

Abstract
Rationale:
A rapid and massive influx of inflammatory cells occurs into ischemic area after myocardial infarction (MI), resulting in local release of cytokines and growth factors. Yet, the mechanisms regulating their production are not fully explored. The release of extracellular vesicles (EVs) in the interstitial space curbs important biological functions, including inflammation, and influences the development of cardiovascular diseases. To date, there is no evidence for in situ release of cardiac EVs after MI.
Objective:
The present study tested the hypothesis that local EV generation in the infarcted heart coordinates cardiac inflammation after MI.
Methods and Results:
Coronary artery ligation in mice transiently increases EV levels in the left ventricle when compared with sham animals. EVs from infarcted hearts were characterized as large vesicles (252±18 nm) expressing cardiomyocyte and endothelial markers and small EVs (118±4 nm) harboring exosomal markers, such as CD (cluster of differentiation) 63 and CD9. Cardiac large EVs generated after MI, but not small EVs or sham EVs, increased the release of IL (interleukin)-6, CCL (chemokine ligand) 2, and CCL7 from fluorescence-activated cell–sorted Ly6C+ cardiac monocytes. EVs of similar diameter were also isolated from fragments of interventricular septum obtained from patients undergoing aortic valve replacement, thus supporting the clinical relevance of our findings in mice.
Conclusions:
The present study demonstrates that acute MI transiently increases the generation of cardiac EVs characterized as both exosomes and microvesicles, originating mainly from cardiomyocytes and endothelial cells. EVs accumulating in the ischemic myocardium are rapidly taken up by infiltrating monocytes and regulate local inflammatory responses.
Keywords: exosomes; humans; inflammation; myocardial infarction; myocytes, cardiac
Acute myocardial infarction (MI) induces a rapid and massive influx of inflammatory cells into ischemic areas, resulting in the release of soluble chemokines, cytokines, and growth factors.1 In murine models of MI, these inflammatory cells consist, in part, of Ly6Chigh and Ly6Clow monocytes.1,2 Whereas Ly6Chigh monocytes are predominant in the early phase of myocardial healing, Ly6Clow monocytes peak at a later time point, but both monocyte subtypes have been shown to modulate post-MI cardiac remodeling and function.2 After infiltrating the inflamed tissues, Ly6Chigh and Ly6Clow monocytes may differentiate into inflammatory or reparative macrophages.1 Inflammatory macrophages are related to proinflammatory processes and adverse left ventricular (LV) remodeling, whereas reparative macrophages are involved in inflammation resolution and tissue repair.
Editorial, see p 15
In This Issue, see p 2
Meet the First Author, see p 3
Extracellular vesicles (EVs) emerge from different subcellular compartments: microvesicles (≈100–1000 nm in diameter) are budding from the plasma membrane whereas exosomes (≈40–100 nm in diameter) are generated from intracellular multivesicular bodies. EV release into the interstitial space regulates important biological functions, including inflammation, which influences the development of cardiovascular diseases.3–5 Presence of exosome-like vesicles in cardiomyocyte cytoplasm was reported earlier in human hearts.6 Culture cardiomyocytes and fibroblasts also release EVs in vitro.7,8 However, we lack in vivo evidence of such phenomenon in the heart, in particular after MI. The present study, therefore, tested the hypothesis that local generation of EVs in the infarcted heart regulates cardiac inflammation after MI.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Mouse Model of MI
Nine- to 12-week-old (Janvier, France) or cardiac α-actin–GFP+ (green fluorescent protein)9 male mice (C57Bl/6J background; n=4–6 per group) were subjected to permanent coronary ligation, according to the ethical committee guidelines for animal experimentation (MESR [Ministère Enseignement Supérieur et Recherche] No. 9228).10,11 Sham-operated mice were subjected to all procedures, excluding permanent coronary artery ligation.
