetrating peptide. Cancer Res. 2013;73:804–812. doi: 10.1158/0008-5472.CAN-12-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alitalo K, Carmeliet P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell. 2002;1:219–227. doi: 10.1016/S1535-6108(02)00051-X. [DOI] [PubMed] [Google Scholar]
- Arap W. Cancer Treatment by Targeted Drug Delivery to Tumor Vasculature in a Mouse Model. Science (80-) 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- Bagri A, Tessier-Lavigne M, Watts RJ. Neuropilins in tumor biology. Clin Cancer Res. 2009;15:1860–1864. doi: 10.1158/1078-0432.CCR-08-0563. [DOI] [PubMed] [Google Scholar]
- Bastian SEP, Walton PE, Ballard FJ, Belford DA. Transport of IGF-I across epithelial cell monolayers. J Endocrinol. 1999;162:361–369. doi: 10.1677/joe.0.1620361. [DOI] [PubMed] [Google Scholar]
- Bernkop-Schnurch A. The use of inhibitory agents to overcome the enzymatic barrier to perorally administered therapeutic peptides and proteins. J Control Release. 1998;52:1–16. doi: 10.1016/S0168-3659(97)00204-6. [DOI] [PubMed] [Google Scholar]
- Bertrand Y, Currie J-C, Poirier J, et al. Influence of glioma tumour microenvironment on the transport of ANG1005 via low-density lipoprotein receptor-related protein 1. Br J Cancer. 2011;105:1697–707. doi: 10.1038/bjc.2011.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Y, Liu L, Lu Y, et al. T7 Peptide-Functionalized PEG-PLGA Micelles Loaded with Carmustine for Targeting Therapy of Glioma. ACS Appl Mater Interfaces. 2016;8:27465–27473. doi: 10.1021/acsami.6b05572. [DOI] [PubMed] [Google Scholar]
- Boegh M, García-Díaz M, Müllertz A, Nielsen HM. Steric and interactive barrier properties of intestinal mucus elucidated by particle diffusion and peptide permeation. Eur J Pharm Biopharm. 2015;95:136–143. doi: 10.1016/j.ejpb.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Boohaker RJ, Lee MW, Vishnubhotla P, et al. The Use of Therapeutic Peptides to Target and to Kill Cancer Cells. Curr Med Chem. 2012;19:3794–3804. doi: 10.2174/092986712801661004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch G, Alsenz J, Van De Waterbeemd H, Folkers G. Estimation of permeability by passive diffusion through Caco-2 cell monolayers using the drugs’ lipophilicity and molecular weight. Eur J Pharm Sci. 1998;6:313–319. doi: 10.1016/S0928-0987(97)10019-7. [DOI] [PubMed] [Google Scholar]
- Ceramide E, Erdreich-epstein A, Shimada H, et al. Integrins avb3 and avb5 Are Expressed by Endothelium of High-Risk Neuroblastoma and Their Inhibition Is Associated with Increased. Endogenous Ceramide. 2000;1:712–721. [PubMed] [Google Scholar]
- Chen C, Duan Z, Yuan Y, et al. Peptide-22 and Cyclic RGD Functionalized Liposomes for Glioma Targeting Drug Delivery Overcoming BBB and BBTB. ACS Appl Mater Interfaces. 2017;9:5864–5873. doi: 10.1021/acsami.6b15831. [DOI] [PubMed] [Google Scholar]
- Connor Y, Tekleab S, Nandakumar S, et al. Physical nanoscale conduit-mediated communication between tumour cells and the endothelium modulates endothelial phenotype. Nat Commun. 2015;6:8671. doi: 10.1038/ncomms9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Zhang M, Zeng F, et al. Dual-Targeting Magnetic PLGA Nanoparticles for Codelivery of Paclitaxel and Curcumin for Brain Tumor Therapy. ACS Appl Mater Interfaces. 2016;8:32159–32169. doi: 10.1021/acsami.6b10175. [DOI] [PubMed] [Google Scholar]
- Cun X, Chen J, Ruan S, et al. A Novel Strategy through Combining iRGD Peptide with Tumor-Microenvironment-Responsive and Multistage Nanoparticles for Deep Tumor Penetration. ACS Appl Mater Interfaces. 2015;7:27458–27466. doi: 10.1021/acsami.5b09391. [DOI] [PubMed] [Google Scholar]
- Curnis F, Gasparri A, Sacchi A, et al. Coupling tumor necrosis factor-alpha with alphaV integrin ligands improves its antineoplastic activity. Cancer Res. 2004;64:565–571. doi: 10.1158/0008-5472.Can-03-1753. [DOI] [PubMed] [Google Scholar]
- Curnis F, Sacchi A, Gasparri A, et al. Isoaspartate-glycine-arginine: A new tumor vasculature-targeting motif. Cancer Res. 2008;68:7073–7082. doi: 10.1158/0008-5472.CAN-08-1272. [DOI] [PubMed] [Google Scholar]
- Dai W, Fan Y, Zhang H, et al. A comprehensive study of iRGD-modified liposomes with improved chemotherapeutic efficacy on B16 melanoma. Drug Deliv. 2014;7544:1–11. doi: 10.3109/10717544.2014.903580. [DOI] [PubMed] [Google Scholar]
- De G, Ko J-K, Tan T, et al. Amphipathic tail-anchoring peptide is a promising therapeutic agent for prostate cancer treatment. Oncotarget. 2014;5:7734–47. doi: 10.18632/oncotarget.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeule M, Régina A, Ché C, et al. Identification and design of peptides as a new drug delivery system for the brain. J Pharmacol Exp Ther. 2008;324:1064–1072. doi: 10.1124/jpet.107.131318. doi: jpet.107.131318 [pii]\r10.1124/jpet.107.131318. [DOI] [PubMed] [Google Scholar]
- Díaz-Perlas C, Sánchez-Navarro M, Teixidó M, Giralt E. Phage display as a tool to discover BBB-shuttle peptides: Panning against a human blood-brain barrier cellular model. J Pept Sci. 2016;22:S107. doi: 10.1002/bip.22928. [DOI] [PubMed] [Google Scholar]
