

Abstract
Antibiotic chloramphenicol (CLM) binds with a moderate affinity at the peptidyl transferase center of the bacterial ribosome and inhibits peptide bond formation. As an approach for modifying and potentially improving properties of this inhibitor, we explored ribosome binding and inhibitory activity of a number of amino-acid analogues of CLM. The L-histidyl analogue binds to the ribosome with the affinity exceeding that of CLM by 10 fold. Several of the newly synthesized analogues were able to inhibit protein synthesis and exhibited the mode of action that was distinct from the action of CLM. However, the inhibitory properties of the semi-synthetic CLM-analogues did not correlate with their affinity and in general, the amino-acid analogues of CLM were less active inhibitors of translation in comparison with the original antibiotic. The X-ray crystal structures of the Thermus thermophilus 70S ribosome in complex with three semi-synthetic analogues showed that CLM derivatives bind at the peptidyl transferase center, where the aminoacyl moiety of the tested compounds established idiosyncratic interactions with rRNA. Although still fairly inefficient inhibitors of translation, the synthesized compounds represent promising chemical scaffolds that target the peptidyl transferase center of the ribosome and potentially are suitable for further exploration.
Keywords: Antibiotic, ribosome, X-ray structure, protein synthesis, peptidyl transferase center
INTRODUCTION
Many antibiotics stop growth of pathogenic bacteria, and thereby cure infections, by selectivelybinding to the bacterial ribosomes and inhibiting protein synthesis. Antibiotics can interfere with translation by interacting with various functional centers of the ribosome and either locking of a particular conformation of the ribosome or hindering the binding of its ligands. The peptidyl transferase center (PTC), located in the large ribosomal subunit, is targeted by a particularly broad array of inhibitors belonging to several distinct chemical classes, such as phenicols, lincosamides, oxazolidinones, pleuromutilins, streptogramins A, and others [1]. From the oldest chloramphenicol (CLM) to the newest FDA-approved retapamulin, PTC-targeting drugs are known as excellent antibacterial agents.
Most of the PTC-targeting compounds inhibit protein synthesis by competing with the positioning of the amino acid side chain of the incoming aminoacyl-tRNA (aa-tRNA) in the A site [2–4]. CLM, a typical example from this family, inhibits translation in a wide range of Gram-positive and Gram-negative bacteria [5]. It binds to the A site of the PTC in a crevice formed by the bases of the conserved nucleotides U2504, A2451, and C2452 of the 23S rRNA and its nitrobenzyl ring forms a π-stacking interaction with the base of C2452 [6, 7]. The aromatic ring of the ribosome-bound CLM overlaps with the placement of the side chains of the incoming aa-tRNAs, thus efficiently preventing the aminoacyl moiety of aa-tRNA from properly accommodating into the PTC active site. CLM was originally viewed as a universal inhibitor of peptide bond formation [8]. However, this model has been recently revised because the newer data revealed CLM as a context-specific inhibitor of translation, whose activity depends on the nature of specific amino acids in the nascent chain and the identity of the residue entering the A site [9].
While CLM does not act upon the eukaryotic cytoplasmic ribosome, it readily binds to the ribosomes of the mammalian mitochondria [10–14]. The interference with mitochondrial translation, which is the cause of the major side effects of CLM, significantly curbed the medical use of this drug in many countries [5, 15, 16]. Amending the CLM structure with the additional chemical entities that would form idiosyncratic interactions specifically with the bacterial ribosome could be one approach for the development of more selective inhibitors. In addition, the rapid spread of antibiotic resistance has significantly limited the medical utility of many available antibiotics, including the PTC-targeting drugs. One of the recently discovered but rapidly spreading resistance mechanisms operates via modification of one of the key PTC nucleotides, A2503, which is located in the binding site of several inhibitors. The rRNA methyltransferase Cfr adds a methyl group to the C8 position of the A2503 and renders bacteria resistant to a wide range of antibiotics targeting the catalytic center of the ribosome [17, 18]. The development of newer derivatives of the existing antibiotic platform, which could avoid a clash with the C8 methyl group of the Cfr-modified A2503, has proven to be a viable approach for overcoming such resistance [19].