EV Isolation
Cardiac-Derived EV Isolation
EVs were isolated from ischemic tissue according to Leroyer et al.12 Briefly, hearts were perfused by ice-cold PBS; apex was minced (30 seconds, 4°C) using fine sterile scissors (2-cm strait blade) in 0.9% NaCl (100 μL per 100-mg tissue; Sham, 48±2 mg; MI, 54±3 mg; n=36). Samples were centrifuged twice at 400 g (15 minutes), and the resulting supernatant was further centrifuged at 20 500 g (45 minutes, 4°C) to pellet large EVs (lEVs). Pilot experiments demonstrated that the 20 500-g centrifugation step pelleted 95±2% of annexin V+ EVs, whereas 5±2% of annexin V+ events remained in the 20 500-g supernatant.
lEV pellets were then suspended in 100 µL 0.9% NaCl and stored at −80°C. lEV supernatant was mixed with the exosome precipitation buffer from miRCURY Exosome Isolation Kit (Exiqon) at 4°C overnight, spinned down at 10 000 g (20°C, 1 hour), and pellets were subjected to 100 000 g (90 min, 4°C). Small EV (sEV) pellets were then suspended in 100 µL 0.9% NaCl and stored at −80°C.
Human Cardiac EV Isolation
Similar procedure was used to isolate EVs from myomectomy fragments of interventricular septum (86±46 mg) obtained from 4 patients (2 of 4 male; 59±4 years old) operated on for aortic valve stenosis and undergoing aortic valve replacement at Institut Mutualiste Montsouris, Paris. Removal of tissue specimens was done during cardioplegic arrest. All patients had given their informed consent before surgery, and procedures were approved by the institutional ethical committee (CEPAR [Comité d’Evaluation des Protocoles et d’Aide a la Recherche] 2016-010).
EV Characterization
Western Blot Analysis
Ischemic LV and EV pellets were prepared in radioimmunoprecipitation assay buffer (3). Five-microgram proteins were loaded onto a 12% SDS-acrylamide gel and transferred on nitrocellulose membrane, incubated with anti-CD (cluster of differentiation) 63, anti-CD9, anti-Hsc70, or anti-cardiac-troponin primary antibodies. Expression of ApoA1 (apolipoprotein A1) and ApoB (apolipoprotein B) was below detection level (not shown).
Integrated Magnetic–Electrochemical Exosome Measurements
Expression of CD63 on lEV and sEV samples was analyzed by an integrated magnetic–electrochemical exosome platform.13 EVs were captured on immunomagnetic beads (specific to CD63). Collected EVs were then labeled with an oxidizing enzyme (horseradish peroxidase) through a second antibody against CD63. Mixing beads with a chromogenic electron mediator (3,3′,5,5′-tetramethylbenzidine) generated electric currents, which were measured by a custom-designed electric reader. About 10 µL of EV samples were used. The measured CD63 level was normalized against EV concentrations per tissue weight.
Flow Cytometry
lEVs were analyzed on a Gallios flow cytometer (Beckman Coulter)12,14 using Megamix-Plus FSC beads (Biocytex) to define events with a 0.3- to 1-μm diameter. Phosphatidylserine externalization was assessed using FITC-conjugated annexin V (Beckman Coulter), with and without CaCl2 (5 mmol/L). Vesicle concentration was calculated using calibrator beads (AccuCount; Spherotech).
lEVs cellular origin was identified as cardiomyocyte (caveolin 3+8), endothelial cell (CD31+CD41−12,14), fibroblast (CD90.2+15), platelet (CD41+12,14), erythrocyte (TER119+12 [glycophorin A–associated antigen]), or leukocyte (CD45+12). Concentrations of each EV subpopulation were given per milligram of tissue. All caveolin 3+ EVs were also annexin V+.
Tunable Resistive Pulse Sensing Measurements
lEV and sEV size distributions were quantified by tunable resistive pulse sensing (qNano; Izon Science, New Zealand) using NP400 and NP150 nanopores and compared with calibration beads CPC400 (mode diameter, 350 nm) and CPC200 (mode diameter, 210 nm), respectively.16,17 EV concentrations were expressed as total particles normalized to LV weight.
Cardiac Immune Infiltrating Cell Analysis
Inflammatory Cell Isolation
Hearts were collected, the LV was isolated, minced with fine scissors, and cells were isolated as described previously.10
Inflammatory Cell Phenotyping Using Flow Cytometry
Total cardiac cells were gated on PerCP-conjugated CD45 (BD Biosciences), and the following antibodies were used: PE-Cy7–conjugated anti-CD11b (BD Pharmingen), PE-conjugated anti-Ly6G (lymphocyte antigen 6 complex, locus G; 1A8; BD Pharmingen), FITC-conjugated anti-Ly6C (lymphocyte antigen 6 complex, locus C; Biolegend), PE-Alexa700–conjugated anti-CD3 (eBioscience) and APC-conjugated anti-F4/80 (Bio-Rad). CD45+/CD11b+ leukocytes were gated on F4/80− monocytes and then further stratified by Ly6G and Ly6C expression. Monocytes were identified as CD11bhighLy6G−Ly6C+. Neutrophils were identified as CD11b+Ly6Ghigh and T cells as CD45+CD3+. The total number of cells was then normalized to LV weight. Cells were analyzed using a flow cytometer (LSR II; BD Biosciences).10,11
Cardiac EV Engulfment by Infiltrating Immune Cells
Cardiac α-actin–GFP+ mice on C57Bl/6J background were subjected to MI. Twenty-four hours post-MI, immune cells were isolated, stained, and analyzed using ImageStream (Amnis Corp) to detect GFP fluorescence in immune cells.
