

Abstract
Brain-related disorders have outmatched cancer and cardiovascular diseases worldwide as the leading cause of morbidity and mortality. The lack of effective therapies and the relatively dry central nervous system (CNS) drug pipeline pose formidable challenge. Superior, targeted delivery of current clinically approved drugs may offer significant potential. Minocycline has shown promise for the treatment of neurological diseases owing to its ability to penetrate the blood–brain barrier (BBB) and potency. Despite its potential in the clinic and in preclinical models, the high doses needed to affect a positive therapeutic response have led to side effects. Targeted delivery of minocycline to the injured site and injured cells in the brain can be highly beneficial. Systemically administered hydroxyl poly(amidoamine) (PAMAM) generation-6 (G6) dendrimers have a longer blood circulation time and have been shown to cross the impaired BBB. We have successfully prepared and characterized the in vitro efficacy and in vivo targeting ability of hydroxyl-G6 PAMAM dendrimer–9-amino-minocycline conjugate (D-mino). Minocycline is a challenging drug to carry out chemical transformations due to its inherent instability. We used a combination of a highly efficient and mild copper catalyzed azide–alkyne click reaction (CuAAC) along with microwave energy to conjugate 9-amino-minocycline (mino) to the dendrimer surface via enzyme responsive linkages. D-mino was further evaluated for anti-inflammatory and antioxidant activity in lipopolysaccharides-activated murine microglial cells. D-mino conjugates enhanced the intracellular availability of the drug due to their rapid uptake, suppressed inflammatory cytokine tumor necrosis factor α (TNF-α) production, and reduced oxidative stress by suppressing nitric oxide production, all significantly better than the free drug. Fluorescently labeled dendrimer conjugate (Cy5–D-mino) was systematically administered (intravenous, 55 mg/kg) on postnatal day 1 to rabbit kits with a clinically relevant phenotype of cerebral palsy. The in vivo imaging study indicates that Cy5–D-mino crossed the impaired blood–brain barrier and co-localized with activated microglia at the periventricular white matter areas, including the corpus callosum and the angle of the lateral ventricle, with significant implications for positive therapeutic outcomes. The enhanced efficacy of D-mino, when combined with the inherent neuroinflammation-targeting capability of the PAMAM dendrimers, may provide new opportunities for targeted drug delivery to treat neurological disorders.
Graphical abstract
INTRODUCTION
Neurodegenerative disorders exert an enormous health burden worldwide and continue to pose most-formidable challenges to the translation of novel therapies.1 Because the average life expectancy around the globe has increased significantly, it is likely that there will be an increase in the number of people with central nervous system (CNS) disorders, which is associated with significant health costs. This challenge is compounded by the fact that more than 98% of the drug candidates for treating CNS disorders are unable to cross the blood–brain barrier (BBB), requiring high doses and thus causing major side effects.2 Because of the high cost, lengthy development periods, and lower clinical success rates as compared with other major disease areas (cardiovascular, cancer, or metabolic disorders), the drug-discovery pipeline for CNS drugs is woefully thin.3 To reduce the risk associated with drug discovery, researchers are shifting their focus to drug repurposing, i.e., identifying and finding new therapeutic targets of already approved drugs and delivering them to target cells in a superior fashion.4,5
Minocycline, a highly lipophilic second-generation semi-synthetic derivative of tetracycline, is a potent broad-spectrum anti-infective drug that has been used widely against Gram-negative and Gram-positive organisms over the last 30 years.6 Recently, there have been numerous reports suggesting the multiple neuroprotective effects of minocycline in various experimental models of neurological diseases such as Alzheimer’s disease,7 Parkinson’s disease,8 Huntington’s disease,9 focal cerebral ischemia,6 traumatic brain injury,10,11 multiple sclerosis, and spinal cord injury.12 Although, the primary mechanism of action of minocycline for the neuroprotective effect is not clear, it exerts biological effect via direct antagonism of multiple mechanisms including: (1) the inhibition of microglial activation, which results in suppression of proinflammatory mediators; (2) the reduction of apoptotic-like neuronal cell death by down-regulating caspase 1 and cytochrome C; (3) the mitigation the p38 MAPK cascade which impairs inflammatory cytokine synthesis; (4) the prevention of glutamate toxicity; and (5) the suppression of the formation of reactive oxygen species.13 The well-established clinical safety and evidence of the blood–brain barrier penetration makes minocycline a promising drug candidate for neurological disorders.13,14 Unfortunately, to attain neuro-protective effects in most CNS models, the required dose of minocycline (~20–100 mg/kg multiple doses) is significantly higher than the dose (~3 mg/kg per day) used clinically to treat infectious and inflammatory diseases, even after accounting for specific surface area differences.15 Literature findings revealed that the high-dose of minocycline leads to deleterious side-effects and may exacerbate neurotoxicity.16 To overcome the side effects at high doses, targeted delivery to the brain is highly desirable, which, in turn, can improve the bioavailability and therapeutic efficacy in desired cells.
Dendrimers17,18 are hyperbranched, monodispersed macromolecules that have shown significant potential in gene delivery,19,20 drug delivery,20-22 imaging,23 and numerous other applications related to nanomedicine.24 Dendrimers offer unique physicochemical features such as (1) highly defined monodisperse structure, (2) nanometer size range, (3) multivalency, and (4) easy tuneability.25,26 Among the different class of dendrimers, commercially available hydroxyl poly-(amidoamine) (PAMAM) dendrimers have been widely investigated for controlled drug-delivery applications.27-32 Our group has investigated the potential of PAMAM dendrimers to transport drugs selectively at the site of injury in the brain across the blood–brain barrier.33-35 We reported that the hydroxyl terminated generation four (G4) PAMAM dendrimers (without any targeting moiety) upon systemic administration target the neuroinflammation mediated by activated microglia, and accumulate at the site of injury in a rabbit model of cerebral palsy (CP) at levels ~20-fold higher than healthy controls.36 We have further validated these findings in various other animal models.33,35,37-40 More recently, we reported that G6 (~6.7 nm) PAMAM hydroxyl dendrimers showed enhanced accumulation in the injured areas of the brain compared to G4 dendrimers (~4.3 nm) due to their prolonged blood circulation times.33,38,41
Targeted delivery of minocycline using dendrimer platform at the site of injury in the brain could be extremely valuable due to its high potency and neuroprotective effects. The prior drug-delivery efforts with minocycline have been primarily focused on the encapsulation of the drug into nanoparticles42 rather than chemical conjugation. Performing chemical transformations on tetracycline platform is a synthetic challenge due to chemical insensitivity of the parent scaffold under a variety of chemical conditions.43,44 Another major obstacle is the unavailability of an easily tunable chemical handle on minocycline, which can be utilized for conjugation with the carrier. Although there are four chemically active hydroxyl groups on minocycline at positions 3, 10, 11, and 12a, the attachment of a linker stereospecifically in a monosubstituted manner is still a strenuous task. We used 9-amino-minocycline (mino) containing a reactive amine group at position 9 and modified it to attach a linker. Prior studies showed that synthetic modifications at position C9 and C7 do not hamper antioxidant, anti-inflammatory, and antibiotic activities of the tetracycline pharmacophore.45
Building on these findings, we here present the rational design of dendrimer-9-amino-minocycline conjugate (D-mino) to attain targeted delivery to the inflamed cells in the injured brain. Herein, we report the design, synthesis, in vitro efficacy, and in vivo targeting evaluation of D-mino conjugate. We used generation 6-PAMAM dendrimer with ~256 hydroxyl surface groups (G6-OH) as the targeting platform, to which free mino was conjugated through a linker via CuAAC click-chemistry using microwave energy.46,47 Since its introduction, CuAAC revolutionized the field of synthetic chemistry, which not only generated huge libraries of new chemical entities but also found widespread use in creating highly complex dendrimer and polymeric nanoconstructs.45,48-50 Other merits of this reaction include its modular and robust nature, chemo-selectivity, high yields, easy purifications, and versatility to be carried out in a range of environments. For more efficient and rapid reactions, we took advantage of microwave energy, which is an invaluable tool for synthetic chemists in recent times. The combination of neuroinflammation targeting potential of PAMAM G6-OH with anti-inflammatory and antioxidant properties of minocycline could provide a potent therapeutic platform for neurological disorders.