Previously, attempts were made to prepare peptide derivatives of CLM containing peptides of up to 10 amino acids long aiming to identify interactions between the peptide chain and the elements of the nascent peptide exit tunnel (NPET) [20, 21]. In the current study, with the goal of paving the way for development newer derivatives of CLM, that would target the PTC, we have prepared several semi-synthetic analogues carrying various individual amino acids attached to the drug. Using competition binding experiments, we investigated the affinities of the newly prepared compounds for their target and found that some of them show improved binding to the bacterial ribosome. We also showed that the mode of action of the newer analogues differs from the site-specific action of CLM. By solving the X-ray crystal structures of the Thermus thermophilus 70S ribosome in complex with several CLM-analogues we observe specific interactions of the amino acid moiety with rRNA thereby rationalizing the improved binding. Although the higher affinity of the derivatives to the vacant E. coli ribosome does not directly translate into stronger inhibition of protein synthesis, the compounds described here could open new directions for improving the medical utility of amphenicol class of the ribosome inhibitors.
RESULTS AND DISCUSSION
Synthesis of CAM-derivatives
Chemical synthesis of CLM-analogues carrying amino acid residues instead of dichloroacetic moiety is based on acylation of CLM amine (CAM), an inactive CLM-derivative, with activated amino acids (Figures 1, S1) [22]. The overall synthesis scheme includes three steps: (i) acid hydrolysis of CLM to yield CAM [23]; (ii) acylation of CAM by succinimide esters of amino acids with protected D-amino group and side-chain amino groups; and (iii) de-protection of the obtained CAM-derivatives to yield aminoacyl-CAM (AA-CAM) (Figures 1, S1). Using this approach, we have prepared CLM analogues aminoacylated with different amino acids, including the N-protected ones (Table 1). Molecular weights and chemical structures of all synthesized CAM-derivatives were confirmed by mass spectrometry and 1H- and 13C-NMR. Although few individual amino-acid analogues of CLM have been studied previously [22, 24, 25], this work represents the first systematic approach to synthesis and functional assessment of more than three dozens of various AA-CAMs.
Figure 1.
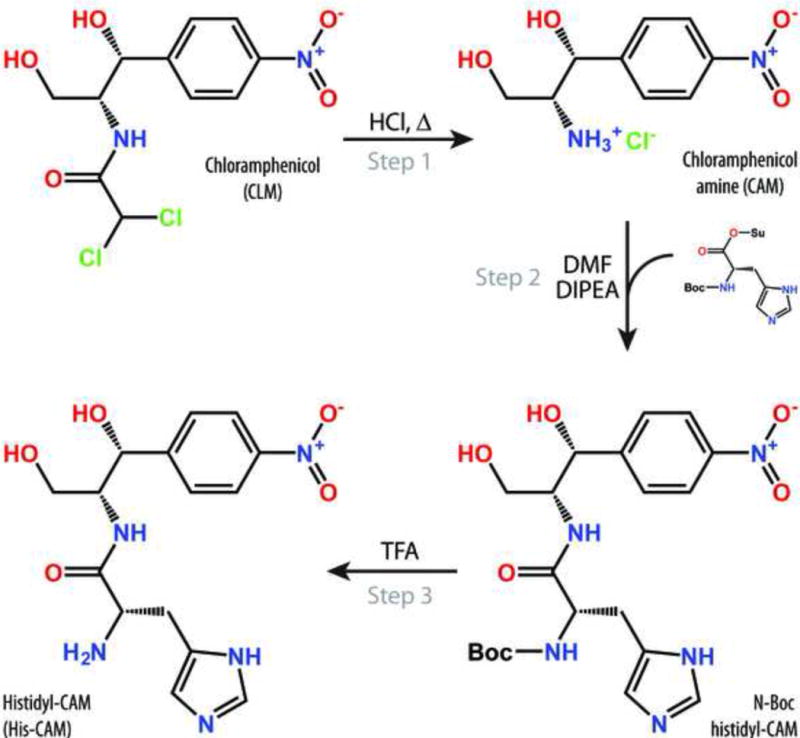
Schematic diagram of chemical synthesis of the histidine analogue of CLM.
Table 1. Apparent dissociation constants (KDapp) of amino-acid CAM-derivatives with the E. coli 70S ribosomes.