EV Effects
Cardiac-derived monocytes were fluorescence-activated cell–sorted from LVs, 24 hours after the onset of MI. Fluorescence-activated cell–sorted CD45+/CD11b+/Ly6G−/Ly6C+ monocytes were counted, seeded at a density of 4.5×105 cells per well, and cultured in complete RPMI (Roswell Park Memorial Institute)-1640 medium (1% exosome-depleted serum; 1% β-mercaptoethanol and penicillin streptomycin). Cells were then stimulated for 24 hours with lEVs or sEVs derived from MI mice at a ratio of 105 lEVs per cell or 103 sEVs per cell to reproduce local concentrations of infiltrating monocytes and those of lEVs and sEVs in the infarcted heart 24 hours postligation. NaCl 0.09% was used as control vehicle. TNFα (tumor necrosis factor alpha), CCL (chemokine ligand) 2, CCL7, IL (interleukin)-4, IL-5, IL-6, IL-10, IL-12, IL-13, and IFNγ (interferon gamma) levels were quantified using ProcartaPlex multiplex immunoassays panels (Affymetrix, eBioscience, France).10,11 Concentrations were calculated from a control standard curve.
Statistics
Results were expressed as mean±SEM. Kruskal–Wallis 1-way ANOVA was used to compare each measure when there were ≥3 independent groups. Comparisons between groups were performed using Dunn multiple comparisons test when the ANOVA test was statistically significant. A P value <0.05 was considered significant. Mann–Whitney U test was used to compare 2 groups. All data were analyzed using Prism 5.0 (GraphPad Software, Inc).
Results
Cardiac EVs were obtained after brief mincing of LVs and isolated by successive centrifugations. Less than 1% of total annexin V+ events were also propidium iodide labeled in the 400-g supernatant, indicating that cardiac EV isolation procedure led to negligible amount of apoptotic bodies. Electron microscopy evidenced the presence of lEVs and sEVs in the 20 500-g pellet and supernatant, respectively (Figure 1A and 1D).
Figure 1.
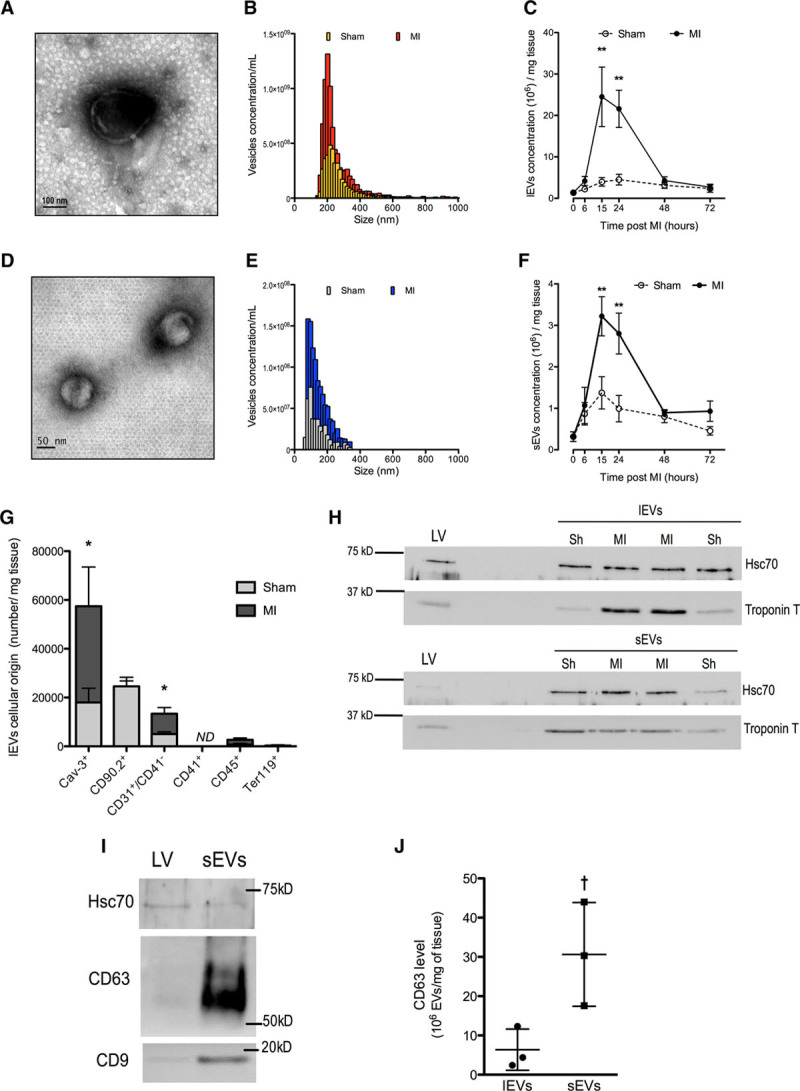
Cardiac extracellular vesicles (EVs) are transiently released in response to myocardial infarction (MI) in mice and originate mainly from cardiomyocyte. A, Typical representative electronic microscopy of large EVs (lEVs). B, Representative size distribution analysis by tunable resistive pulse sensing (TRPS) of lEVs from ischemic and sham murine cardiac tissue 24 h postligation. C, lEVs release during 72 h after the onset of ischemia in sham (open circles) and