RESULTS AND DISCUSSION
Preparation and Characterization of Intermediates, D-mino, and Cy5–D-mino Conjugates
9-Amino-minocycline was conjugated to the surface hydroxyl groups of the G6-OH via enzyme sensitive amide linkages. One of the most significant challenges while constructing dendrimer–drug conjugates is selecting the highly efficient chemical transformation for ligation of both appropriately functionalized reaction partners. It has been observed that even the highly robust synthetic protocols provide inefficient reaction rates, leading to low drug loadings, and become sluggish due to steric crowding. To successfully conjugate 9-amino-minocycline in a convenient manner, we used a highly facile CuAAC click-reaction in combination with microwave energy. Microwave energy is known to provide remarkable accessibility to the reactive sites, resulting in enhanced reaction rates, shorter reaction time (from days to minutes), high reaction yields, and cleaner products in comparison with conventional methods. There has been a significant increase in the number of scientific reports in which microwave-assisted click reactions conveniently generated highly complex polymeric and dendritic architectures.51
The limited choice of available chemical transformations for tetracyclines presents significant challenges to the drug modification. Most of the tetracycline scaffolds degrade in the presence of acid, base, light, or heat. Therefore, we used milder reaction conditions to attach an orthogonal linker having an amide linkage on free mino and a clickable functional group on the other side, which could be exploited later on for dendrimer conjugation. Multiple attempts were made and different chemical transformations were explored to create a linker on the free drug. We first tried to acylate the amino group on mino using coupling agents (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate, HATU and N,N-diisopropylethylamine, DIPEA) by reacting it with 6-azido hexanoic acid, which yielded no success. We further tried to acylate using highly reactive bromoacetyl bromide in the presence of 4-dimethylaminopyridine (DMAP), which was unsuccessful. Moreover, under the above-stated reaction conditions, the color of mino turned black. Finally, we decided to explore acylation reaction using symmetrical anhydrides due to their excellent reactivities with aromatic amines. First, we aimed at synthesizing symmetrical anhydride 3 (Figure 1A), and the synthesis started with the nucleophilic substitution of 6-bromohexanoic acid 1 using sodium azide in dimethylformamide (DMF) at 50 °C to yield compound 2, in which bromide moiety was replaced by azido terminal functionality. Compound 2 was further treated with the coupling reagent dicyclohexylcarbodiimide (DCC) to yield 6-azidohexanoic anhydride 3 in quantitative yields. 6-Azidohexanoic anhydride 3 was subsequently reacted with mino 4 using sodium bicarbonate in DMF to afford compound 5 having azido functionality at the terminal to participate in click reaction. Compound 5 was purified using reverse-phase high-performance liquid chromatography (HPLC) as a light-yellow solid. Detailed 1H nuclear magnetic resonance (NMR) spectroscopic analysis confirmed the formation of compound 5 (Figure 2A,B). Once the linker is attached, the peak corresponding to H8 at δ 6.96 ppm moved to δ 8.14 ppm. Methylene protons corresponding to linker appeared at δ = 3.31, 2.50, and 1.80–1.40 ppm. The purity of the compound was greater than 99% as confirmed from HPLC.
Figure 1.
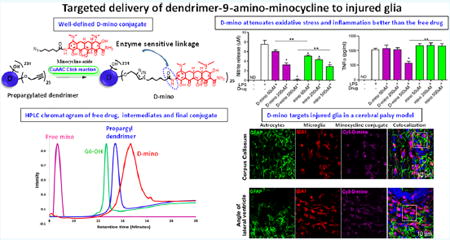
Key steps in the synthetic pathway of (A) azido-functionalized 9-amino-minocycline, 5; (B) dendrimer–9-amino-minocycline conjugate (D-mino), 8; and (C) Cy5-labeled dendrimer–9-amino-minocycline conjugate (Cy5-D-mino), 12. Reagents and conditions: (i) NaN3, DMF, 60 °C, 18 h 60%; (ii) DCC, DMF, rt, 12 h, 75%; (iii) NaHCO3, DMF, 6 h, 62%; (iv) propargyl bromide, NaH, DMF, 0 °C–rt, 12 h quantitative yield; (v) CuSO4·5H2O, Na ascorbate, DMF, THF, H2O, microwave, 40 °C, 6 h, quantitative yield; (vi) ethylene dichloride (EDC)·HCl, DMAP, Boc−GABA−OH, DMF, rt, 12 h; (vii) dichloromethane, trifluoroacetic acid; (viii) CuSO4·5H2O, Na ascorbate, DMF, THF, H2O, microwave, 40 °C; (ix) Cy5 NHS ester, DIPEA, pH 7.5, DMF, rt, 24 h.
Figure 2.
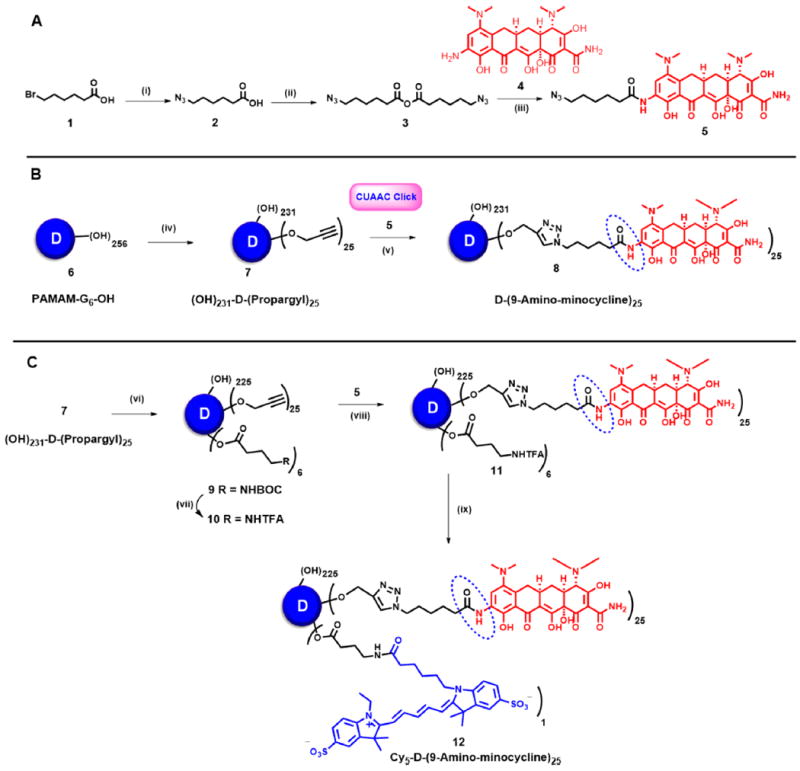
Comparison of 1H NMR spectra (panels A and B in CD3OD and C and D in DMSO) of 4, 5, 7, and 8 with the appearance and disappearance of characteristic signals toward the construction of D-mino (8).
Once the linker was successfully installed on mino, the modifications on the dendrimer were carried out (Figure 1B). G6-OH (6) was subjected to partial O-alkylation using sodium hydride in DMF and reacted with 80% propargyl bromide solution in toluene at 0 °C for overnight and provided compound 7 with 25 propargyl arms. The appearance of two sharp signals (P1 and P2) in 1H NMR at δ 4.12 and 3.75 ppm, corresponding to O−CH2 of the propargyl arm, confirmed the complete and neat transformation of the product (Figure 2C). 13C NMR further affirmed product formation with characteristic signals of acetylene around 80 ppm. Finally, the conjugation of propargylated dendrimer (7) with azide-functionalized mino (5) using a click reaction in the presence of copper sulfate pentahydrate and sodium ascorbate in the microwave afforded our desired D-mino conjugate (8). The conjugate was subjected to extensive dialysis against DMF followed by water to ensure the removal of any free drug entrapped within the dendrimer.
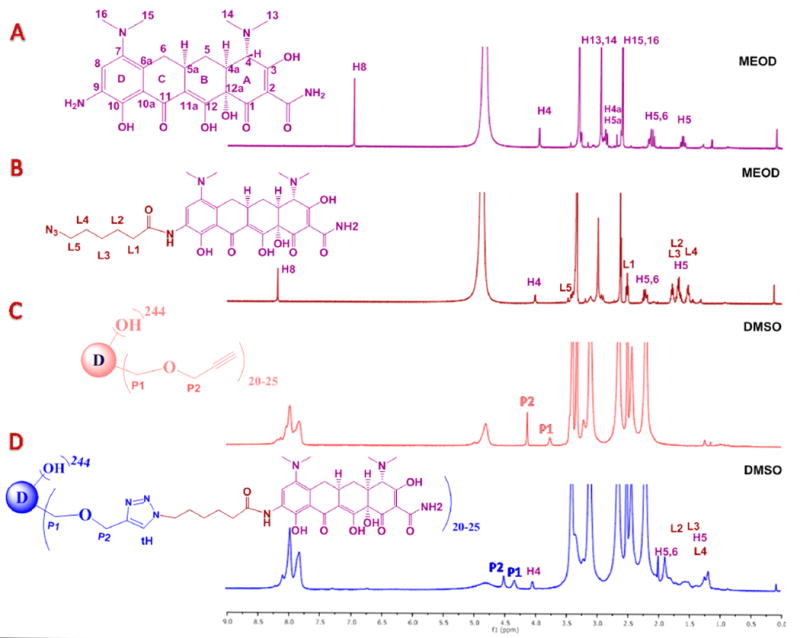
The structure of the conjugate was characterized using 1H NMR spectroscopy, Matrix-assisted laser desorption–ionization time-of-flight (MALDI-TOF) and HPLC. The peaks associated with drug protons in between δ 2.0–1.0 (ppm), and a characteristic H4 proton of minocycline at δ 4.0 (ppm) were clearly seen after the attachment of linker on the drug and after the conjugation on the dendrimer surface (Figure 2A–D). To see the peak corresponding to triazole and H8 of mino, which were hidden under the peaks of internal amide protons of the dendrimer in between 7 and 8 ppm, the NMR was taken in deuterated methanol. The internal amides protons of dendrimer got exchanged and disappeared, thus showing both of the peaks in the same region (Figure S11). The theoretical molecular weight of the conjugate 8 with 25 mino molecules corresponds to 74 554 Da, and the MALDI-TOF spectrum revealed a peak at 74,795 further confirming the formation of the conjugate (Figure S12). The purity of all the intermediates and the final conjugate by HPLC was more than 95%. HPLC profiles of the dendrimers starting from G6-OH (RT = 12.6 min) shows a clear shift in the retention times toward the hydrophobic region after the propargylation on the dendrimer surface (compound 7, RT= 13.3 min) and the final conjugation reaction (D-mino, RT= 14.6 min, Figure 3A). We further evaluated the size of the conjugate using dynamic light scattering (DLS). The mean hydrodynamic diameter of both PAMAM−G6−OH (6) and D-mino (8) in water was ~6 and ~8.4 nm, respectively (Figure 3B). The increase in the size of the conjugate as compared to PAMAM−G6−OH once again suggests a successful conjugation of the drugs on the dendrimer surface. Although minocycline is a lipophilic drug with poor aqueous solubility, the conjugation on the surface of water-soluble PAMAM dendrimer provides remarkable aqueous solubility to the resulting conjugate, which is highly desired for drug-delivery applications. Moreover, we attached only 20–25 molecules of mino on the dendrimer capping approximately 10% of the surface to keep the inherent properties of the dendrimer intact.