Numbers in the parenthesis correspond to the particular compound from the synthesis scheme shown in Figure S1.
| N-unprotected Compounds | N-Protected Compounds | ||
|---|---|---|---|
| Compound | KDapp (μM) | Compound | KDapp (μM) |
| CLM | 2.8 ± 0.5 | Ac-Pro-CAM (7a) | 10 ± 1.5 |
| His-CAM (1) | 0.24 ± 0.06 | Ac-Arg-CAM (3a) | 26 ± 10 |
| Lys-CAM (2) | 1.6 ± 0.5 | Ac-His-CAM (1a) | 34 ± 5 |
| Arg-CAM (3) | 3.2 ± 1.4 | Ac-Lys-CAM (2a) | 43 ± 12 |
| Ala-CAM (4) | 5.3 ± 0.9 | Ac-Ala-CAM (4a) | 55 ± 13 |
| Gly-CAM (5) | 5.7 ± 0.9 | Ac-Gly-CAM (5a) | 69 ± 14 |
| Gln-CAM (6) | 6.3 ± 5.1 | Ac-Phe-CAM (11a) | 92 ± 22 |
| Pro-CAM (7) | 7.5 ± 1.6 | Ac-Trp-CAM (10a) | 106 ± 36 |
| Tyr-CAM (8) | 8.2 ± 1.5 | Ac-Tyr-CAM (8a) | 140 ± 50 |
| Ser-CAM (9) | 8.4 ± 2.4 | Boc-His-CAM (1d) | 19 ± 5 |
| Trp-CAM (10) | 12 ± 2 | Boc-Gly-CAM (5d) | 45 ± 8 |
| Phe-CAM (11) | 19 ± 8 | Boc-Phe-CAM (11d) | 160 ± 120 |
| Val-CAM (12) | 34 ± 8 | Boc-Ala-CAM (4d) | 170 ± 60 |
| Leu-CAM (13) | 36 ± 6 | Boc-Pro-CAM (7d) | 440 ± 230 |
| Ile-CAM (14) | 36 ± 10 | f-Gly-CAM (5b) | 45 ± 6 |
| Asn-CAM (15) | 40 ± 26 | ||
| D-His-CAM (1b) | 2.9 ± 0.8 | ||
| D-Ala-CAM (4b) | 7.9 ± 1.4 | ||
| D-Pro-CAM (7b) | 13 ± 2 | ||
| β-Ala-CAM (16) | 6.3 ± 1.3 | ||
AA-CAM compounds bind tightly to the bacterial ribosome
We used the competition-binding assay, exploiting BODIPY-labeled erythromycin (BODIPYERY) to assess the affinity of the synthesized compounds for the ribosome [26, 27]. While CLM binds in the A site of the PTC [6, 28], ERY binds in the upper part of the NPET [7] but their binding sites sufficiently overlap so that even unmodified CLM and ERY compete for their binding to the ribosome [29] (Figure S2). The overlap of the AA-CAMs with ERY is expected to be even more pronounced. The apparent dissociation constant KDapp of CLM obtained using competition with BODIPY-ERY (2.8 ± 0.5 μM) is consistent with the previously published data determined by direct [14C]-CLM binding (2.3 μM [30]). Using this approach, we found that many of the synthesized CAM derivatives exhibited considerable affinity for the ribosome (KDapp in the low micromolar range) (Table 1). Interestingly, all of the AA-CAM derivatives carrying free α-amino groups bind to the ribosome with higher affinities than the corresponding compounds in which the α-amino group was modified by acetylation, formylation or protected by the Boc group (Figure S3; Table 1). This result suggests that either the positive charge, the small size of the α-amino group, or both, contribute to the efficient ribosome binding of AA-CAMs. Importantly, one AA-CAM variant, His-CAM, binds to the ribosome with a more than 10-fold higher affinity than CLM (His-CAM, KDapp = 0.24 ± 0.06 μM) (Figure 2A; Table 1).
Figure 2. Binding and inhibitory properties of AA-CAM-derivatives.