- Du R, Zhong T, Zhang WQ, et al. Antitumor effect of iRGD-modified liposomes containing conjugated linoleic acid-paclitaxel (CLA-PTX) on B16-F10 melanoma. Int J Nanomedicine. 2014;9:3091–3105. doi: 10.2147/IJN.S65664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr DM, White SJ, Schluesener HJ. Identification of peptide sequences that induce the transport of phage across the gastrointestinal mucosal barrier. J Virol Methods. 2004;116:177–180. doi: 10.1016/j.jviromet.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Eliceiri BP, Cheresh DA. Adhesion events in angiogenesis. Curr Opin Cell Biol. 2001;13:563–568. doi: 10.1016/S0955-0674(00)00252-0. [DOI] [PubMed] [Google Scholar]
- Fan X, Venegas R, Fey R, et al. An in vivo approach to structure activity relationship analysis of peptide ligands. Pharm Res. 2007;24:868–879. doi: 10.1007/s11095-007-9238-z. [DOI] [PubMed] [Google Scholar]
- Fievez V, Plapied L, Plaideau C, et al. In vitro identification of targeting-ligands of human M cells by Phage Display. Int J Pharm. 2010;394:35–42. doi: 10.1016/j.ijpharm.2010.04.023. [DOI] [PubMed] [Google Scholar]
- Fittipaldi A, Giacca M. Transcellular protein transduction using the Tat protein of HIV-1. Adv Drug Deliv Rev. 2005;57:597–608. doi: 10.1016/j.addr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Fogal V, Zhang L, Krajewski S, Ruoslahti E. Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 2008;68:7210–7218. doi: 10.1158/0008-5472.CAN-07-6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey A, Giannasca KT, Weltzin R, et al. Role of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting. J Exp Med. 1996;184:1045–59. doi: 10.1084/jem.184.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard PJ, Appeldoorn CCM, Dorland R, et al. Pharmacokinetics, brain delivery, and efficacy in brain tumor-bearing mice of glutathione pegylated liposomal doxorubicin (2B3-101) PLoS One. 2014 doi: 10.1371/journal.pone.0082331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Qian J, Cao S, et al. Precise glioma targeting of and penetration by aptamer and peptide dual-functioned nanoparticles. Biomaterials. 2012;33:5115–5123. doi: 10.1016/j.biomaterials.2012.03.058. [DOI] [PubMed] [Google Scholar]
- Gao H, Yang Z, Zhang S, et al. Study and evaluation of mechanisms of dual targeting drug delivery system with tumor microenvironment assays compared with normal assays. Acta Biomater. 2014;10:858–867. doi: 10.1016/j.actbio.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Georgieva JV, Brinkhuis RP, Stojanov K, et al. Peptide-mediated blood-brain barrier transport of polymersomes. Angew Chemie - Int Ed. 2012;51:8339–8342. doi: 10.1002/anie.201202001. [DOI] [PubMed] [Google Scholar]
- Goldberg M, Gomez-Orellana I. Challenges for the oral delivery of macromolecules. Nat Rev Drug Discov. 2003;2:289–295. doi: 10.1038/nrd1067. [DOI] [PubMed] [Google Scholar]
- Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- Gu G, Gao X, Hu Q, et al. The influence of the penetrating peptide iRGD on the effect of paclitaxel-loaded MT1-AF7p-conjugated nanoparticles on glioma cells. Biomaterials. 2013;34:5138–5148. doi: 10.1016/j.biomaterials.2013.03.036. [DOI] [PubMed] [Google Scholar]
- Guixer B, Arroyo X, Belda I, et al. Chemically synthesized peptide libraries as a new source of BBB shuttles. Use of mass spectrometry for peptide identification. J Pept Sci. 2016:577–591. doi: 10.1002/psc.2900. [DOI] [PubMed] [Google Scholar]
- Hamzeh-Mivehroud M, Mahmoudpour A, Rezazadeh H, Dastmalchi S. Non-specific translocation of peptide-displaying bacteriophage particles across the gastrointestinal barrier. Eur J Pharm Biopharm. 2008;70:577–581. doi: 10.1016/j.ejpb.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and Emerging Mechanisms Review of the Angiogenic Switchduring Tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/S0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Heldin C-H, Rubin K, Pietras K, Östman A. High interstitial fluid pressure — an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- Higgins LM, Lambkin I, Donnelly G, et al. In vivo phage display to identify M cell-targeting ligands. Pharm Res. 2004;21:695–705. doi: 10.1023/B:PHAM.0000022418.80506.9a. [DOI] [PubMed] [Google Scholar]
- Hu Gu G, Liu Z, Jiang M, Kang T, Miao D, Tu Y, Pang Z, Song Q, Yao L, Xia H, Chen H, Jiang X, Gaob X, Chen JQ. F3 peptide-functionalized PEG-PLA nanoparticles co-administrated with tLyp-1 peptide for anti-glioma drug delivery. Biomaterials. 2013;34:1135–1145. doi: 10.1016/j.biomaterials.2012.10.048. [DOI] [PubMed] [Google Scholar]
- Hwang SR, Byun Y. Advances in oral macromolecular drug delivery. Expert Opin Drug Deliv. 2014;11:1955–1967. doi: 10.1517/17425247.2014.945420. [DOI] [PubMed] [Google Scholar]
- Jain RK. Transport of Molecules, Particles, and Cells in Solid Tumors. Annu Rev Biomed Eng. 1999;1:241–263. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- Jain RK. Determinants of Tumor Blood Flow: A Review. Cancer Res. 1988;48:2641–2658. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- Jain RK, Joshi R, Byrav DP, et al. Delivery of molecular and cellular medicine to solid tumors1PII of original article: S0169-409X(97)00027-6. The article was originally published in Advanced Drug Delivery Reviews 26 (1997) 71–90.1. Adv Drug Deliv Rev. 2001;46:149–168. doi: 10.1016/S0169-409X(00)00131-9. [DOI] [PubMed] [Google Scholar]