Figure 3.
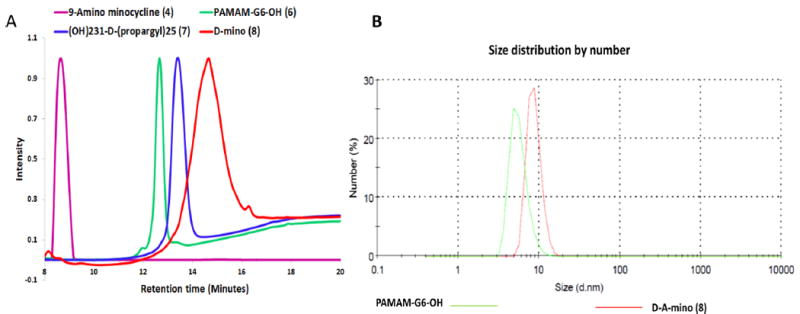
(A) Comparison of HPLC profiles of free drug, intermediates, and final D-mino conjugate. (B) Comparison of size distribution of PAMAM-G6-OH (6) and D-mino conjugate (8) using DLS.
To investigate the cellular uptake of the D-mino, we conjugated a near-infrared (IR) fluorescence tag (Cy5) to it (Figure 1C). For the synthesis of Cy5-labeled conjugate (Cy5–D-mino) (12), we coupled Boc−GABA−OH to propargylated dendrimer (7), employing Steglich esterification in the presence of ethylene dichloride (EDC)·HCl and DMAP in DMF to afford protected bifunctional dendrimer (9). 1H NMR confirmed the attachment of five to six molecules of Boc–GABA. The Boc group was deprotected in the presence of a trifluoroacetic acid/dichloromethane (DCM) (3:1) mixture. The peak attributed to the tert-butyl group of the Boc group in between (1.4–1.3 ppm) disappeared in NMR confirming the formation of dendrimer 10, which was further reacted with mino 5 using CuAAC click conditions in the microwave to afford dendrimer 11. Finally, compound 11 was coupled with activated ester of Cy5 at pH 7.5 in DMF to provide Cy5–D-mino (12) in quantitative yield. All the intermediates and Cy5 conjugated dendrimer were characterized using 1H NMR, and the purity was evaluated using HPLC.
In Vitro Release Studies
The release profile of the free drug from the conjugate was assessed using HPLC both at physiological pH (phosphate-buffered saline [PBS] buffer, pH 7.4) for extracellular environment and in the presence of enzyme (carboxyl-esterase at pH 5.5) for intracellular lysosomal environment (Figure 4).52 We did not observe any drug release in PBS buffer at a pH 7.4, suggesting that the D-mino conjugate is stable under extracellular conditions. Although aliphatic amide bonds are quite stable for aqueous hydrolysis at physiological pH and enzymatic hydrolysis by esterase, aromatic amides can be substrates for esterase. PEGylated prodrugs employing acyl aromatic amide linkages, which are susceptible to esterase, have been designed.53 Because the dendrimer hydroxyl groups and mino have been conjugated via acyl aromatic amide linkages, the drug release was studied in the presence of esterase. Moreover, previous studies by our groups have revealed that the cellular uptake of PAMAM dendrimer conjugates is mediated mainly by fluid-phase endocytosis, and thus, the conjugates are present in the lysosomes for considerable time periods;54 therefore, we used citrate buffer along with the enzyme to investigate the release characteristics of the drug from the conjugate. Evaluating the release of free drug from D-mino was very challenging due to the drug’s instability under acidic conditions. The dendrimer–drug conjugate showed a sustained release of free drug. Less than 10% of the drug was released in 48 h, reaching to around 35% drug release over a period of 8 days (Figure 4). We could not evaluate the release of the free drug from the conjugate after this period as D-mino conjugate was not stable at later time points. The release study over a period of 8 days gives us enough information based on the treatment and time points of our in vitro and in vivo studies.
Figure 4.
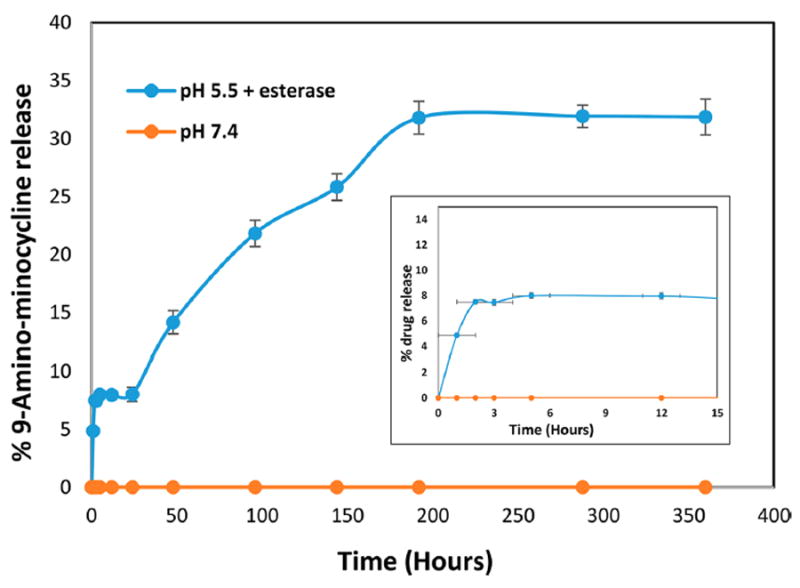
Drug-release profile from D-mino at physiological pH and in the presence of esterase (pH 5.5). Inset shows the drug-release profile at initial hours.
Enhanced Cellular Uptake of D-mino Conjugate by Activated Microglial Cells
Flow cytometry and confocal microscopy were performed to access the cellular uptake kinetics of D-mino conjugate. We used a near-IR dye Cy5 as a fluorescent probe to label the D-mino conjugate for imaging purposes. The flow cytometry studies enabled us to determine if the conjugation of drug alters the uptake kinetics of the dendrimers and establishes the treatment window (time) to evaluate the efficacy. The microglial cells were preactivated using lipopolysaccharides (LPS) for 3 h, followed by treatment of the cells with 2.5 μM Cy5–D-mino. At 3 h after Cy5–D-mino treatment, ~99% of the cell population were positive for Cy5 in the FL4 channel, indicating rapid uptake of D-mino by microglial cells (Figure 5A), consistent with previous studies on free dendrimers without drug.55 This suggests that the conjugation of ~25 molecules of mino to the dendrimer surface did not change the uptake kinetics of the dendrimer, and 3 h drug treatment and wash-out would work for functional study. Confocal microscopy confirmed the relatively rapid cellular uptake of D-mino into preactivated BV-2 microglia, which is consistent with our previous findings with Cy5-labeled hydroxyl PAMAM dendrimer (Figure 5B).56 The punctuate signals of Cy5–D-mino (red) were widely distributed throughout the cytoplasm of the microglia after 3 h treatment.
Figure 5.
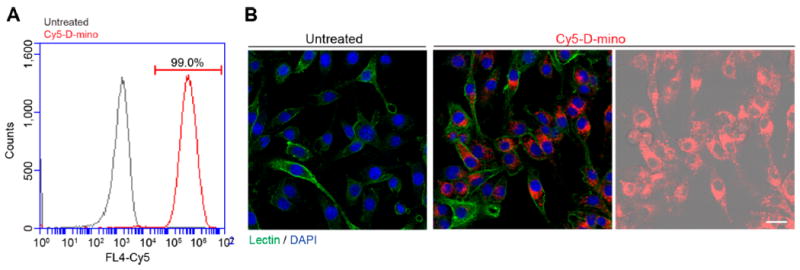
Cellular uptake of Cy5−D-mino. Preactivated BV-2 microglia by LPS at 100 ng/mL for 3 h were treated with Cy5−D-mino (2.5 μM) for 3 h. (A) Flow cytometry after treatment. Cy5 intensity on x-axis is plotted against the cell counts on y-axis. (B) Confocal microscopy of Cy5−D-mino (red) cell uptake into BV-2 microglia. The cells were stained with Isolectin-488 (green). In the right panel, Cy5 signal is overlaid on DIC image of cells. Scale bar: 25 μm.
Antioxidant and Anti-Inflammatory Activity of D-mino Conjugate in Microglial Cells
Before comparing the in vitro efficacy of D-mino and free mino, we first evaluated their toxicity to cells at the selected treatment concentration range. We performed a cell viability assay based on calorimetric method using soluble tetrazolium salt reagent. At the concentration range of 50–500 μM, free and D-mino (containing equivalent amount of mino) did not exert any cell cytotoxicity at the end of all procedures for efficacy studies (Figure 6A,D). To access the anti-inflammatory and antioxidant activity of D-mino, we used LPS-induced pro-inflammatory murine BV-2 microglial cells. LPS activates protein kinases in microglial cells through the toll-like receptors (TLR) pathway, thereby stimulating the release of immune-regulated proin-flammatory cytokines and induces oxidative stress.55,56 The cells were activated with 100 ng/mL of LPS followed by 3 h of treatment with different concentrations of free mino and D-mino. After 3 h of treatment, the cells were washed with warm PBS quickly and replenished with LPS containing medium for next 24 h. Therefore, the efficacy studies reffect the amount of free mino or D-mino taken up by the cells over a limited time of 3 h and in the presence of LPS for 24 h. The antioxidant activity of D-mino was measured by the nitric oxide (NO) release from activated microglial cells at 24 h post-treatment. LPS activation resulted in significant increase in NO production (~6 fold) (Figure 6B). Both free mino and D-mino treatment inhibited NO release from activated microglia in a dose-dependent manner. At 500 μM concentrations, D-mino treatment inhibited almost all NO production significantly better compared to free mino, 0.12 ± 0.026 μM versus 2.92 ± 0.21 μM (p < 0.001). D-mino treatment of 200 μM concentration achieved a similar effect on the NO suppression compared to the free drug at 500 μM. This may be attributed to the fact that the dendrimer increased the intracellular uptake of D-mino. This was true even in the case of 50 μM concentration suggesting enhanced efficacy of D-mino.