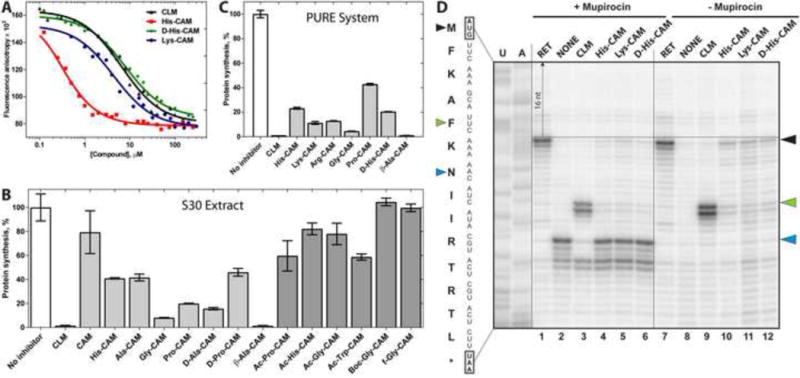
(A) Competition binding assay to test the inhibition of BODIPY-ERY binding to the E. coli ribosomes in the presence of increasing concentrations of AA-CAM derivatives measured by fluorescence anisotropy. (B, C) Inhibition of in vitro synthesis of firefly luciferase by AA-CAM derivatives in the E. coli S30 cell extract (B) or in the PURE system (C). All the inhibitors were present in the reaction at 30 μM. All the reactions were performed in triplicates and error bars represent confidence interval (α= 0.05). Inhibitory activity of AA-CAM compounds with free α-amino groups are shown as light grey bars, N-protected AA-CAM compounds – dark grey, positive control CLM – white bars. (D) Primer extension inhibition (toe-printing) analysis of site specificity of action of CLM and AA-CAMs. The synthetic mini-gene was translated in the cell-free translation (‘PURE’) system and sites of antibiotic-induced translation arrest were analyzed by primer extension. The reactions loaded onto lanes 1-6 contained mupirocin, an inhibitor of isoleucyl-tRNA synthetase. The sample in lane 2 (labeled ‘NONE’) contained no other antibiotics besides mupirocin. The control antibiotic retapamulin (RET) inhibits translation initiation and arrests the ribosome at the start codon (black arrowheads). Bands corresponding to the CLM-induced translation arrest at the 5th codon are indicated by the green arrowheads. Stalling of ribosomes at the 7th codon of the ORF due to the presence of mupirocin that causes depletion of isoleucyl-tRNA (lanes 1-6) is indicated by the blue arrowheads. U- and A-specific sequencing lanes are indicated. The nucleotide sequence of the gene and the corresponding encoded amino acids are indicated on the left. Note that the reverse transcriptase stops 15-16 nucleotides downstream from the first nucleotide of the P-site codon as indicated by the dashed arrow.
AA-CAM compounds are less efficient inhibitors of translation than CLM and do not show the same context-specificity
To check whether the binding strength of AA-CAMs correlates with inhibition of translation we tested their ability to interfere with in vitro protein synthesis. Addition of 30 μM CLM to the cell-free transcription-translation system based on the E. coli cell extract resulted in a near-complete inhibition of synthesis of firefly luciferase reporter (Figure 2B). Consistent with previously published results, removal of the dichloroacetyl moiety from CLM results in nearly complete loss of its biological activity (Figure 2B, CLM vs. CAM) [23], likely due to the loss of interactions of the dichloroacetic moiety with the ribosome [6]. However, similar to the previous studies employing peptide derivatives of CLM [5, 20, 31], the inhibitory activity of CAM can be partially restored by incorporation of specific amino acids (Figure 2B), indicating that the contacts of the aminoacyl side chain of AA-CAMs with the ribosome may compensate for the interactions lost upon the removal of the dichloroacetic group. However, most of the tested AA-CAM-derivatives were less potent inhibitors of translation than CLM (Figure 2B). Even His-CAM, which exhibited higher than CLM affinity for the ribosome, reduced the yield of the functional luciferase by only 60%. Only the activity of β-Ala-CAM approached that of CLM even though this derivative demonstrated a 2-fold weaker binding to the ribosome compared to the parental compound. The modification of the α-amino group of AA-CAMs (with formyl, acetyl, or Boc groups) leads to a nearly complete loss of their inhibitory potency (Figure 2B). The inability of most of the AA-CAM compounds to efficiently inhibit translation could be potentially explained by the susceptibility of AA-CAMs to the action of peptidases present in the bacterial extract-based cell-free translation system. Therefore, we re-tested the inhibitory activity of some of the compounds in the PURE cell-free translation system reconstituted from purified components and thus, lacking peptidases (Figure 2C) [32]. Although the overall inhibitory activity of AA-CAM derivatives improved in the PURE system compared to their effect upon translation in the cell extract, it still did not reach the level of inhibition elicited by the CLM, suggesting that stability of our compounds is unlikely to be the key issue and indicating that the equilibrium binding of the inhibitors to a vacant ribosome often does not directly correlate with their inhibitory properties. Several peptide derivatives of CAM have been prepared and tested previously in the binding and inhibition assays [20, 31]. Similar to our findings, those derivatives were found to be poor inhibitors of translation.