- Järver P, Mäger I, Langel Ü. In vivo biodistribution and efficacy of peptide mediated delivery. Trends Pharmacol Sci. 2010;31:528–535. doi: 10.1016/j.tips.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Jiang T, Olson ES, Nguyen QT, et al. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101:17867–72. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Song Y, Zhu X, et al. Biomaterials Goblet cell-targeting nanoparticles for oral insulin delivery and the influence of mucus on insulin transport. Biomaterials. 2012;33:1573–1582. doi: 10.1016/j.biomaterials.2011.10.075. [DOI] [PubMed] [Google Scholar]
- Kamei N, Morishita M, Eda Y, et al. Usefulness of cell-penetrating peptides to improve intestinal insulin absorption. J Control Release. 2008a;132:21–25. doi: 10.1016/j.jconrel.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Kamei N, Morishita M, Ehara J, Takayama K. Permeation characteristics of oligoarginine through intestinal epithelium and its usefulness for intestinal peptide drug delivery. J Control Release. 2008b;131:94–99. doi: 10.1016/j.jconrel.2008.07.016. [DOI] [PubMed] [Google Scholar]
- Kamei N, Morishita M, Takayama K. Importance of intermolecular interaction on the improvement of intestinal therapeutic peptide/protein absorption using cell-penetrating peptides. J Control Release. 2009;136:179–186. doi: 10.1016/j.jconrel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- Kang SK, Woo JH, Kim MK, et al. Identification of a peptide sequence that improves transport of macromolecules across the intestinal mucosal barrier targeting goblet cells. J Biotechnol. 2008;135:210–216. doi: 10.1016/j.jbiotec.2008.01.021. [DOI] [PubMed] [Google Scholar]
- Kang T, Jiang M, Jiang D, et al. Enhancing Glioblastoma-Specific Penetration by Functionalization of Nanoparticles with an Iron-Mimic Peptide Targeting Transferrin/Transferrin Receptor Complex. Mol Pharm. 2015;12:2947–2961. doi: 10.1021/acs.molpharmaceut.5b00222. [DOI] [PubMed] [Google Scholar]
- Kannan R, Chakrabarti R, Tang D, et al. GSH transport in human cerebrovascular endothelial cells and human astrocytes: Evidence for luminal localization of Na+-dependent GSH transport in HCEC. Brain Res. 2000;852:374–382. doi: 10.1016/S0006-8993(99)02184-8. [DOI] [PubMed] [Google Scholar]
- Karmali PP, Kotamraju VR, Kastantin M, et al. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomedicine Nanotechnology, Biol Med. 2009;5:73–82. doi: 10.1016/j.nano.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman B, Fuselier T, He J, Wimley W. Mechanism Matters: A taxonomy of cell penetrating peptides. Trends Biochem Sci. 2015;40:749–764. doi: 10.3109/10253890.2015.1094689.Post-Traumatic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenngott EE, Cole S, Hein WR, et al. Identification of Targeting Peptides for Mucosal Delivery in Sheep and Mice. Mol Pharm. 2016;13:202–210. doi: 10.1021/acs.molpharmaceut.5b00635. [DOI] [PubMed] [Google Scholar]
- Khafagy ES, Morishita M. Oral biodrug delivery using cell-penetrating peptide. Adv Drug Deliv Rev. 2012;64:531–539. doi: 10.1016/j.addr.2011.12.014. [DOI] [PubMed] [Google Scholar]
- Kinsella JM, Jimenez RE, Karmali PP, et al. X-ray computed tomography imaging of breast cancer by using targeted peptide-labeled bismuth sulfide nanoparticles. Angew Chemie - Int Ed. 2011;50:12308–12311. doi: 10.1002/anie.201104507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen E, Wang B, Ruoslahti E. Phage libraries displaying cyclic peptides with different ring sizes: ligand specificities of the RGD-directed integrins. Biotechnology (N Y) 1995;13:265–270. doi: 10.1038/nbt0395-265. [DOI] [PubMed] [Google Scholar]
- Komin A, Russell LM, Hristova KA, Searson PC. Peptide-based strategies for enhanced cell uptake, transcellular transport, and circulation: Mechanisms and challenges. Adv Drug Deliv Rev. 2017:110–111. 52–64. doi: 10.1016/j.addr.2016.06.002. [DOI] [PubMed] [Google Scholar]
- Kompella UB, Lee VHL. Delivery systems for penetration enhancement of peptide and protein drugs: Design considerations. Adv Drug Deliv Rev. 2001;46:211–245. doi: 10.1016/S0169-409X(00)00137-X. [DOI] [PubMed] [Google Scholar]
- Laakkonen P, Porkka K, Hoffman JA, Ruoslahti E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat Med. 2002;8:751–755. doi: 10.1038/nm720. [DOI] [PubMed] [Google Scholar]
- Langguth P, Bohner V, Heizmann J, et al. The challenge of proteolytic enzymes in intestinal peptide delivery. J Control Release. 1997;46:39–57. doi: 10.1016/S0168-3659(96)01586-6. [DOI] [Google Scholar]
- Larhed AW, Artursson P, Björk E. The influence of intestinal mucus components on the diffusion of drugs. Pharm Res. 1998;15:66–71. doi: 10.1023/A:1011948703571. [DOI] [PubMed] [Google Scholar]
- Larhed AW, Artursson P, Gråsjö J, Björk E. Diffusion of drugs in native and purified gastrointestinal mucus. J Pharm Sci. 1997;86:660–665. doi: 10.1021/js960503w. [DOI] [PubMed] [Google Scholar]