Figure 6.
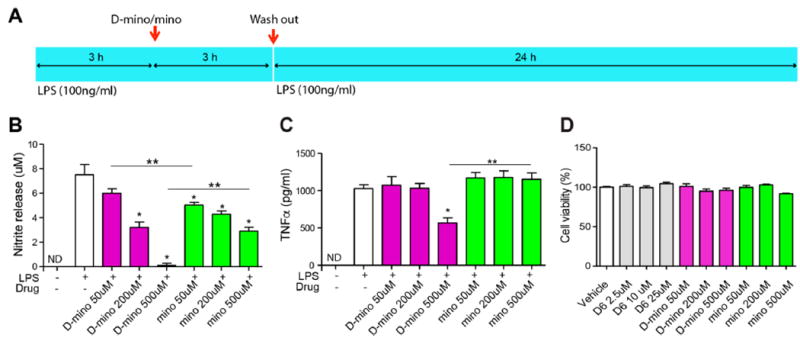
Antioxidant and anti-inflammatory activity of D-mino in LPS-activated BV-2 microglial cells and cytotoxicity assay. (A) Schematic image describes the drug treatment procedure. BV-2 microglia were activated with LPS at 100 ng/mL concentration before and after 3 h of drug treatment, and the culture supernatants were assayed for nitric oxide production by the Griess reaction and tumor necrosis factor α (TNF-α) release by ELISA at 24 h after the second LPS treatment. (B) Effects of free and D-mino on NO release. The concentration was indicated on the basis of free 9-amino-minocycline. (C) Effects of free and D-mino on TNF-α release. (D) BV-2 microglial viability was measured by CellTiter AQ proliferation assay. Preactivated BV-2 microglia were treated with free and D-mino for 3 h before second LPS activation. The cell viability was assayed at 24 h after the second LPS treatment. Data were expressed as mean ± standard error of the mean; the asterisk indicates p < 0.05 significant differences from the LPS alone group, and double asterisks indicate p < 0.05 significant differences between free and dendrimer conjugate.
The anti-inflammatory activity of D-mino was evaluated by measuring the tumor necrosis factor α (TNF-α) cytokine released by the activated microglial cells using a mouse TNF-α ELISA kit. LPS activation resulted in a ~100-fold increase TNF-α levels. D-mino treatment at 500 μM concentration inhibited LPS-induced TNF-α production significantly, 566.5 ± 82.1 pg/mL versus 1150.2 ± 172.6 pg/mL (p < 0.01), while the free drug treatment did not reduce TNF-α level at any of the concentrations (Figure 6C). Interestingly, according to in vitro release studies (Figure 4), less than 10% of the free drug is released from conjugate over this time period, which suggests that the conjugate is delivering a higher amount of the drug inside the cells. In vitro, relative to free drug, the conjugate is showing comparable (at low concentrations) or superior (at higher concentration) efficacy on both antioxidant and anti-inflammatory activity despite releasing only a small fraction of the drug inside the cells during the measurement period. All together, these efficacy data in LPS-activated BV-2 microglia suggest possible advantages of D-mino in comparison to free drug, especially when combined with the ability of this dendrimer to target activated microglia in vivo for therapeutic use against CNS pathologies.
In Vivo Co-Localization of Cy5–D-mino with Activated Microglia in the White Matter of Cerebral Palsy Rabbit Kits
To evaluate the brain cell targeting ability of D-mino conjugate, we intravenously injected the fluorescently labeled Cy5−D-mino conjugate in endotoxin kits with cerebral palsy (CP) on PND1. At 24 h post-injection, we analyzed the Cy5−D-mino distribution in the periventricular white matter area in both the corpus callosum (Figure 7, top), and the periventricular region (Figure 7, bottom), where most severe brain injury happens in this model, with significant microglial activation.33,57 Our results suggest that Cy5−D-mino is co-localized with activated microglia (IBA1 positive cells) in CP kits (Figure 7). The activation of microglia can induce neuronal damage through the release of proinflammatory cytokines and chemokines, reactive oxygen species, and free radicals. Therefore, the cell-specific delivery of minocycline to activated microglia will potentially suppress microglia-mediated inflammation. Interestingly, the astrocytes in these regions did not appear to be activated and did not show appreciable uptake of the conjugate. The uptake in activated microglia of the conjugate is similar to that of dendrimer−Cy5 (without the drug conjugated), suggesting that drug conjugation did not negatively impact the uptake by the activated microglia, which are the key targets for the drug in the treatment of neuroinflammation.
Figure 7.
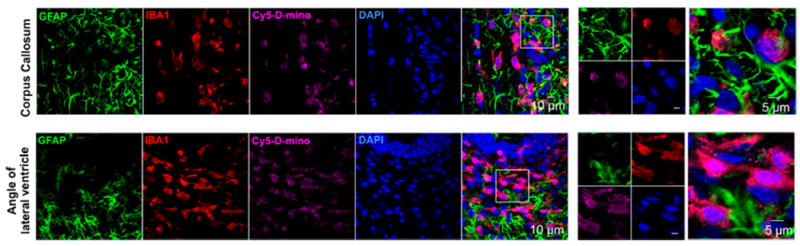
In vivo cellular localization of Cy5−D-mino. The endotoxin kits with CP (n = 3) received Cy5−D-mino (55 mg/kg, intravenous) on PND1 and were sacrificed 24 h post-injection. Brain slices contain Cy5−D-mino (magenta) were co-stained with GFAP (astrocyte marker, green), IBA1 (microglial marker, red), and DAPI (blue). Cy5−D-mino is mainly co-localized with activated microglia at the corpus callosum and the angle of lateral ventricle. The images on the right panels are the higher magnification of the region of interest marked with the boxes on the left panels.
CONCLUSIONS
In conclusion, we have successfully conjugated 9-amino-minocycline on dendrimer surface via the efficient and robust CuAAC click reaction as a ligation tool. In addition, we employed microwave energy to enhance the reaction rate and to reduce the reaction time for this challenging drug with stability and reactivity issues. The milder reaction protocols were developed and followed to introduce a linker on the drug as well as to finally conjugate it on to the dendrimer surface to avoid any degradation. In addition, D-mino conjugate is highly water-soluble unlike the free drug. The free drug released from the conjugate in a sustained fashion with release of ~35% over a period of 1 week. D-mino shows excellent cellular uptake into the activated microglia and exhibits significant higher efficacy in suppressing NO and TNF-α release from LPS-stimulated BV-2 cells, in comparison to the free drug. Our in vivo imaging study demonstrates that dendrimer conjugate facilitates minocycline to cross the impaired BBB and to target activated microglia, the major neuroinflammation mediators, at the site of injury. Combining the brain targeting ability of hydroxyl PAMAM dendrimer with high potency of minocycline in this platform can provide a potential therapeutic tool to treat neuro-inflammation. Moreover, the facile synthetic protocol developed here will be a foundation to construct novel dendrimer-conjugates with other tetracycline drugs, which is a huge synthetic challenge.
EXPERIMENTAL PROCEDURES
Materials and Methods
All reactions in the organic medium were performed in standard oven-dried glassware under an inert nitrogen atmosphere using freshly distilled solvents. CH2Cl2 and DMF were distilled from CaH2 and ninhydrin, respectively, and were kept over molecular sieves. Solvents and reagents were deoxygenated when necessary by purging with nitrogen. PAMAM dendrimer (biomedical-grade generation 6 consisting of 256 hydroxyl end-groups) (G6-OH) was purchased from Dendritech Inc. Dialysis membrane (MWCO: 1000–10 000 Da) was purchased from Spectrum Laboratories Inc. As-received dendrimer was purified before use. Methanol was removed under reduced pressure to yield a viscous oil. G6-OH dendrimer was redissolved in DI water, transferred to a 10 000 MWCO dialysis membrane, and dialyzed against 2 gallons of Nanopure water for 36 h while being stirred. Water was changed six or seven times at regular intervals. Purified dendrimer was lyophilized and dried to yield hygroscopic white sticky material. Dendrimer was stored at −20 °C under argon atmosphere. All reagents were used as supplied without prior purification unless otherwise stated and obtained from Sigma-Aldrich Chemical Company Ltd. Cy5−mono-NHS ester was purchased from Amersham Biosciences-GE Healthcare. LPS were purchased from Sigma. Reactions were monitored by analytical thin-layer chromatography (TLC) using silica gel 60 F254 precoated plates (E. Merck), and compounds were visualized by 254 nm light, a mixture of iodine and silica gel, a mixture of ceric ammonium molybdate solution (100 mL of H2SO4, 900 mL of H2O, 25 g of (NH4)6Mo7O24H2O, and 10 g of Ce(SO4)2), or both, and subsequent development was performed by gentle warming with a heat-gun. Purifications were performed by combi-flash column chromatography system from Biotage using prepacked silica gel columns with the indicated eluent.