The inhibition of protein synthesis by CLM depends on the amino-acid sequence of the nascent peptide and the identity of the aminoacyl-tRNA with the most efficient arrests occurring after alanine (and to a lesser extent after serine or threonine) that appear at the penultimate position of the nascent chain [9]. To test whether the specificity of action of AA-CAM compounds matches that of CLM, we used primer extension inhibition assay (toe-printing) to check whether the derivatives that exhibited inhibitory activity in the PURE system (Figure 2C), would arrest the ribosome at the same codon(s) where translation is arrested by CLM. In these experiments, we used a short synthetic open reading frame (ORF) encoding the peptide MFKAFKNIIRTRTL, where CLM efficiently arrests the ribosome at the 5th codon after the MFKAF peptide has been synthesized (Figure 2D, lanes 3 and 9, green arrowheads). Some translation reactions were additionally supplemented with mupirocin, an inhibitor of isoleucyl-tRNA synthetase. The depletion of isoleucyl-tRNA efficiently traps the ribosomes at the 7th codon of the ORF if they were not arrested at any of the prior codons by the action of the ribosome inhibitor (Figure 2D, lanes 1–6, blue arrowheads) helping to evaluate the efficiency of the antibiotic-dependent arrest [33]. Only a small fraction of ribosomes pre-incubated with CLM was able to reach the ‘trap’ codon (Figure 2D, lane 3) because most of the translation complexes were arrested at the 5th codon. In contrast, the intensity of the trap-codon band was only slightly diminished in samples supplemented with AA-CAM analogues and no strong arrest was observed at the 5th codon (Figure 2D, lanes 4–6). This was in spite of the fact that the concentrations of AA-CAM compounds in the toe-printing experiments (100 uM) were several-fold higher than those used in the cell-free translation experiments (Figure 2B, C), where the same derivatives were able to inhibit expression of the luciferase reporter with a considerable activity. We noted, however, that the toe-print bands corresponding to the initiating ribosome were slightly enhanced compared to the no-drug (+/− mupirocin) control (Figure 2D, lanes 2 and 8 vs. lanes 4–6 and 10–12, respectively, black arrowheads), indicating that AA-CAM analogues weakly interfered with translation initiation, likely by inhibiting the formation of the first peptide bond. We concluded that AA-CAM derivatives do not show the same site-specificity of action as the parent compound CLM, apparently due to idiosyncratic interactions of the aminoacyl moieties of AA-CAMs with the ribosome.
Aminoacyl moieties of AA-CAMs mediate specific interactions with the ribosome
To unambiguously determine the mode of binding of AA-CAM derivatives to the ribosome, we crystallized Thermus thermophilus (Tth) 70S ribosomes in the presence of mRNA, deacylated A-, P-, and E-site tRNAs, and His-CAM and solved the structure of the complex at 2.8Å resolution. However, the other complexes prepared in this way did not diffract sufficiently well. Therefore, we amended our crystallization approach by using ribosome complexed with the protein Y (PY) as a tool to obtain structures of higher resolution [34, 35]. This approach is based on our recent finding that binding of PY to a vacant 70S ribosome stabilizes it by locking the head of the 30S subunit in an unrotated state, which leads to a better diffraction [34, 35]. Since PY binds in the mRNA channel on the 30S subunit far away from the CLM binding site in the PTC of the 50S subunit, it precludes the presence of mRNA or A- and P-tRNAs [34]. However, this does not interfere with the binding of AA-CAMs. Using this approach, we were able to solve two additional structures of D-His-CAM and Lys-CAM in complex with the PY-bound 70S ribosome at 2.7Å and 2.6Å resolution, respectively (Table S1). An unbiased difference Fourier map, calculated using the amplitudes from the crystals and phases derived from a model of the ribosome without the bound compound, revealed positive electron density resembling characteristic features of each of the compounds (Figure 3). A single binding site for each of the AA-CAMs is observed in the ribosome within the PTC of the large ribosomal subunit (Figure 4).