- Lee JH, Engler JA, Collawn JF, Moore BA. Receptor mediated uptake of peptides that bind the human transferrin receptor. Eur J Biochem. 2001;268:2004–2012. doi: 10.1046/j.1432-1327.2001.02073.x. [DOI] [PubMed] [Google Scholar]
- Lee VHL, Yamamoto A. Penetration and enzymatic barriers to peptide and protein absorption. Adv Drug Deliv Rev. 1989;4:171–207. doi: 10.1016/0169-409X(89)90018-5. [DOI] [Google Scholar]
- Li J, Feng L, Fan L, et al. Targeting the brain with PEG-PLGA nanoparticles modified with phage-displayed peptides. Biomaterials. 2011;32:4943–4950. doi: 10.1016/j.biomaterials.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang C, Li J, et al. Brain delivery of NAP with PEG-PLGA nanoparticles modified with phage display peptides. Pharm Res. 2013;30:1813–1823. doi: 10.1007/s11095-013-1025-4. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang Q, Pang Z, et al. Identification of peptide sequences that target to the brain using in vivo phage display. Amino Acids. 2012;42:2373–2381. doi: 10.1007/s00726-011-0979-y. [DOI] [PubMed] [Google Scholar]
- Li M, Tang Z, Zhang D, et al. Doxorubicin-loaded polysaccharide nanoparticles suppress the growth of murine colorectal carcinoma and inhibit the metastasis of murine mammary carcinoma in rodent models. Biomaterials. 2015a;51:161–172. doi: 10.1016/j.biomaterials.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Li X, Wang C, Liang R, et al. The glucose-lowering potential of exenatide delivered orally via goblet cell- targeting nanoparticles. Pharm Res. 2015b;32:1017–1027. doi: 10.1007/s11095-014-1513-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Zheng X, Gong M, Zhang J. Delivery of a peptide-drug conjugate targeting the blood brain barrier improved the efficacy of paclitaxel against glioma. 2016 doi: 10.18632/oncotarget.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JF, Yang VC. Insulin-cell penetrating peptide hybrids with improved intestinal absorption efficiency. Biochem Biophys Res Commun. 2005;335:734–738. doi: 10.1016/j.bbrc.2005.07.142. [DOI] [PubMed] [Google Scholar]
- Lindqvist A, Rip J, Van Kregten J, et al. In vivo Functional Evaluation of Increased Brain Delivery of the Opioid Peptide DAMGO by Glutathione-PEGylated Liposomes. Pharm Res. 2016;33:177–185. doi: 10.1007/s11095-015-1774-3. [DOI] [PubMed] [Google Scholar]
- Lindsay MA. Peptide-mediated cell delivery: Application in protein target validation. Curr Opin Pharmacol. 2002;2:587–594. doi: 10.1016/S1471-4892(02)00199-6. [DOI] [PubMed] [Google Scholar]
- Liu GW, Livesay BR, Kacherovsky NA, et al. Efficient Identification of Murine M2 Macrophage Peptide Targeting Ligands by Phage Display and Next-Generation Sequencing. Bioconjug Chem. 2015;26:1811–1817. doi: 10.1021/acs.bioconjchem.5b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist P, Artursson P. Oral absorption of peptides and nanoparticles across the human intestine: Opportunities, limitations and studies in human tissues. Adv Drug Deliv Rev. 2016;106:256–276. doi: 10.1016/j.addr.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Ma H, Yu P, Shen S, Xu B. A dual functional fluorescent probe for glioma imaging mediated by BBB penetration and glioma cell targeting. Biochem Biophys Res Commun. 2014;449:44–48. doi: 10.1016/j.bbrc.2014.04.148. [DOI] [PubMed] [Google Scholar]
- Mahmood A, Prüfert F, Efiana NA, et al. Cell-penetrating self-nanoemulsifying drug delivery systems (SNEDDS) for oral gene delivery. Expert Opin Drug Deliv. 2016;13:1503–1512. doi: 10.1080/17425247.2016.1213236. [DOI] [PubMed] [Google Scholar]
- Malcor JD, Payrot N, David M, et al. Chemical Optimization of New Ligands of the Low-Density Lipoprotein Receptor As Potential Vectors for Central Nervous System Targeting. J Med Chem. 2012 doi: 10.1021/jm2014919. [DOI] [PubMed] [Google Scholar]
- Matochko WL, Derda R. Next-generation sequencing of phage-displayed peptide libraries. Methods Mol Biol. 2015;1248:249–266. doi: 10.1007/978-1-4939-2020-4_17. [DOI] [PubMed] [Google Scholar]
- Maussang D, Rip J, van Kregten J, et al. Glutathione conjugation dose-dependently increases brain-specific liposomal drug delivery in vitro and in vivo. Drug Discov Today Technol. 2016;20:59–69. doi: 10.1016/j.ddtec.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- Mohamadzadeh M, Duong T, Sandwick SJ, et al. Dendritic cell targeting of Bacillus anthracis protective antigen expressed by Lactobacillus acidophilus protects mice from lethal challenge. Proc Natl Acad Sci. 2009;106:4331–4336. doi: 10.1073/pnas.0900029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita M, Kamei N, Ehara J, et al. A novel approach using functional peptides for efficient intestinal absorption of insulin. J Control Release. 2007;118:177–184. doi: 10.1016/j.jconrel.2006.12.022. [DOI] [PubMed] [Google Scholar]
- Morishita M, Morishita I, Takayama K, et al. Site-dependent effect of aprotinin, sodium caprate, Na2EDTA and sodium glycocholate on intestinal absorption of insulin. Biol Pharm Bull. 1993;16:68–72. doi: 10.1248/bpb.16.68. [DOI] [PubMed] [Google Scholar]
- Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11:905–910. doi: 10.1016/j.drudis.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Moroz E, Matoori S, Leroux JC. Oral delivery of macromolecular drugs: Where we are after almost 100 years of attempts. Adv Drug Deliv Rev. 2016;101:108–121. doi: 10.1016/j.addr.2016.01.010. [DOI] [PubMed] [Google Scholar]
- Murphy EA, Majeti BK, Barnes LA, et al. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc Natl Acad Sci U S A. 2008;105:9343–8. doi: 10.1073/pnas.0803728105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase I, Niwa M, Takeuchi T, et al. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol Ther. 2004;10:1011–1022. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Nguyen J, Hossain SS, Cooke JRN, et al. Flow arrest intra-arterial delivery of small TAT-decorated and neutral micelles to gliomas. J Neurooncol. 2017;133:1–9. doi: 10.1007/s11060-017-2429-5. [DOI] [PubMed] [Google Scholar]
- Nielsen DS, Shepherd NE, Xu W, et al. Orally Absorbed Cyclic Peptides. Chem Rev. 2017;117:8094–8128. doi: 10.1021/acs.chemrev.6b00838. [DOI] [PubMed] [Google Scholar]
- Nielsen EJB, Yoshida S, Kamei N, et al. In vivo proof of concept of oral insulin delivery based on a co-administration strategy with the cell-penetrating peptide penetratin. J Control Release. 2014;189:19–24. doi: 10.1016/j.jconrel.2014.06.022. [DOI] [PubMed] [Google Scholar]
- O’Sullivan CC, Lindenberg M, Bryla C, et al. ANG1005 for breast cancer brain metastases: correlation between 18F-FLT–PET after first cycle and MRI in response assessment. Breast Cancer Res Treat. 2016;160:51–59. doi: 10.1007/s10549-016-3972-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill MJ, Bourre L, Melgar S, O’Driscoll CM. Intestinal delivery of non-viral gene therapeutics: Physiological barriers and preclinical models. Drug Discov Today. 2011;16:203–218. doi: 10.1016/j.drudis.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science (80-) 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paasonen L, Sharma S, Braun GB, et al. New p32/gC1qR Ligands for Targeted Tumor Drug Delivery. ChemBioChem. 2016;17:570–575. doi: 10.1002/cbic.201500564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang H-B, Braun GB, Friman T, et al. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat Commun. 2014;5:4904. doi: 10.1038/ncomms5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM. Molecular Trojan horses for blood-brain barrier drug delivery. Curr Opin Pharmacol. 2006;6:494–500. doi: 10.1016/j.coph.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Drug Transport across the Blood–Brain Barrier. J Cereb Blood Flow Metab. 2012;32:1959–1972. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualini R, Koivunen E, Kain R, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60:722–727. doi: #. [PMC free article] [PubMed] [Google Scholar]
- Pasqualini R, Koivunen E, Ruoslahti E. Alpha v integrins as receptors for tumor targeting by circulating ligands. Nat Biotechnol. 1997;15:542–546. doi: 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- Pasqualini R, Ruoslahti E. Organ targeting In vivo using phage display peptide libraries. Nature. 1996;380:364–366. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- Pauletti GM, Gangwar S, Knipp GT, et al. Structural requirements for intestinal absorption of peptide drugs. J Control Release. 1996;41:3–17. doi: 10.1016/0168-3659(96)01352-1. [DOI] [Google Scholar]
- Pelaseyed T, Bergström JH, Gustafsson JK, et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol Rev. 2014;260:8–20. doi: 10.1111/imr.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- Porkka K, Laakkonen P, Hoffman JA, et al. A fragment of the HMGN2 protein homes to the nuclei of tumor cells and tumor endothelial cells in vivo. Proc Natl Acad Sci U S A. 2002;99:7444–7449. doi: 10.1073/pnas.062189599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prados MD, Schold SCJRSC, Fine HA, et al. A randomized, double-blind, placebo-controlled, phase 2 study of RMP-7 in combination with carboplatin administered intravenously for the treatment of recurrent malignant glioma. Neuro Oncol. 2003;5:96–103. doi: 10.1215/S1522851702000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Saus C, Rojas L, Laborda E, et al. iRGD tumor-penetrating peptide-modified oncolytic adenovirus shows enhanced tumor transduction, intratumoral dissemination and antitumor efficacy. Gene Ther. 2014;21:767–774. doi: 10.1038/gt.2014.52. [DOI] [PubMed] [Google Scholar]
- Qosa H, Mohamed LA, Alqahtani S, et al. Transporters as Drug Targets in Neurological Diseases. Clin Pharmacol Ther. 2016;100:441–453. doi: 10.1002/cpt.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Régina A, Demeule M, Ché C, et al. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br J Pharmacol. 2008;155:185–97. doi: 10.1038/bjp.2008.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regina A, Demeule M, Tripathy S, et al. ANG4043, a novel brain-penetrant peptide-mAb conjugate, is efficacious against HER2-positive intracranial tumors in mice. Mol Cancer Ther. 2015;14:129–40. doi: 10.1158/1535-7163.MCT-14-0399. [DOI] [PubMed] [Google Scholar]