Characterization via Nuclear Magnetic Resonance
NMR spectra were recorded on a Bruker 500 MHz spectrometer at ambient temperatures. Proton and carbon chemical shifts (δ) are reported in ppm. The resonance multiplicity in the 1H NMR spectra are described as “s” (singlet), “d” (doublet), “t” (triplet), and “m” (multiplet) and broad resonances are indicated by “b”. Residual protic solvent of CDCl3 (1H, δ 7.27 ppm; 13C, δ 77.0 ppm (central resonance of the triplet)), D2O (1H, δ4.79 ppm), and MeOD (1H, δ3.31 ppm and 13C, δ 49.0 ppm) were used for chemical shifts calibration.
Mass Spectroscopy
Accurate mass measurements (high-resolution mass spectrometry, HRMS) were performed on Bruker microTOF-II mass spectrometer using ESI in the positive mode and direct flow sample introduction in CH3CN/H2O (9:1) solvent system. Either protonated molecular ions [M + nH]n+ or adducts [M + nX]n+ (X = Na, K, or NH4) were used for empirical formula confirmation. MALDI-TOF experiments were performed on Bruker Daltonics MALDI instrument. The conjugate was dissolved in ultrapurified water at 5 mg/mL and 2,5-dihydroxybenzoic acid (DHB) matrix was dissolved in a 50:50 (v/v) acetonitrile/water mixture at 10 mg/ mL concentration. The samples were prepared by mixing 10 μL of conjugate solution with 10 μL of DHB solution, and 3 μL of the sample was spotted on a Bruker Daltonics MALDI plate.
High-Performance Liquid Chromatography
The purity of intermediates and final dendrimer conjugate were analyzed using HPLC. The chromatograph (Waters Corporation, Milford, Massachusetts) was equipped with a 1525 binary pump, an in-line degasser AF, a 717 plus autosampler, a 2998 photodiode array detector, a 2475 multi λ fluorescence detector interfaced with Waters Empower software, and a Symmetry C18 reverse-phase column having 5 μm particle size, 25 cm length, and 4.6 mm internal diameter. The HPLC chromatograms of the starting dendrimer (G6-OH) and all the intermediates were monitored at 210 nm. D-mino was monitored at both 210 and 277 nm. The fluorescently (Cy5) labeled dendrimer was monitored at both 650 and 210 nm using PDA and fluorescence detectors. For G6-OH, intermediates and D-mino conjugate, a gradient flow was used in HPLC starting with 90:10 (H2O/ACN), gradually increasing to 25:75 (H2O/ACN) in 10 min, to 10:90 (H2O/ACN) in 25 min and returning to 90:10 (H2O/ACN) in 35 min maintaining a flow rate of 1 mL/min. For Cy5-labeled dendrimer, a different HPLC method was used starting with 90:10 (H2O/ACN), gradually increasing to 10:90 (H2O/ACN) in 4 min, maintaining 10:90 (H2O/ACN) up to 20 min and returning to 90:10 (H2O/ACN) in 25 min with a flow rate of 1 mL/min.
Dynamic Light Scattering and ζ Potential
The particle size and ζ-potential of G6-OH and their respective conjugates were determined by using a Zetasizer Nano ZS (Malvern Instrument Ltd. Worchester, U.K) equipped with a 50 mW He–Ne laser (633 nm). The conjugates (G6-OH and D-mino) were dissolved in deionized water (18.2 Ω) to make solutions with a final concentration of 0.2 mg/mL. The solution was filtered through a cellulose acetate membrane (0.45 μm, PALL Life Science), and DLS measurements were performed in triplicate, at 25 °C with a scattering angle of 173°.
Cell Culture
BV-2 microglia were maintained in Dulbecco’s Modiffed Eagles Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidiffed incubator with atmosphere of 5% CO2. The cells were subcultured twice a week using 0.05% trypsin–ethylenediamine tetra-acetic acid (EDTA).
Flow Cytometry
BV-2 microglia were seeded at a density of 1 × 105 cells in a 12-well plate (Costar, Cambridge, MA). After being cultured overnight, the cells were activated with 100 ng/mL LPS for 3 h and treated with 2.5 μM of Cy5–D-mino for 3 h in the presence of LPS. The cells were harvested with trypsin–EDTA, centrifuged, and washed twice with PBS, and then finally resuspended in 2% paraformaldehyde (PFA) PBS. Cells were scanned with AccuriC6 (BD Biosciences, San Jose, CA) by counting 50 000 events. The mean Cy5 fluorescence intensity of cells was graphed in the histogram plot.
Immunocytochemistry
BV-2 microglia were grown overnight after seeding at a density of 4 × 104 cells in an 8-well slide chamber (IbidiGmbh, Martinsried, Germany). After LPS activation at a concentration of 100 ng/mL for 3 h, the cells were treated with 2.5 μM of Cy5–D-mino for 3 h. After washing with PBS, the cells were fixed with 4% PFA in PBS for 10 min, washed twice, and incubated with blocking solution (1% bovine serum albumin [BSA] and 0.1% Triton X-100 in PBS) for 30 min and then Alexa-488 conjugated Isolectin IB4 (Invitrogen, Carlsbad, CA) for 3 h. 4,6-Diamidino-2-phenyl-indole (DAPI, Vectorlabs, San Diego, CA) was applied for nucleus counter staining. Images were captured using confocal microscope of Zeiss 510 (Carl Zeiss, Meditec, Oberkochen, Germany).
Drug Treatment
All procedures of drug treatment and supernatant harvest were followed as previously described.58 For oxidative stress and inflammation cytokine assays, cells were seeded at a density of 1 × 104 cells in 96-well plates (200 μL culture medium per well) and activated with 100 ng/mL LPS for 3 h before drug treatment. The preactivated BV-2 microglia were treated with free and dendrimer-conjugated 9-amino-minocycline minocycline (containing equivalent amount of drug in conjugated form) for 3 h. Afterward, the cells were washed out and restimulated with 100 ng/mL LPS in fresh media for 24 h. The supernatants were harvested for oxidative stress and inflammation cytokine assays.
Nitrite Measurement
NO release was measured by accumulated nitrite in BV-2 culture supernatants using Griess reagent system (Promega, Madison, WI) as per the manufacturer’s protocol. All experiments were performed at least three times, and data are expressed as mean ± standard error of the mean (SEM).
Tumor Necrosis Factor α Assay
Cell culture media were centrifuged in 96-well plates. The collected supernatants were used at a 5-fold dilution for the measurement of TNF-α using a mouse TNF-α Quantikine ELISA kit (R & D Systems, Minneapolis, MN).
Cytotoxic Assay
Cell viability was measured at the end of cell culture for nitrite and TNF-α assays using MTS cell proliferation assay kit (Promega, Minneapolis, MN). MTS stock solution was diluted with fresh cell culture media, and 100 μL of MTS–media mixture was added to each 96-well plate and incubated at 37 °C in a humidiffed incubator with atmosphere of 5% CO2 for 1 h. Absorbance was read at 450 nm using fluorescence microplate reader (BioTek Instruments, Winooski, VT).
Statistics
GraphPad Prism5 was used for statistical analyses. One-way ANOVA (host hoc Tukey’s test) was used to determine significance between groups, and values at P < 0.05 were considered significantly different. Results were expressed as mean ± SEM value.
Synthetic Protocols and Characterization
The compound 2 was synthesized by the adaptation of the previously published procedure.21
Synthesis of Compound 2
To a solution of 6-bromohexanoic acid 1 (5 g, 25.63 mmol) in DMF, sodium azide (4.99 g, 76.90 mmol) was added, and the resulting solution was stirred at 50 °C for 18 h. The reaction was monitored with the help of thin-layer chromatography. Upon completion, the reaction mixture was diluted with DCM (100 mL) and washed with ice cold water (3 × 20 mL) followed by washing with brine (3 × 10 mL). The organic layer was then dried using anhydrous sodium sulfate, filtered, and evaporated. The crude mixture was purified through silica gel combi-flash chromatography system using an ethyl acetate/Hexanes mixture to afford 2 as transparent oil (yield: 60%).
Synthesis of Compound 3
6-Azidohexanoic acid 2 (2.5 g, 15.91 mmol) was dissolved in DCM (8 mL). The solution was cooled to 0 °C, and a solution of DCC (1.64 g, 7.95 mmol) in DCM (1 mL) was slowly added to the reaction mixture. The reaction was left to proceed at room temperature for 12 h. On completion, reaction mixture was kept at ice bath for 30 min. Dicyclohexylurea was precipitated as a white solid. The reaction mixture was filtered and washed with ether. DCM was evaporated using a rotary evaporator, and the transparent viscous oil was redissolved in toluene. The product was kept in solution in an ice bath for another 60 min and filtered again. Desired product 3 was obtained as transparent oil (yield: 3.3 g, 70%). 1H NMR (500 MHz, CDCl3) δ): 3.29 (t, J = 6.8 Hz, 1H), 2.48 (t, J = 7.3 Hz, 1H), 1.71 (dd, J = 15.2, 7.6 Hz, 1H), 1.66–1.58 (m, 1H), 1.50–1.40 (m, 1H). 13C (1H) NMR (126 MHz, CDCl3) δ: 169.0, 51.0, 34.9, 28.40, 25.8, 23.6. HRMS (ESI+) m/z: [M + Na]+ calcd for C12H20N6O3Na, 319.3197; found, 319.1489.