Figure 3. Chemical structures and electron density maps of CAM-derivatives.
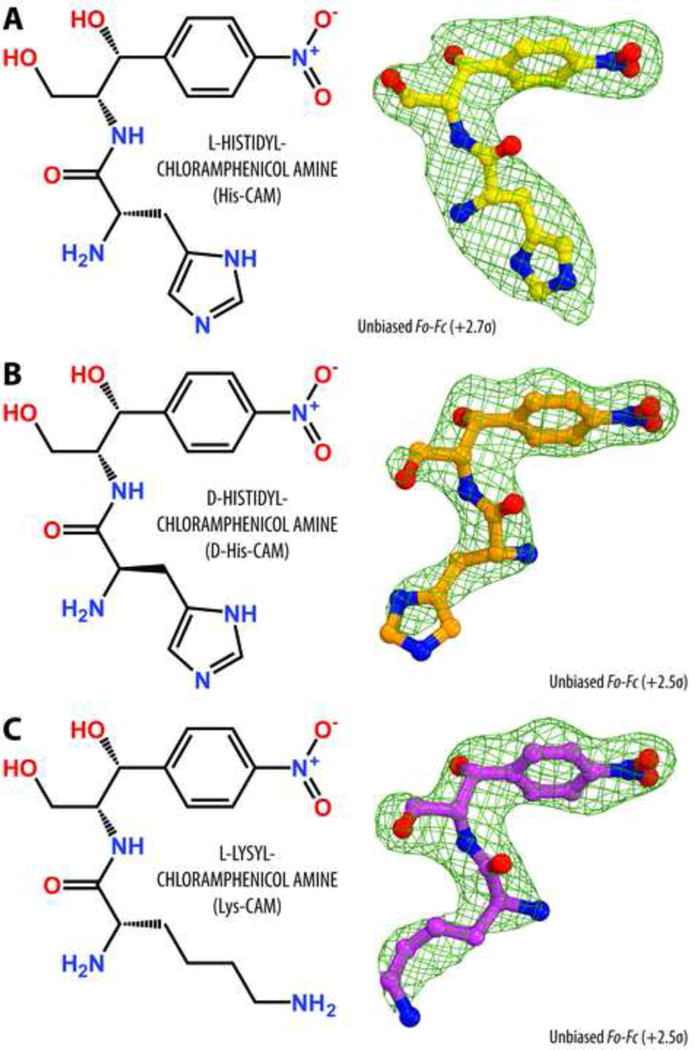
Chemical structures and difference Fourier maps of His-CAM (A), D-His-CAM (B), and Lys-CAM (C) in complex with the T. thermophilus 70S ribosome. The refined model of each compound is displayed in its respective electron density before the refinement (green mesh). Carbon atoms are colored yellow for His-CAM, orange for D-His-CAM, magenta for Lys-CAM, nitrogens are blue, oxygens are red.
Figure 4. Structure of His-CAM in complex with the 70S ribosome and A- and P-tRNAs.