MI mice (dark circles). D, Typical representative electronic microscopy of small EVs (sEVs). E, Representative size distribution analysis by TRPS of sEVs from ischemic and sham murine cardiac tissue 24 h postligation. F, sEVs release during 72 h after the onset of ischemia in shams (open circles) and MI mice (dark circles). G, Cellular origin of lEVs isolated from infarcted hearts 24 h postligation. H, Representative Western blots for Hsc70 and troponin T in ischemic left ventricle (LV) in lEV and sEVs derived from sham and MI. I, Representative Western blots for Hsc70, CD63, and CD9 in ischemic LV and sEVs. J, CD63 levels measured using integrated magnetic–electrochemical exosome and normalized to EV concentrations per tissue weight. Data are mean±SEM. n=4 to 7 animals per time point. ND indicates not detectable. **P<0.01 vs corresponding sham, *P<0.05 vs sham, †P<0.05 vs lEVs.
The release of lEVs in murine heart increased significantly at 15 and 24 hours after coronary artery ligation when compared with sham animals and then returned to basal levels (Figure 1B and 1C). Coronary artery ligation transiently increased the release of annexin V+ lEVs from 2.6 (sham) to 4.7 105 vesicles per milligram tissue (24 hours post-MI; P=0.01; n=5). This response was decreased by 95±1% (n=3) after 0.05% Triton exposure. The local release of sEVs in the infarcted heart followed a similar pattern, with a significant increase at 15 and 24 hours postligation (Figure 1D through 1F) and then returned to levels similar to sham conditions. sEV diameter (118±4 nm; n=45) was significantly lesser than that of lEVs (252±18 nm; n=45; Figure 1B and 1E).
lEVs originated mainly from cardiomyocyte (caveolin-3+; Troponin T+) and also from cardiac fibroblast (CD90.2+) and endothelial cells (CD31+CD41−; Figure 1G and 1H). A small population of leukocyte-derived lEVs expressing CD45 was detected. Platelet- (CD41+) and erythrocyte-derived (Ter119+) vesicles, if any, were below detectable levels (Figure 1G). When compared with sham, MI significantly increased the intracardiac release of lEVs derived from cardiomyocytes and endothelial cells (Figure 1G and 1H). However, MI did not affect the release of lEVs from fibroblasts, leukocytes, platelets, and erythrocytes (Figure 1G). sEV harbored cardiomyocyte troponin T (Figure 1H) and also expressed exosomal markers CD63 and CD918,19 (Figure 1I and 1J).