Synthesis of Compound 5
9-Amino minocycline hydrochloride 4 (300 mg, 0.589 mmol, 1eq), and NaHCO3 (99 mg, 1.178 mmol, 2eq) were dissolved in 10 mL of DCM and stirred for 30 min at room temperature under argon atmosphere. 6-Azidohexanoic anhydride 3 (157 mg, 0.530 mmol, 0.9 eq) was dissolved in 2 mL of DMF and added drop-wise to the reaction mixture. The reaction mixture was stirred in the dark for 4 h. The reaction was monitored with the help of HPLC. Upon completion, the reaction mixture was diluted with 40 mL of DCM, and the organic layer was washed with brine solution (3 × 20 mL). The organic layer was dried using anhydrous sodium sulfate then filtered and concentrated under reduced pressure. The crude mixture was purified by reverse-phase HPLC to afford the title compound as a yellow solid in 52% yield (200 mg). 1H NMR (500 MHz, MeOD) δ: 8.14 (s, 1H), 3.97 (s, 1H), 3.48–3.32 (m, 4H), 3.07 (s, 1H), 3.03–2.80 (m, 7H), 2.58 (d, J = 5.7 Hz, 6H), 2.48 (t, J = 7.4 Hz, 2H), 2.25–2.13 (m, 2H), 1.79–1.70 (m, 2H), 1.64 (dd, J = 15.1, 7.0 Hz, 4H), 1.55–1.44 (m, 2H). 13C (1H) NMR (126 MHz, DMSO) δ: 170.18, 152.37, 118.11, 108.94, 74.18, 73.43, 64.82, 25.79, 25.41. HRMS (ESI+) m/z: [M + H]+ calcd for C29H37N7O8, 611.6560; found, 612.2776 . HPLC purity: 100%; retention time: 15.0 min
Synthesis of Compound 7
To a solution of generation-6 hydroxyl-terminated PAMAM dendrimer 6 (256 end groups) (2.5 g, 0.043 mmol) in DMF (35 mL) at 0 °C, sodium hydride (1.24 g, 51.72 mmol) was added. The mixture was stirred at 0 °C for 15 min followed by the addition of propargyl bromide (0.154 mL, 1.72 mmol) drop-wise. The mixture was stirred at room temperature overnight. The reaction was quenched by the drop-wise addition of saturated NH4Cl sol. at 0 °C. The pH was adjusted around 7–7.5 using ammonium chloride, and the reaction mixture was transferred to a 3000 MWCO dialysis membrane and dialyzed against 4 gallons of Nanopure water for 36 h. Stirring was applied to dialyze the mixture, and the water was changed six or seven times at regular intervals. The water was lyophilized, and desired product 7 was obtained as off-white hygroscopic solid material in quantitative yield. 1H NMR (500 MHz, DMSO) δ: 7.94 (m, amidic H), 4.80 (m, OH), 4.12 (s, 50H), 3.75 (s, 40H), 3.36 (m, dendrimer), 3.11 (dendrimer), 2.64 (m, dendrimer), 2.43 (m, dendrimer), 2.20 (m, dendrimer). HPLC purity: 99.7%; retention time: 13.3 min.
Synthesis of Compound 8 (D-mino)
An acetylene-terminated dendrimer 7 (250 mg, 0.0042 mmol) and azide-terminated 9-amino-minocycline linker 5 (103 mg, 0.168 mmol) were suspended in a 3:1 mixture of DMF and water (5 mL/mmol) in a 5 mL glass vial equipped with a small magnetic stirring bar. To this, copper sulfate (27 mg, 0.105 mmol, 0.2 eq/acetylene) and sodium ascorbate (21 mg, 0.105 mmol, 0.2 eq/acetylene) were added, and the vial was tightly sealed with an aluminum and Teflon crimp top. The mixture was then irradiated for 6 h at 40 °C using an irradiation power of 100 W. After completion of the reaction, the vial was cooled to 25 °C by gas jet cooling before it was opened. The reaction mixture was then transferred to 10 000 MWCO dialysis tube and dialyzed against DMF for 6 h. DMF was replaced three or four times at regular intervals. It was further dialyzed for 12 h against a 15% solution of EDTA. The reaction mixture was finally stirred against DI water for another 12 h. The aqueous layer was lyophilized to provide dark brownish powder in a quantitative yield. 1H NMR (500 MHz, DMSO) δ: 8.20–7.60 (m, amidic H, triazole H, H8 minocycline), 5.10–4.55 (m, OH), 4.48–4.38 (m, −O−CH2−triazole, 50H), 4.36–4.18 (m, −O−CH2−dendrimer), 4.00–3.90 (m, H4 minocycline, 25H) 3.54–3.15 (m, dendrimer), 3.12–2.96 (m, dendrimer), 2.66–2.50 (m, dendrimer), 2.40–2.31(m, dendrimer), 2.19–2.06 (m, dendrimer), 1.93–1.90(m, H5,6 minocycline), 1.86–1.70 (m, H5, H6 minocycline, linker), 1.60–0.97 (m, linker H, H5 minocycline). HPLC purity: 98.8%; retention time: 14.6 min
Synthesis of Compound 9
A reaction mixture containing acetylene-terminated dendrimer 7 (2 g, 0.033 mmol) was dissolved in anhydrous DMF (5 mL), and the addition of EDC (151 mg, 0.79 mmol), DMAP (97 mg, 0.46 mmol) and Boc−GABA−OH (81 mg, 0.39 mmol) was performed. The mixture was stirred for 24 h. The reaction mixture was then transferred to a dialysis tube (2000 MWCO) and dialyzed against DMF for 6 h. DMF was changed three times at regular intervals followed by the dialysis against DI water for another 24 h, changing water in between. Finally, the product 9 was lyophilized to provide off-white solid. 1H NMR (500 MHz, DMSO) δ: 8.42–7.52 (m, amide H), 4.77 (s, OH), 4.15–4.10 (m, O−CH2−propargyl), 3.85–3.70 (m, O–CH2), 3.46–3.36 (m, dendrimer), 3.24–3.04 (m, dendrimer), 2.90–2.88 (m, dendrimer), 2.70 (s, dendrimer), 2.47–2.10 (m, dendrimer), 1.36 (s, BOC−H).
Synthesis of Compound 10
To the dendrimer 9 (1 g, 0.015 mmol) in a 50 mL round-bottom flask, anhydrous DCM (4 mL) was added, which was followed by the addition of trifluoroacetic acid (2 mL), and the reaction mixture was stirred for 6 h. The solvent was evaporated thoroughly. Methanol was added to the compound and evaporated using rotary evaporator. This step was repeated five or six times. The desired compound 10 was obtained as off-white solid. The crude material was used as such for next step.
Synthesis of Compound 11
An acetylene terminated dendrimer 10 (250 mg, 0.0042 mmol) and azide terminated 9-amino-minocycline linker (5) (103 mg, 0.168 mmol) were suspended in a 3:1 mixture of DMF and water (5 mL/mmol) in a 5 mL glass vial equipped with a small magnetic stirring bar. To this solution, copper sulfate (27 mg, 0.105 mmol, 0.2 equiv of acetylene) and sodium ascorbate (21 mg, 0.105 mmol, 0.2 equiv of acetylene) were added, and the vial was tightly sealed with an aluminum and Teflon crimp top. The mixture was then irradiated in microwave for 6 h at 40 °C using an irradiation power of 100 W. After the completion of the reaction, the vial was cooled to 25 °C by gas jet cooling before it was opened. The reaction mixture was then transferred to 3000 MWCO dialysis tube and dialyzed against DMF for 6 h. DMF was replaced three or four times in between. It was then dialyzed for 12 h against 15% solution of EDTA. Reaction mixture was finally stirred against DI water for another 12 h. The aqueous layer was lyophilized to provide dark brownish powder 11 in quantitative yield. 1H NMR (500 MHz, DMSO) δ: 8.18–7.70 (m, amidic H, triazole H, H8 minocycline), 5.05–4.55 (m, OH), 4.55–4.45 (m, −O−CH2−triazole H), 4.40–4.28 (m,−O−CH2−dendrimer), 4.10–3.90 (m, H4 minocycline,) 3.46–3.38 (m, dendrimer), 3.25–3.20 (m, dendrimer), 3.14–3.06 (m, dendrimer), 2.69–2.55 (m, dendrimer), 2.46–2.41(m, dendrimer), 2.25–2.15 (m, dendrimer), 1.93–0.97(m, H5,6 minocycline, GABA-linker). HPLC purity: 93.7%; retention time: 7.4 min
Synthesis of Compound 12
To a stirred solution of compound 11 (200 mg, 0.003 mmol) in anhydrous DMF at room temperature, N,N-di-isopropylethylamine was added slowly to adjust the pH around 7.5 under argon atmosphere. Cy5 NHS ester (4.6 mg, 0.007 mmol) was dissolved in 2 mL of DMF and added drop-wise to the reaction flask. The solution was stirred for 24 h. Upon completion, the reaction mixture was transferred to a 10 000 MWCO dialysis tube and dialyzed against DMF for 12 h. DMF was replaced three or four times at regular intervals. It was then dialyzed for 6 h against DI water. The aqueous layer was lyophilized to provide dark blue powder in quantitative yield. 1H NMR (500 MHz, DMSO) δ: 8.40–8.32 (m, Cy5 H), 8.22–7.75 (m, internal amide H dendrimer, triazole and minocycline), 7.65–7.61 (m, cy5 H), 7.40–7.15 (m, cy5 H), 6.75–6.55 (m, cy5 H), 6.35–6.27 (m, cy5 H), 4.55–4.45 (m, dendrimer linker H), 4.40–4.25 (m, dendrimer linker H), 4.16–4.05 (m, dendrimer linker H), 3.46–3.36 (m, dendrimer), 3.30–3.20 (m, dendrimer), 3.14–3.02 (m, dendrimer), 2.75–2.55 (m, dendrimer), 2.46–2.41(m, dendrimer), 2.26–2.15 (m, dendrimer), 1.93–0.97(m, H5,6 minocycline, GABA linker). HPLC purity: 99.3%; retention time: 12.8 min
In Vitro Drug-Release Studies
In vitro drug release from D-mino conjugate was studied at two different conditions: (1) physiological pH using phosphate-buffered saline (pH 7.4) and (2) in the presence of esterase in citrate buffer (pH 5.5). The conjugate was dissolved in PBS (4 mL × 3) and citrate buffer (4 mL × 3) at a concentration of 1 mg/mL. The esterase was added as 2 equiv per amide to the citrate buffer solution. The solutions were continuously stirred and maintained at 37 °C. The enzyme was replenished every 2 days to maintain the activity. The samples were withdrawn at appropriate time points in triplicates, and the samples containing enzyme were quenched with methanol to precipitate the enzyme. The samples were then centrifuged for 5 min at 10 000 rpm, and the clear supernatant was collected. The final samples were stored at −80°C until HPLC analysis.