- Ren Y, Cheung HW, von Maltzhan G, et al. Targeted Tumor-Penetrating siRNA Nanocomplexes for Credentialing the Ovarian Cancer Oncogene ID4. Sci Transl Med. 2012;4:147ra112-147ra112. doi: 10.1126/scitranslmed.3003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renukuntla J, Vadlapudi AD, Patel A, et al. Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm. 2013;447:75–93. doi: 10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard JP, Melikov K, Vives E, et al. Cell-penetrating peptides: A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- Roth L, Agemy L, Kotamraju VR, et al. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2012;31:3754–3763. doi: 10.1038/onc.2011.537. [DOI] [PubMed] [Google Scholar]
- Rotman M, Welling MM, Bunschoten A, et al. Enhanced glutathione PEGylated liposomal brain delivery of an anti-amyloid single domain antibody fragment in a mouse model for Alzheimer’s disease. J Control Release. 2015;203:40–50. doi: 10.1016/j.jconrel.2015.02.012. [DOI] [PubMed] [Google Scholar]
- Rousselle C, Clair P, Temsamani J, Scherrmann JM. Improved brain delivery of benzylpenicillin with a peptide-vector-mediated strategy. J Drug Target. 2002 Jun;10(4):309–15. doi: 10.1080/10611860290031886. 2002. [DOI] [PubMed] [Google Scholar]
- Rubas W, Cromwell MEM, Shahrokh Z, et al. Flux measurements across Caco-2 monolayers may predict transport in human large intestinal tissue. J Pharm Sci. 1996;85:165–169. doi: 10.1021/js950267+. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv Mater. 2012;24:3747–3756. doi: 10.1002/adma.201200454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E. Tumor penetrating peptides for improved drug delivery. Adv Drug Deliv Rev. 2017:110–111. 3–12. doi: 10.1016/j.addr.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E. Specialization of Tumour Vasculature. Nat Rev Cancer. 2002;2:83–90. doi: 10.1038/nrc724. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Rgd and Other Recognition Sequences for Integrins. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. The RGD story: A personal account. Matrix Biol. 2003;22:459–465. doi: 10.1016/S0945-053X(03)00083-0. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Navarro M, Teixidó M, Giralt E. Jumping Hurdles: Peptides Able To Overcome Biological Barriers. Acc Chem Res acs.accounts.7b00204. 2017 doi: 10.1021/acs.accounts.7b00204. [DOI] [PubMed] [Google Scholar]
- Sato AK, Viswanathan M, Kent RB, Wood CR. Therapeutic peptides: technological advances driving peptides into development. Curr Opin Biotechnol. 2006;17:638–642. doi: 10.1016/j.copbio.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Schmithals C, Köberle V, Korkusuz H, et al. Improving drug penetrability with iRGD leverages the therapeutic response to sorafenib and doxorubicin in hepatocellular carcinoma. Cancer Res. 2015;75:3147–3154. doi: 10.1158/0008-5472.CAN-15-0395. [DOI] [PubMed] [Google Scholar]
- Schwarze SR. In Vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. 1999;285:1569–1573. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Scott Swenson E, Curatolo WJ. (C) Means to enhance penetration. (2) Intestinal permeability enhancement for proteins, peptides and other polar drugs: mechanisms and potential toxicity. Adv Drug Deliv Rev. 1992;8:39–92. [Google Scholar]
- Sha H, Zou Z, Xin K, et al. Tumor-penetrating peptide fused EGFR single-domain antibody enhances cancer drug penetration into 3D multicellular spheroids and facilitates effective gastric cancer therapy. J Control Release. 2015;200:188–200. doi: 10.1016/j.jconrel.2014.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah RB, Ahsan F, Khan MA. Oral delivery of proteins: progress and prognostication. Crit Rev Ther Drug Carrier Syst. 2002;19:135–69. doi: 10.1615/CritRevTherDrugCarrierSyst.v19.i2.20. [DOI] [PubMed] [Google Scholar]
- Shen J, Meng Q, Sui H, et al. IRGD conjugated TPGS mediates codelivery of paclitaxel and survivin shRNA for the reversal of lung cancer resistance. Mol Pharm. 2014;11:2579–2591. doi: 10.1021/mp400576f. [DOI] [PubMed] [Google Scholar]
- Shi Y, Jiang X, Zhang L, et al. Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1621174114. 201621174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MW, Al-Jayyoussi G, Gumbleton M. Peptide sequences mediating tropism to intact blood-brain barrier: An in vivo biodistribution study using phage display. Peptides. 2012;38:172–180. doi: 10.1016/j.peptides.2012.06.019. [DOI] [PubMed] [Google Scholar]
- Staquicini FI, Ozawa MG, Moya CA, et al. Systemic combinatorial peptide selection yields a non-canonical iron-mimicry mechanism for targeting tumors in a mouse model of human glioblastoma. J Clin Invest. 2011;121:161–173. doi: 10.1172/JCI44798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell. 2009;16:510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara KN, Teesalu T, Karmali PP, et al. Coadministration of a Tumor-Penetrating Peptdei Enhances the Efficacy of Cancer Drugs. Science (80-) 2010;328:1031–1038. doi: 10.1007/s13398-014-0173-7.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambley WNH TW. Is anticancer drug development heading in the right direction? Cancer Res. 2009;69:1259–1262. doi: 10.1158/0008-5472.CAN-08-3786. [DOI] [PubMed] [Google Scholar]
- Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci. 2009;106:16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teesalu T, Sugahara KN, Ruoslahti E. Tumor-Penetrating Peptides. Front Oncol. 2013 doi: 10.3389/fonc.2013.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixidó M, Belda I, Zurita E, et al. Evolutionary combinatorial chemistry, a novel tool for SAR studies on peptide transport across the blood-brain barrier. Part 2. Design, synthesis and evaluation of a first generation of peptides. J Pept Sci. 2005;11:789–804. doi: 10.1002/psc.679. [DOI] [PubMed] [Google Scholar]
- Tian Y, Li S, Song J, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- Toome K, Willmore A-MAA, Paiste Päärn, et al. Ratiometric in vivo auditioning of targeted silver nanoparticles. Nanoscale. 2017:1–9. doi: 10.1039/C7NR04056C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozaki H, Odoriba T, Iseki T, et al. Use of protease inhibitors to improve calcitonin absorption from the small and large intestine in rats. J Pharm Pharmacol. 1998;50:913–920. doi: 10.1111/j.2042-7158.1998.tb04008.x. [DOI] [PubMed] [Google Scholar]
- Tremmel R, Uhl P, Helm F, Wupperfeld D, et al. Delivery of Copper-chelating Trientine (TETA) to the central nervous system by surface modified liposomes. Int J Pharm. 2016 Oct 15;512(1):87–95. doi: 10.1016/j.ijpharm.2016.08.040. 2016. [DOI] [PubMed] [Google Scholar]
- Tuma PL, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev. 2003;83:871–932. doi: 10.1152/physrev.00001.2003. [DOI] [PubMed] [Google Scholar]
- Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 Tat Requires Cell Surface Heparan Sulfate Proteoglycans. J Biol Chem. 2001;276:3254–3261. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- Urich E, Schmucki R, Ruderisch N, et al. Cargo Delivery into the Brain by in vivo identified Transport Peptides. Sci Rep. 2015;5:14104. doi: 10.1038/srep14104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rooy I, Cakir-Tascioglu S, Couraud PO, et al. Identification of peptide ligands for targeting to the blood-brain barrier. Pharm Res. 2010;27:673–682. doi: 10.1007/s11095-010-0053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rooy I, Hennink WE, Storm G, et al. Attaching the phage display-selected GLA peptide to liposomes: Factors influencing target binding. Eur J Pharm Sci. 2012;45:330–335. doi: 10.1016/j.ejps.2011.11.015. [DOI] [PubMed] [Google Scholar]
- Vela Ramirez JE, Sharpe LA, Peppas NA. Current state and challenges in developing oral vaccines. Adv Drug Deliv Rev. 2017 doi: 10.1016/j.addr.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter E, Kissel T, Amidon GL. The intestinal peptide carrier: A potential transport system for small peptide derived drugs. Adv Drug Deliv Rev. 1996;20:33–58. doi: 10.1016/0169-409X(95)00129-U. [DOI] [Google Scholar]
- Wang C-FC-F, Sarparanta MP, Makila EM, et al. Multifunctional porous silicon nanoparticles for cancer theranostics. Biomaterials. 2015a;48:108–118. doi: 10.1016/j.biomaterials.2015.01.008. [DOI] [PubMed] [Google Scholar]
- Wang J, Yadav V, Smart AL, et al. Toward oral delivery of biopharmaceuticals: An assessment of the gastrointestinal stability of 17 peptide drugs. Mol Pharm. 2015b;12:966–973. doi: 10.1021/mp500809f. [DOI] [PubMed] [Google Scholar]
- Wang L, Hao Y, Li H, et al. Co-delivery of doxorubicin and siRNA for glioma therapy by a brain targeting system: angiopep-2-modified poly(lactic-co-glycolic acid) nanoparticles. J Drug Target. 2015c;2330:1–15. doi: 10.3109/1061186X.2015.1025077. [DOI] [PubMed] [Google Scholar]
- Wang N, Jin X, Guo D, et al. Iron Chelation Nanoparticles with Delayed Saturation as an Effective Therapy for Parkinson Disease. Biomacromolecules. 2017;18:461–474. doi: 10.1021/acs.biomac.6b01547. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhen X, Wang J, et al. Doxorubicin delivery to 3D multicellular spheroids and tumors based on boronic acid-rich chitosan nanoparticles. Biomaterials. 2013;34:4667–4679. doi: 10.1016/j.biomaterials.2013.03.008. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhao Y, Jiang Y, et al. Enhanced anti-ischemic stroke of ZL006 by T7-conjugated PEGylated liposomes drug delivery system. Sci Rep. 2015d;5:12651. doi: 10.1038/srep12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wängler C, Nada D, Höfner G, et al. In vitro and initial in vivo evaluation of 68Ga-labeled transferrin receptor (TfR) binding peptides as potential carriers for enhanced drug transport into TfR expressing cells. Mol Imaging Biol. 2011;13:332–341. doi: 10.1007/s11307-010-0329-6. [DOI] [PubMed] [Google Scholar]
- Wickham TJ. Targeting adenovirus. Gene Ther. 2000;7:110–114. doi: 10.1038/sj.gt.3301115. [DOI] [PubMed] [Google Scholar]