The transient nature of EV release after coronary artery ligation prompted us to examine whether immune cells infiltrating the ischemic heart might be responsible for EV disappearance from injured ventricles. In agreement with previous findings,2 we observed that neutrophils (CD45+/CD11b+/Ly6GHigh/Ly6C+; Figure 2A) transiently infiltrated ischemic hearts, whereas monocytes (CD45+/CD11b+/Ly6G−/Ly6C+; Figure 2B) accumulated with time in the infarcted area. T lymphocytes (CD45+/CD3+; Figure 2C) also rapidly infiltrated ischemic tissues; their levels remained elevated at 72 hours postligation, but to a lower extent than neutrophils and monocytes. Because cardiac EVs generated after MI originated mostly from cardiomyocytes, we performed coronary artery ligation in α-actin–GFP+ mice expressing GFP specifically in cardiomyocytes. We subsequently assessed GFP labeling in inflammatory cells invading the ischemic myocardium. Both neutrophils and monocytes exhibited GFP fluorescence, indicating that GFP+ material derived from GFP+ cardiomyocytes had been taken up (Figure 2D). However, GFP fluorescence was preferentially associated with monocytes (+85% versus neutrophils, P<0.05; Figure 2E). GFP signal was also detected at a low level in CD3+ cells.
Figure 2.
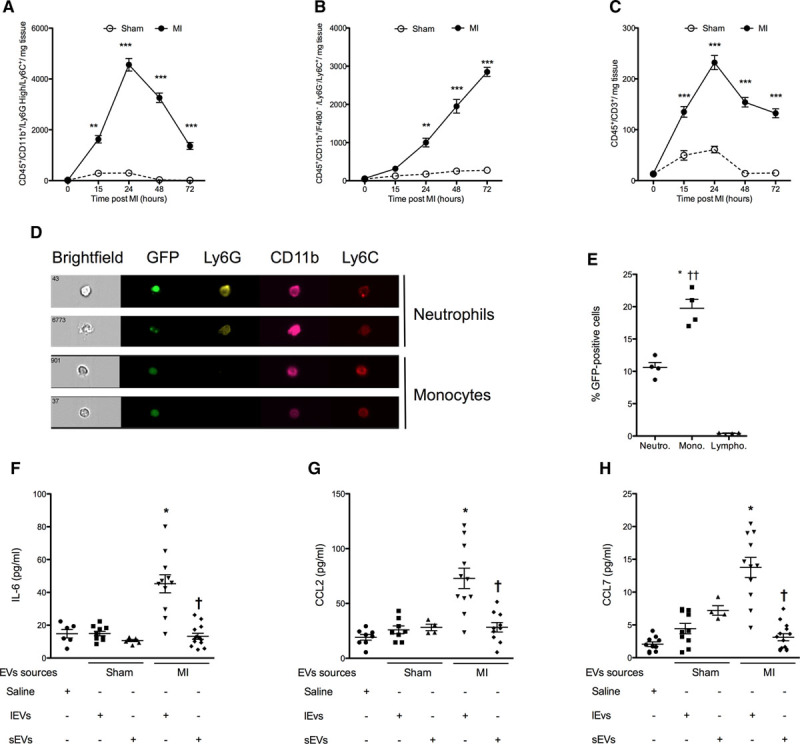
Cardiac extracellular vesicles (EVs) are engulfed by monocytes and shape the inflammatory response. A–C, Infiltration of neutrophils (Neutro; A), monocytes (Mono; B), and lymphocytes (Lympho; C) in sham (open circles) and myocardial infarction mice (dark circles) for 72 h after coronary artery ligation. Data are mean±SEM. n=4 to 7 animals per time point. **P<0.01 vs corresponding sham-operated mice at the same time point. D, Representative images of engulfment of GFP+ material by infiltrating immune cells (CD45+/CD11b+ leukocytes were gated on F4/80− monocytes and then further stratified by Ly6G and Ly6C expression) and (E) corresponding quantification. Data are mean±SEM. n=4 animals. *P<0.05 vs GFP+ cardiac neutrophils, ††P<0.01 vs GFP+ cardiac T lymphocytes. F–H, IL-6, CCL2, and CCL7 release by monocytes stimulated with cardiac-derived large (lEVs) or small EVs (sEVs). Data are mean±SEM. n=4 to 11 per experimental conditions. *P<0.05, **P<0.01 vs saline control.