In Vivo Cy5−D-mino Localization
Time pregnant New Zealand white rabbits were purchased from Robinson Services Inc. Time represents the specific dates the rabbits were bred in order to calculate the gestational days. Experimental procedures were approved by the Johns Hopkins University Animal Care and Use Committee (IACUC). After 1 week of acclimation, the pregnant rabbits underwent laparotomy on gestational day 28 (G28) and received a total of 3200 EU of lipopolysaccharides (Escherichia coli serotype O127:B8, Sigma-Aldrich, Saint Louis, MO) injection along the wall of the uterus as previously described.54,55 The kits from LPS-injected dams (defined as endotoxin kits) received intravenous administration of Cy5-D-mino (55 mg/kg) on postnatal day 1 (PND1) and were sacrificed 24 h post-injection. The animals were perfused with PBS, and their brains were post-fixed in 10% formalin for 48 h and then cryo-protected in 30% sucrose. To evaluate the co-localization of Cy5-D-mino and astrocytes or microglia, brain sections (30 μm) were incubated overnight at 4 °C with chicken anti-glial fibrillary acidic protein (1:250, Abcam) and goat anti-IBA1 (1:250, Abcam). Sections were subsequently washed and incubated with fluorescent secondary antibodies (1:250; Life Technologies) for 2 h at room temperature. Next, the sections were incubated with DAPI (1:1000, Invitrogen) for 15 min. After washing, the slides were dried and cover-slipped with mounting media (Dako, Carpinteria, CA). Confocal images were acquired with a Zeiss ZEN LSM 710 and processed with ZEN software.
Supplementary Material
Acknowledgments
This study was partly funded by the NIBIB grant no. R01EB018306 (R.M.K.) and NICHD grant no. R01HD076901 (R.M.K.). The authors thank the Wilmer Core Module for Microscopy and Imaging for allowing us to use LSM710 confocal microscope
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.7b00569.
Complete characterization data including NMR, ESI-TOF-MS, MALDI-TOF MS, and analytical HPLC. (PDF)
Notes
The authors declare no competing financial interest.
ORCID
Rangaramanujam M. Kannan: 0000-0002-3712-7836
References
- 1.Heemels M-T. Neurodegenerative diseases. Nature. 2016;539:179–179. doi: 10.1038/539179a. [DOI] [PubMed] [Google Scholar]
- 2.Pardridge WM. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wegener G, Rujescu D. The current development of CNS drug research. Int J Neuropsychopharmacol. 2013;16:1687–1693. doi: 10.1017/S1461145713000345. [DOI] [PubMed] [Google Scholar]
- 4.Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discovery. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 5.Lee H-M, Kim Y. Drug Repurposing Is a New Opportunity for Developing Drugs against Neuropsychiatric Disorders. Schizophr Res Treat. 2016;2016:1. doi: 10.1155/2016/6378137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi Y, Kim H-S, Shin KY, Kim E-M, Kim M, Kim H-S, Park CH, Jeong YH, Yoo J, et al. Minocycline Attenuates Neuronal Cell Death and Improves Cognitive Impairment in Alzheimer’s Disease Models. Neuropsychopharmacology. 2007;32:2393–2404. doi: 10.1038/sj.npp.1301377. [DOI] [PubMed] [Google Scholar]
- 8.Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas M, Ashizawa T, Jankovic J. Minocycline in Huntington’s disease: A pilot study. Mov Disord. 2004;19:692–695. doi: 10.1002/mds.20018. [DOI] [PubMed] [Google Scholar]
- 10.Abdel Baki SG, Schwab B, Haber M, Fenton AA, Bergold PJ. Minocycline Synergizes with N-Acetylcysteine and Improves Cognition and Memory Following Traumatic Brain Injury in Rats. PLoS One. 2010;5:e12490. doi: 10.1371/journal.pone.0012490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovesdi E, Kamnaksh A, Wingo D, Ahmed F, Grunberg NE, Long JB, Kasper CE, Agoston DV. Acute Minocycline Treatment Mitigates the Symptoms of Mild Blast-Induced Traumatic Brain Injury. Front Neurol. 2012;3:111. doi: 10.3389/fneur.2012.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wells JEA, Hurlbert RJ, Fehlings MG, Yong VW. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126:1628–1637. doi: 10.1093/brain/awg178. [DOI] [PubMed] [Google Scholar]
- 13.Plane JM, Shen Y, Pleasure DE, Deng W. Prospects for Minocycline Neuroprotection. Arch Neurol. 2010;67:1442–1448. doi: 10.1001/archneurol.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garrido-Mesa N, Zarzuelo A, Gálvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169:337–352. doi: 10.1111/bph.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z, Wang Z, Nong J, Nix CA, Ji H-F, Zhong Y. Metal ion-assisted self-assembly of complexes for controlled and sustained release of minocycline for biomedical applications. Biofabrication. 2015;7:015006–015006. doi: 10.1088/1758-5090/7/1/015006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang L, Sugama S, Chirichigno JW, Gregorio J, Lorenzl S, Shin DH, Browne SE, Shimizu Y, Joh TH, Beal, et al. Minocycline enhances MPTP toxicity to dopaminergic neurons. J Neurosci Res. 2003;74:278–285. doi: 10.1002/jnr.10709. [DOI] [PubMed] [Google Scholar]
- 17.Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S, Roeck J, Ryder J, Smith P. A New Class of Polymers: Starburst-Dendritic Macromolecules. Polym J. 1985;17:117–132. [Google Scholar]
- 18.Newkome GR, Yao Z, Baker GR, Gupta VK. Micelles. Part 1. Cascade molecules: a new approach to micelles A [27]-arborol. J Org Chem. 1985;50:2003–2004. [Google Scholar]
- 19.Sharma R, Zhang I, Shiao TC, Pavan GM, Maysinger D, Roy R. Low generation polyamine dendrimers bearing flexible tetraethylene glycol as nanocarriers for plasmids and siRNA. Nanoscale. 2016;8:5106–5119. doi: 10.1039/c5nr06757j. [DOI] [PubMed] [Google Scholar]
- 20.Caminade AM, Turrin CO. Dendrimers for drug delivery. J Mater Chem B. 2014;2:4055–4066. doi: 10.1039/c4tb00171k. [DOI] [PubMed] [Google Scholar]
- 21.Sharma A, Mejia D, Maysinger D, Kakkar A. Design and synthesis of multifunctional traceable dendrimers for visualizing drug delivery. RSC Adv. 2014;4:19242–19245. [Google Scholar]
- 22.Wang M, Li Y, HuangFu M, Xiao Y, Zhang T, Han M, Xu D, Li F, Ling D, Jin Y, Gao J. Pluronic-attached polyamidoamine dendrimer conjugates overcome drug resistance in breast cancer. Nanomedicine. 2016;11:2917–2934. doi: 10.2217/nnm-2016-0252. [DOI] [PubMed] [Google Scholar]
- 23.Tomalia DA, Reyna LA, Svenson S. Dendrimers as multi-purpose nanodevices for oncology drug delivery and diagnostic imaging. Biochem Soc Trans. 2007;35:61–67. doi: 10.1042/BST0350061. [DOI] [PubMed] [Google Scholar]
- 24.Kannan RM, Nance E, Kannan S, Tomalia DA. Emerging concepts in dendrimer-based nanomedicine: from design principles to clinical applications. J Intern Med. 2014;276:579–617. doi: 10.1111/joim.12280. [DOI] [PubMed] [Google Scholar]
- 25.Mintzer MA, Grinstaff MW. Biomedical applications of dendrimers: a tutorial. Chem Soc Rev. 2011;40:173–190. doi: 10.1039/b901839p. [DOI] [PubMed] [Google Scholar]
- 26.Madaan K, Kumar S, Poonia N, Lather V, Pandita D. Dendrimers in drug delivery and targeting: Drug-dendrimer interactions and toxicity issues. J Pharm BioAllied Sci. 2014;6:139–150. doi: 10.4103/0975-7406.130965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esfand R, Tomalia DA. Poly(amidoamine) (PAMAM) dendrimers: from biomimicry to drug delivery and biomedical applications. Drug Discovery Today. 2001;6:427–436. doi: 10.1016/s1359-6446(01)01757-3. [DOI] [PubMed] [Google Scholar]
- 28.Xu L, Cooper RC, Wang J, Yeudall WA, Yang H. Synthesis and Application of Injectable Bioorthogonal Dendrimer Hydrogels for Local Drug Delivery. ACS Biomater Sci Eng. 2017;3(8):1641–1653. doi: 10.1021/acsbiomaterials.7b00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong PT, Chen D, Tang S, Yanik S, Payne M, Mukherjee J, Coulter A, Tang K, Tao K, Sun K, et al. Modular Integration of Upconverting Nanocrystal-Dendrimer Composites for Folate Receptor-Specific NIR Imaging and Light-Triggered Drug Release. Small. 2015;11:6078–6090. doi: 10.1002/smll.201501575. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Cai X, Hu J, Shao N, Wang F, Zhang Q, Xiao J, Cheng Y. Glutathione-Triggered “Off-On” Release of Anticancer Drugs from Dendrimer-Encapsulated Gold Nanoparticles. J Am Chem Soc. 2013;135:9805–9810. doi: 10.1021/ja402903h. [DOI] [PubMed] [Google Scholar]