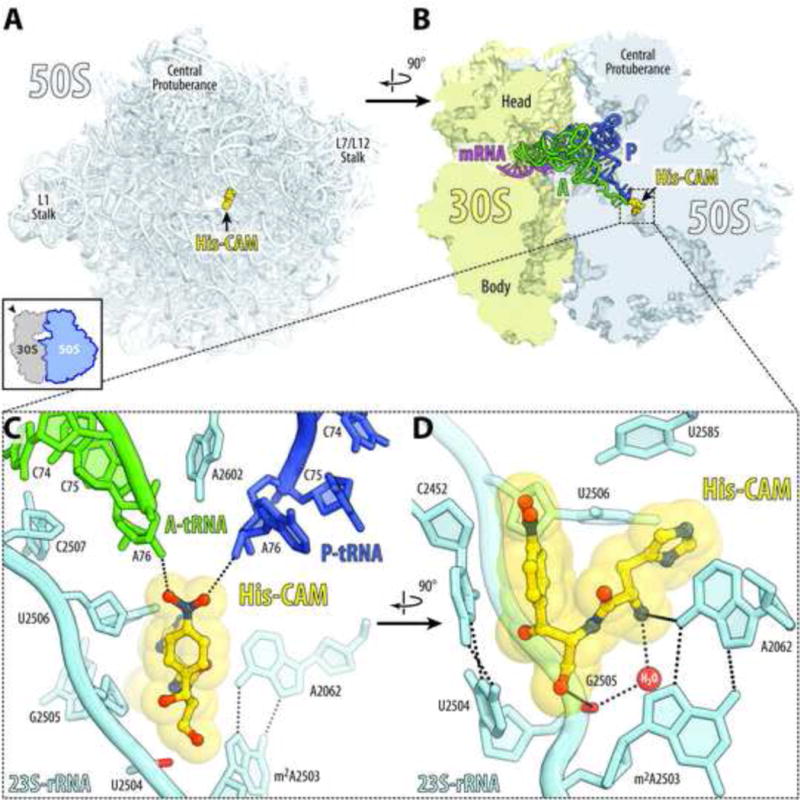
(A, B) Overview of the His-CAM binding site (yellow) in the T. thermophilus 70S ribosome viewed from the PTC down the tunnel as indicated by the inset (A), or as a cross-cut section through the ribosome (B). The 30S subunit is shown in light yellow, the 50S subunit is in light blue, the mRNA is magenta and the A- and P-site tRNAs are green and dark blue, respectively. The E-site tRNA is omitted for clarity. (C, D) Close-up views of the His-CAM bound in the PTC. The E. coli nucleotide numbering is used throughout. Potential H-bond interactions are indicated with dashed lines. Note that side chain of His-CAM compound forms tilted edge-to-face Π-stacking with the nucleobase of U2506 of the 23S rRNA (shown as spheres).
The binding position of the amphenicol parts of AA-CAMs is identical to those observed previously for parent CLM in the Tth [7] or Eco [28] ribosomes in the absence of mRNA and tRNAs (Figure S4). The amino acid moieties of AA-CAMs are oriented towards the NPET and exhibit unique compound-specific interactions at the PTC (Figures 4, 5). Histidine side chain of His-CAM forms tilted edge-to-face π-stacking with the nucleobase of U2506 (Figures 4D and 5A). In addition, the α-amino group of His-CAM forms hydrogen bond (H-bond) via a water molecule with the phosphate of nucleotide G2505 (Figure 5A). These additional interactions of the aminoacyl moiety likely account for its increased affinity to the ribosome compared to CLM.
Figure 5. Side-chain-specific interactions of AA-CAMs with the ribosome.
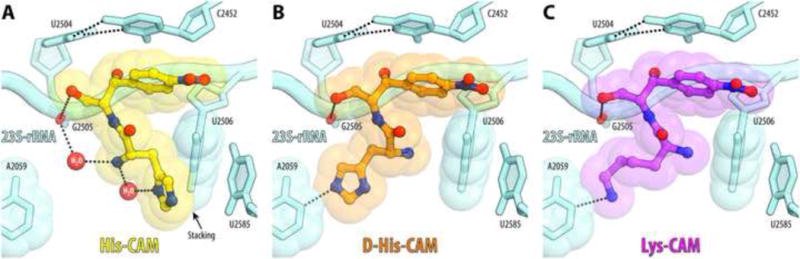
Compound-specific H-bond interactions of His-CAM (A), D-His-CAM (B), or Lys-CAM (C) with the nucleotides of the 23S rRNA are indicated with dashed lines. Stacking interactions of His-CAM are shown with the black arrow.
Contrary to His-CAM and consistent with its significantly lower affinity, α-amino group of the D-His-CAM stereoisomer does not form the water-mediated H-bond with G2505 (Figure 5B). Surprisingly, the orientation of the amino acid side chain of L-Lys-CAM is similar to that of D-His-CAM (Figure 5C). The lysine moiety of Lys-CAM extends towards the wall of the NPET and, similar to D-His-CAM, forms a single H-bond with the nucleobase of A2059 explaining why their binding is less tight in comparison with His-CAM.