Because infiltrating monocytes were the major cell type engulfing GFP+ material from ischemic myocardium of mice expressing GFP in cardiomyocytes, we then examined the functional effects of sEVs and lEVs isolated from ischemic hearts on cardiac monocytes. Ly6C+ monocytes were isolated from ischemic hearts 24 hours after the onset of ischemia and exposed to lEVs or sEVs at their respective in situ concentrations in ischemic hearts. lEVs from ischemic hearts significantly increased the release of IL-6 and chemokines CCL2 and CCL7 (P<0.05 versus saline; Figure 2F through 2H), but not tissue necrosis factor α, IL-4, IL-5, IL-10, IL-12, IL-13, or interferon γ levels (data not shown), whereas sham EVs or sEVs did not modify cytokine release (Figure 2I). Further studies will determine whether EV uptake or binding to membrane receptor mediates these proinflammatory effects.
To investigate the clinical relevance of our findings obtained in murine MI, we searched for the presence of EVs in human hearts. We identified cardiac EVs in fragments of the interventricular septum obtained from 4 patients undergoing extracorporeal circulation for aortic valve replacement (Figure 3A and 3B). As observed for the murine EVs, the mean diameter of human lEVs was significantly greater than that of sEVs (n=4; Figure 3C). Integrated magnetic–electrochemical exosome CD63 expression averaged 8.2±1.3 and 14.8±3.2 arbitrary units per 106 vesicles per milligram tissue in lEV and sEV fractions, respectively (data not shown). Intracardiac concentration of lEVs was greater than that of sEVs (Figure 3D).
Figure 3.
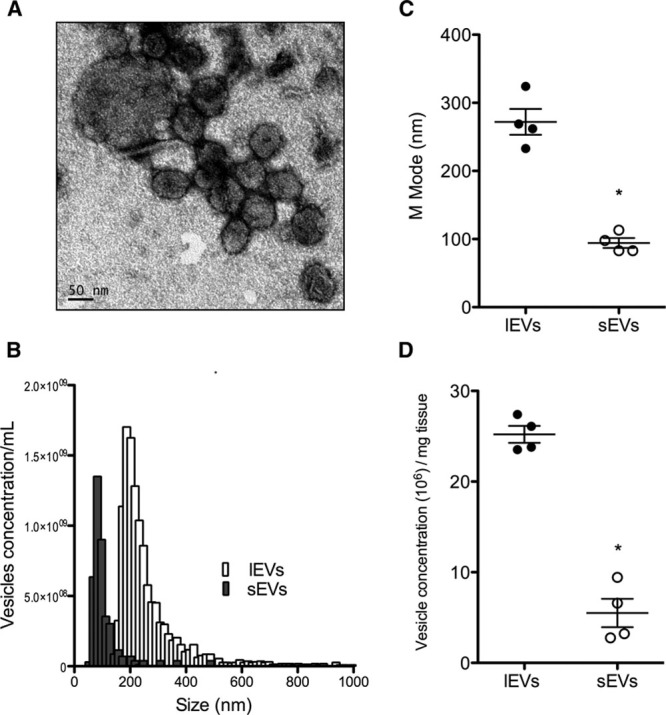
Intracardiac extracellular vesicles (EVs) in human myomectomy biopsies. A, Typical representative electronic microscopy of cardiac EVs from human biopsies indicating the presence of large (lEVs) and small EVs (sEVs). B, Representative size distribution analysis by tunable resistive pulse sensing (TRPS) of lEVs (in white) and sEVS (in grey). C, M-mode analysis of both lEVs and sEVs. D, EV concentration determined by TRPS. Data are mean±SEM. n=4 samples per fraction. *P<0.05 vs lEVs.
Discussion
Here, we report that coronary artery ligation transiently increases lEV and sEV levels in a murine model of MI, in comparison with control sham animals. Vesicles of similar diameter were also isolated from human interventricular septum fragments, attesting the pathophysiological relevance of our findings in the murine model.
Calcium-dependent annexin V labeling and sensitivity to Triton both demonstrated that lEVs harbored microvesicle characteristics.4,14 Furthermore, the presence of CD63 and CD9 in sEV is in favor of the exosome nature of these fractions.18,19 lEVs expressed detectable levels of caveolin-3 and troponin T, attesting their cardiomyocyte origin.8 They also harbored CD31 but not the platelet marker CD41, indicating that some of these lEVs were also of endothelial origin.12 Taken all together, these findings demonstrate that MI triggers the transient cardiac release of EVs of cardiomyocyte and endothelial origin. lEVs locally generated after MI increased the release of chemokines and inflammatory cytokines from Ly6C+ monocytes infiltrating the infarcted heart, whereas lEVs from sham animals and sEVs were without effects.