- Winer I, Wang S, Lee YEK, et al. F3-targeted cisplatin-hydrogel nanoparticles as an effective therapeutic that targets both murine and human ovarian tumor endothelial cells in vivo. Cancer Res. 2010;70:8674–8683. doi: 10.1158/0008-5472.CAN-10-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodley JF. Enzymatic barriers for GI peptide and protein delivery. Crit Rev Ther Drug Carrier Syst. 1994;11:61–95. [PubMed] [Google Scholar]
- Wu Y, Luo X, Liu X, et al. Intraperitoneal Administration of a Novel TAT-BDNF Peptide Ameliorates Cognitive Impairments via Modulating Multiple Pathways in Two Alzheimer’s Rodent Models. Sci Rep. 2015;5:15032. doi: 10.1038/srep15032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Anderson B, Mao Q, Davidson BL. Recombinant human adenovirus: targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium. J Virol. 2000;74:11359–66. doi: 10.1128/jvi.74.23.11359-11366.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Ito S, Kurogi-Hirayama M, Ohtsuki S. Identification of cyclic peptides for facilitation of transcellular transport of phages across intestinal epithelium in vitro and in vivo. J Control Release. 2017;262:232–238. doi: 10.1016/j.jconrel.2017.07.037. [DOI] [PubMed] [Google Scholar]
- Yan C, Gu J, Hou D, et al. Improved tumor targetability of Tat-conjugated PAMAM dendrimers as a novel nanosized anti-tumor drug carrier. Drug Dev Ind Pharm. 2015;41:617–622. doi: 10.3109/03639045.2014.891127. [DOI] [PubMed] [Google Scholar]
- Yan F, Wu H, Liu H, et al. Molecular imaging-guided photothermal/photodynamic therapy against tumor by iRGD-modified indocyanine green nanoparticles. J Control Release. 2016;224:217–228. doi: 10.1016/j.jconrel.2015.12.050. [DOI] [PubMed] [Google Scholar]
- Yao VJ, D’Angelo S, Butler KS, et al. Ligand-targeted theranostic nanomedicines against cancer. J Control Release. 2016;240:267–286. doi: 10.1016/j.jconrel.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo MK, Kang SK, Choi JH, et al. Targeted delivery of chitosan nanoparticles to Peyer’s patch using M cell-homing peptide selected by phage display technique. Biomaterials. 2010;31:7738–7747. doi: 10.1016/j.biomaterials.2010.06.059. [DOI] [PubMed] [Google Scholar]
- Yu KF, Zhang WQ, Luo LM, et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: In vitro and in vivo evaluation. Int J Nanomedicine. 2013;8:2473–2485. doi: 10.2147/IJN.S46962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun Y, Cho YW, Park K. Nanoparticles for oral delivery: Targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev. 2013;65:822–832. doi: 10.1016/j.addr.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Sun X, Mei H, et al. LDLR-mediated peptide-22-conjugated nanoparticles for dual-targeting therapy of brain glioma. Biomaterials. 2013a;34:9171–9182. doi: 10.1016/j.biomaterials.2013.08.039. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wan X, Zheng X, et al. Biomaterials Dual-functional nanoparticles targeting amyloid plaques in the brains of Alzheimer ’ s disease mice. Biomaterials. 2013b;35:1–10. doi: 10.1016/j.biomaterials.2013.09.063. [DOI] [PubMed] [Google Scholar]
- Zhang C, Zheng X, Wan X, et al. The potential use of H102 peptide-loaded dual-functional nanoparticles in the treatment of Alzheimer’s disease. J Control Release. 2014;192:317–324. doi: 10.1016/j.jconrel.2014.07.050. [DOI] [PubMed] [Google Scholar]
- Zhang L, Song L, Zhang C, Ren Y. Improving intestinal insulin absorption efficiency through coadministration of cell-penetrating peptide and hydroxypropyl-β-cyclodextrin. Carbohydr Polym. 2012;87:1822–1827. doi: 10.1016/j.carbpol.2011.10.002. [DOI] [Google Scholar]
- Zhang Q, Zhang Y, Li K, et al. A novel strategy to improve the therapeutic efficacy of Gemcitabine for non-small cell lung cancer by the tumor-penetrating peptide iRGD. PLoS One. 2015 doi: 10.1371/journal.pone.0129865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhai M, Chen Z, et al. Dual-modified liposome codelivery of doxorubicin and vincristine improve targeting and therapeutic efficacy of glioma. Drug Deliv. 2017;24:1045–1055. doi: 10.1080/10717544.2017.1344334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Chen S, Gao Y, et al. Enhanced oral bioavailability of insulin using PLGA nanoparticles co-modified with cell-penetrating peptides and Engrailed secretion peptide (Sec) Drug Deliv. 2015;0:1–12. doi: 10.3109/10717544.2015.1043472. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Xie C, Liu Q, et al. The effect of hydrophilic chain length and iRGD on drug delivery from poly(ε-caprolactone)-poly(N-vinylpyrrolidone) nanoparticles. Biomaterials. 2011;32:9525–9535. doi: 10.1016/j.biomaterials.2011.08.072. [DOI] [PubMed] [Google Scholar]
- Zong T, Mei L, Gao H, et al. Synergistic dual-ligand doxorubicin liposomes improve targeting and therapeutic efficacy of brain glioma in animals. Mol Pharm. 2014;11:2346–2357. doi: 10.1021/mp500057n. [DOI] [PubMed] [Google Scholar]
- Zupančič O, Bernkop-Schnürch A. Lipophilic peptide character – What oral barriers fear the most. J Control Release. 2017;255:242–257. doi: 10.1016/j.jconrel.2017.04.038. [DOI] [PubMed] [Google Scholar]
- Zuris JA, Thompson DB, Shu Y, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2014;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]