- 31.Yiyun C, Na M, Tongwen X, Rongqiang F, Xueyuan W, Xiaomin W, Longping W. Transdermal Delivery of Nonsteroidal Anti-Inflammatory Drugs Mediated by Polyamidoamine (PAMAM) Dendrimers. J Pharm Sci. 2007;96:595–602. doi: 10.1002/jps.20745. [DOI] [PubMed] [Google Scholar]
- 32.Kurtoglu YE, Mishra MK, Kannan S, Kannan RM. Drug release characteristics of PAMAM dendrimer-drug conjugates with different linkers. Int J Pharm. 2010;384:189–194. doi: 10.1016/j.ijpharm.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Kannan S, Dai H, Navath RS, Balakrishnan B, Jyoti A, Janisse J, Romero R, Kannan RM. Dendrimer-Based Postnatal Therapy for Neuroinflammation and Cerebral Palsy in a Rabbit Model. Sci Transl Med. 2012;4:130ra46–130ra46. doi: 10.1126/scitranslmed.3003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nance E, Zhang F, Mishra MK, Zhang Z, Kambhampati SP, Kannan RM, Kannan S. Nanoscale effects in dendrimer-mediated targeting of neuroinflammation. Biomaterials. 2016;101:96–107. doi: 10.1016/j.biomaterials.2016.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mishra MK, Beaty CA, Lesniak WG, Kambhampati SP, Zhang F, Wilson MA, Blue ME, Troncoso JC, Kannan S, Johnston MV, et al. Dendrimer Brain Uptake and Targeted Therapy for Brain Injury in a Large Animal Model of Hypothermic Circulatory Arrest. ACS Nano. 2014;8:2134–2147. doi: 10.1021/nn404872e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lesniak WG, Mishra MK, Jyoti A, Balakrishnan B, Zhang F, Nance E, Romero R, Kannan S, Kannan RM. Biodistribution of Fluorescently Labeled PAMAM Dendrimers in Neonatal Rabbits: Effect of Neuroinflammation. Mol Pharmaceutics. 2013;10:4560–4571. doi: 10.1021/mp400371r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dai H, Navath RS, Balakrishnan B, Guru BR, Mishra MK, Romero R, Kannan RM, Kannan S. Intrinsic targeting of inflammatory cells in the brain by polyamidoamine dendrimers upon subarachnoid administration. Nanomedicine (London, U K) 2010;5:1317–1329. doi: 10.2217/nnm.10.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Y, Johnson MA, Mehrabian Z, Mishra MK, Kannan R, Miller NR, Bernstein SL. Dendrimers Target the Ischemic Lesion in Rodent and Primate Models of Nonarteritic Anterior Ischemic Optic Neuropathy. PLoS One. 2016;11:e0154437. doi: 10.1371/journal.pone.0154437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kambhampati SP, Clunies-Ross AJ, Bhutto I, Mishra MK, Edwards M, McLeod DS, Kannan RM, Lutty G. Systemic and intravitreal delivery of dendrimers to activated microglia/ macrophage in ischemia/reperfusion mouse retina. Invest Ophthalmol Visual Sci. 2015;56:4413–4424. doi: 10.1167/iovs.14-16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nance E, Porambo M, Zhang F, Mishra MK, Buelow M, Getzenberg R, Johnston M, Kannan RM, Fatemi A, Kannan S. Systemic dendrimer-drug treatment of ischemia-induced neonatal white matter injury. J Controlled Release. 2015;214:112–120. doi: 10.1016/j.jconrel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang F, Trent Magruder J, Lin Y-A, Crawford TC, Grimm JC, Sciortino CM, Wilson MA, Blue ME, Kannan S, Johnston MV, et al. Generation-6 hydroxyl PAMAM dendrimers improve CNS penetration from intravenous administration in a large animal brain injury model. J Controlled Release. 2017;249:173–182. doi: 10.1016/j.jconrel.2017.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao W, Xu P, Pang Z, Zhao J, Chai Z, Li X, Li H, Jiang M, Cheng H, Zhang B, Cheng N. Local delivery of minocycline-loaded PEG-PLA nanoparticles for the enhanced treatment of periodontitis in dogs. Int J Nanomed. 2014;9:3963–3970. doi: 10.2147/IJN.S67521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barton DHR, Magnus PD. Experiments on the synthesis of tetracycline. Part I. Introduction to the series. J Chem Soc C. 1971:2164–2166. doi: 10.1039/j39710002164. [DOI] [PubMed] [Google Scholar]
- 44.Muxfeldt HH, Michael J. US3947517 A. Stereoselective introduction of tetracyclines hydroxyl group at 12(a) position in synthesis of tetracyclines Google Patents. 1976
- 45.Wu P, Malkoch M, Hunt JN, Vestberg R, Kaltgrad E, Finn MG, Fokin VV, Sharpless KB, Hawker CJ. Multivalent, bifunctional dendrimers prepared by click chemistry. Chem Commun. 2005:5775–5777. doi: 10.1039/b512021g. [DOI] [PubMed] [Google Scholar]
- 46.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 47.Sharma A, Neibert K, Sharma R, Hourani R, Maysinger D, Kakkar A. Facile Construction of Multifunctional Nanocarriers Using Sequential Click Chemistry for Applications in Biology. Macromolecules. 2011;44:521–529. [Google Scholar]
- 48.Neibert K, Gosein V, Sharma A, Khan M, Whitehead MA, Maysinger D, Kakkar A. Click” Dendrimers as Anti-inflammatory Agents: With Insights into Their Binding from Molecular Modeling Studies. Mol Pharmaceutics. 2013;10:2502–2508. doi: 10.1021/mp4000508. [DOI] [PubMed] [Google Scholar]
- 49.Sharma A, Mejía D, Regnaud A, Uhlig N, Li C-J, Maysinger D, Kakkar A. Combined A3 Coupling and Click Chemistry Approach for the Synthesis of Dendrimer-Based Biological Tools. ACS Macro Lett. 2014;3:1079–1083. doi: 10.1021/mz5006298. [DOI] [PubMed] [Google Scholar]
- 50.Sharma R, Naresh K, Chabre YM, Rej R, Saadeh NK, Roy R. Onion peel dendrimers: a straightforward synthetic approach towards highly diversified architectures. Polym Chem. 2014;5:4321–4331. [Google Scholar]
- 51.Sharma R, Zhang I, Abbassi L, Rej R, Maysinger D, Roy R. A fast track strategy toward highly functionalized dendrimers with different structural layers: an onion peel approach. Polym Chem. 2015;6:1436–1444. [Google Scholar]
- 52.Sk UH, Kambhampati SP, Mishra MK, Lesniak WG, Zhang F, Kannan RM. Enhancing the Efficacy of Ara-C through Conjugation with PAMAM Dendrimer and Linear PEG: A Comparative Study. Biomacromolecules. 2013;14:801–810. doi: 10.1021/bm3018615. [DOI] [PubMed] [Google Scholar]
- 53.D’Souza AJM, Topp EM. Release from polymeric prodrugs: Linkages and their degradation. J Pharm Sci. 2004;93:1962–1979. doi: 10.1002/jps.20096. [DOI] [PubMed] [Google Scholar]
- 54.Perumal OP, Inapagolla R, Kannan S, Kannan RM. The effect of surface functionality on cellular trafficking of dendrimers. Biomaterials. 2008;29:3469–3476. doi: 10.1016/j.biomaterials.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 55.Wang B, Navath RS, Romero R, Kannan S, Kannan R. Anti-inflammatory and anti-oxidant activity of anionic dendrimer-N-acetyl cysteine conjugates in activated microglial cells. Int J Pharm. 2009;377:159–168. doi: 10.1016/j.ijpharm.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kambhampati SP, Mishra MK, Mastorakos P, Oh Y, Lutty GA, Kannan RM. Intracellular delivery of dendrimer triamcinolone acetonide conjugates into microglial and human retinal pigment epithelial cells. Eur J Pharm Biopharm. 2015;95:239–249. doi: 10.1016/j.ejpb.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saadani-Makki F, Kannan S, Lu X, Janisse J, Dawe E, Edwin S, Romero R, Chugani D. Intrauterine administration of endotoxin leads to motor deficits in a rabbit model: a link between prenatal infection and cerebral palsy. Am J Obstet Gynecol. 2008;199:651e1–651e7. doi: 10.1016/j.ajog.2008.06.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z, Wang J, Gao F, Zhang J, Tian H, Shi X, Lian C, Sun Y, Li W, Xu JY, et al. Human Adipose-Derived Stem Cells Delay Retinal Degeneration in Royal College of Surgeons Rats Through Anti-Apoptotic and VEGF-Mediated Neuroprotective Effects. Curr Mol Med. 2016;16:553–566. doi: 10.2174/1566524016666160607090538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.