Taken altogether, these findings suggest that lEVs from ischemic hearts could serve as a spatiotemporal rheostat to fine-tune inflammation after MI.2
Acknowledgments
We are indebted to Camille Brunaud for help with cell sorting and Bamba Gaye for tunable resistive pulse sensing data analysis. X. Loyer, I. Zlatanova, M. Yin, K.-Y. Howangyin, C. Devue, P. Klaihmon, C.L. Guerin, M. Kheloufi, J. Vilar, D.W. Hwang, and J. Park performed experiments and collected the data. X. Loyer, J.-S. Silvestre, H. Lee, and C.M. Boulanger designed and supervised the research. B.K. Fleischmann, P. Menasché, and K. Zannis provided resources. X. Loyer, J.-S. Silvestre, and C.M. Boulanger wrote the manuscript.
Sources of Funding
This work was supported by Institut National de la Santé et de la Recherche Médicale, Agence National de la Recherche (ANR 11-META-004-02 MiRA, ANR-16-CE92-0032-01, ANR-16-CE92-0032-02, and CARDINAL [Cardiomyocyte-Derived Vesicular Non Coding RNAs and Post-Ischemic Remodeling]), Fondation de France (Engt2012-00029497 and FDF00066471), Fondation pour la Recherche Médicale (DEQ20160334910), and Aviesan/AstraZeneca. M. Yin was supported by University Paris Diderot and Fondation de France, M. Kheloufi by University Paris Descartes and Fondation pour la Recherche Médicale (FDT20160435690), I. Zlatanova by Région île-de-France and Fondation pour la Recherche Médicale (FDT20160435312), and P. Klaihmon by Thailand Research Fund.
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- CCL
- chemokine ligand
- CD
- cluster of differentiation
- EV
- extracellular vesicles
- GFP
- green fluorescent protein
- IL
- interleukin
- lEV
- large extracellular vesicle
- LV
- left ventricle
- MI
- myocardial infarction
- sEV
- small extracellular vesicle
In February 2018, the average time from submission to first decision for all original research papers submitted to Circulation Research was 12 days.
Novelty and Significance
What Is Known?
Microvesicles and exosomes are extracellular membrane vesicles mediating new type of cell–cell communication.
Cultured vascular and cardiac cells release extracellular vesicles that are able to curb important biological functions, such as inflammation, in target cells.
Presence of extracellular vesicles is observed in the heart.
What New Information Does This Article Contribute?
In a murine model of myocardial infarction, coronary artery ligation transiently increased the release of extracellular vesicles of cardiomyocyte and endothelial origin in the ischemic heart.
Cardiac extracellular vesicles locally produced after myocardial infarction are taken up by monocytes infiltrating the ischemic heart and subsequently increased their proinflammatory response.
In this article, we tested the hypothesis that myocardial infarction stimulates the release of extracellular vesicles within the infarcted heart to modulate the subsequent cardiac inflammatory response. Using a murine model of coronary artery permanent ligation, we demonstrate in the infarcted heart the transient release of extracellular vesicles expressing markers from cardiomyocytes and endothelial cells. Similar experiments performed in mice expressing green fluorescent cardiomyocytes identified infiltrating monocytes as the prevailing inflammatory cell type incorporating fluorescent extracellular vesicles of cardiomyocyte origin after coronary artery ligation. We then examined in vitro the functional consequences of exposing monocytes to extracellular vesicles, which were all isolated from infarcted or control cardiac ventricles. Extracellular vesicles isolated from mice with myocardial infarction stimulate the release of proinflammatory cytokines from monocytes invading the ischemic myocardium, whereas vesicles isolated from control animals had no effects. Taken all together, these findings demonstrate that extracellular vesicles accumulating in the ischemic myocardium are rapidly taken up by infiltrating monocytes to regulate the local proinflammatory response.
References
- 1.Silvestre JS, Smadja DM, Lévy BI. Postischemic revascularization: from cellular and molecular mechanisms to clinical applications. Physiol Rev. 2013;93:1743–1802. doi: 10.1152/physrev.00006.2013. doi: 10.1152/physrev.00006.2013. [DOI] [PubMed] [Google Scholar]
- 2.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loyer X, Vion AC, Tedgui A, Boulanger CM. Microvesicles as cell-cell messengers in cardiovascular diseases. Circ Res. 2014;114:345–353. doi: 10.1161/CIRCRESAHA.113.300858. doi: 10.1161/CIRCRESAHA.113.300858. [DOI] [PubMed] [Google Scholar]
- 4.Boulanger CM, Loyer X, Rautou PE, Amabile N. Extracellular vesicles in coronary artery disease. Nat Rev Cardiol. 2017;14:259–272. doi: 10.1038/nrcardio.2017.7. doi: 10.1038/nrcardio.2017.7. [DOI] [PubMed] [Google Scholar]
- 5.Robbins PD, Dorronsoro A, Booker CN. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J Clin Invest. 2016;126:1173–1180. doi: 10.1172/JCI81131. doi: 10.1172/JCI81131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahoo S, Losordo DW. Exosomes and cardiac repair after myocardial infarction. Circ Res. 2014;114:333–344. doi: 10.1161/CIRCRESAHA.114.300639. doi: 10.1161/CIRCRESAHA.114.300639. [DOI] [PubMed] [Google Scholar]
- 7.Bang C, Batkai S, Dangwal S, et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136–2146. doi: 10.1172/JCI70577. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waldenström A, Gennebäck N, Hellman U, Ronquist G. Cardiomyocyte microvesicles contain DNA/RNA and convey biological messages to target cells. PLoS One. 2012;7:e34653. doi: 10.1371/journal.pone.0034653. doi: 10.1371/journal.pone.0034653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleischmann M, Bloch W, Kolossov E, Andressen C, Müller M, Brem G, Hescheler J, Addicks K, Fleischmann BK. Cardiac specific expression of the green fluorescent protein during early murine embryonic development. FEBS Lett. 1998;440:370–376. doi: 10.1016/s0014-5793(98)01476-8. [DOI] [PubMed] [Google Scholar]
- 10.Ngkelo A, Richart A, Kirk JA, et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J Exp Med. 2016;213:1353–1374. doi: 10.1084/jem.20160081. doi: 10.1084/jem.20160081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howangyin KY, Zlatanova I, Pinto C, Ngkelo A, Cochain C, Rouanet M, Vilar J, Lemitre M, Stockmann C, Fleischmann BK, Mallat Z, Silvestre JS. Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation. 2016;133:826–839. doi: 10.1161/CIRCULATIONAHA.115.020857. doi: 10.1161/CIRCULATIONAHA.115.020857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leroyer AS, Ebrahimian TG, Cochain C, Récalde A, Blanc-Brude O, Mees B, Vilar J, Tedgui A, Levy BI, Chimini G, Boulanger CM, Silvestre JS. Microparticles from ischemic muscle promotes postnatal vasculogenesis. Circulation. 2009;119:2808–2817. doi: 10.1161/CIRCULATIONAHA.108.816710. doi: 10.1161/CIRCULATIONAHA.108.816710. [DOI] [PubMed] [Google Scholar]
- 13.Jeong S, Park J, Pathania D, Castro CM, Weissleder R, Lee H. Integrated magneto-electrochemical sensor for exosome analysis. ACS Nano. 2016;10:1802–1809. doi: 10.1021/acsnano.5b07584. doi: 10.1021/acsnano.5b07584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robert S, Lacroix R, Poncelet P, Harhouri K, Bouriche T, Judicone C, Wischhusen J, Arnaud L, Dignat-George F. High-sensitivity flow cytometry provides access to standardized measurement of small-size microparticles–brief report. Arterioscler Thromb Vasc Biol. 2012;32:1054–1058. doi: 10.1161/ATVBAHA.111.244616. doi: 10.1161/ATVBAHA.111.244616. [DOI] [PubMed] [Google Scholar]
- 15.Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, Conway SJ, Nagai R. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest. 2010;120:254–265. doi: 10.1172/JCI40295. doi: 10.1172/JCI40295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osteikoetxea X, Németh A, Sódar BW, Vukman KV, Buzás EI. Extracellular vesicles in cardiovascular disease: are they Jedi or Sith? J Physiol. 2016;594:2881–2894. doi: 10.1113/JP271336. doi: 10.1113/JP271336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coumans FA, van der Pol E, Böing AN, Hajji N, Sturk G, van Leeuwen TG, Nieuwland R. Reproducible extracellular vesicle size and concentration determination with tunable resistive pulse sensing. J Extracell Vesicles. 2014;3:25922. doi: 10.3402/jev.v3.25922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, Sahoo S, Tahara H, Wauben MH, Witwer KW, Théry C. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913. doi: 10.3402/jev.v3.26913. [DOI] [PMC free article] [PubMed] [Google Scholar]