In the structure of the ribosome/His-CAM complex, the oxygens of the nitro group in the CAM moiety form H-bonds with the A76 ribose hydroxyls of deacylated A- and P-site tRNAs (Figure 4C). It is unclear whether such interactions are possible in the translating ribosome when the P-site tRNA is attached to the aminoacyl or peptidyl groups. Nevertheless, our structures clearly demonstrate that amphenicol part of the AA-CAMs is capable of anchoring the derivatives in the PTC active site directing the attached amino acids in the direction corresponding to the growing peptide chain.
Resistance to many peptidyl transferase inhibitors (including CLM) can be conferred by Cfr, a methyltransferase that methylates A2503 of the 23S rRNA at the C8 atom [18]. The methyl group added to A2503 by Cfr would invade the CLM binding pocket if the placement of the modified A2503 remains unchanged (Figure 6A). In silico modeling shows, however, that the ribosome-bound His-CAM whose placement in the PTC is somewhat shifted relative to CLM, would avoid the collision with C8-methyl group of A2503 (Figure 6B), suggesting that this compound could retain certain activity against the Cfr-modified ribosome. Likewise, the resistance to CLM conferred by the mutation of A2503 to G [36], is likely to be less pronounced in the case of His-CAM due to a more remote position of the amino acid side chain of His-CAM relative to the site of the mutation. Therefore, the structure of the ribosome with the AA-CAM derivatives suggest that some of the amino acid analogues of CLM could be less susceptible to the action of specific resistance mechanisms. Further experiments would be required to check whether any of the synthesized CLM-analogues are active against ribosomes rendered resistant to CLM.
Figure 6. Structural basis for resistance to CLM (A) and His-CAM (B).
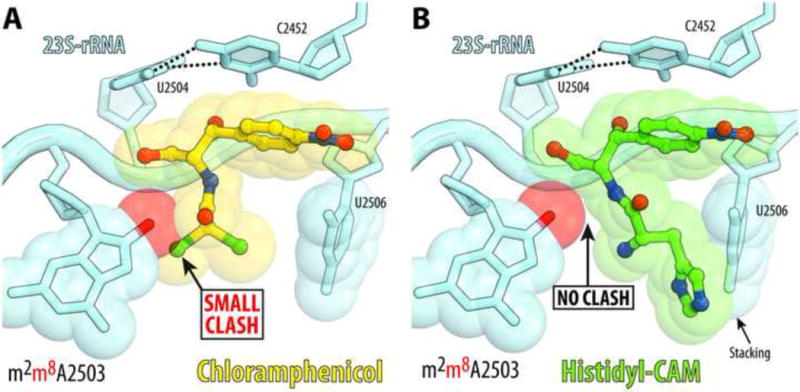
Molecular modeling of the C8-methylation of A2503 (red sphere) catalyzed by the Cfr-methyltransferase reveals a small clash with CLM (A) but not with histidine analogue of CLM (B). Note that the path of the His-CAM in the PTC is located further away from the C8-position of A2503 (compared to CLM) that should allow it to avoid a possible steric clash with this position carrying methyl group in the Cfr-modified ribosome.
The structure of PTC is highly conserved among evolutionary distant ribosomes, including the ribosomes of mitochondria which are known to be targeted by CLM [10–14]. However, even minor variations in the placement of the nucleotides directly interacting with the drug, which could be influenced by the less conserved second-shell nucleotides, could account for the selectivity of the antibiotics that bind in the PTC active site as well as for the differential effect of these drugs upon ribosomes from different species. Because the compounds examined in our study have fairly low activity against the bacterial ribosome, we have not pursued studies of their effects upon mitochondrial translation. However, we believe that adding an amino acid or its analogue to the drug core could potentially influence the spectrum of action of this class of the ribosomal antibiotics opening a possibility of making them more selective inhibitors of bacterial translation.
CONCLUSIONS
The goal of the current study was to develop amino-acid analogues of CLM as an approach for exploring this line of derivatives as new inhibitors of translation. We used organic synthesis to generate a diverse set of amino-acid analogues of native CLM and examined their ribosome binding and inhibitory properties. We have demonstrated that His-CAM, a histidine analogue of CLM, exhibits 10 times higher affinity for the bacterial ribosome as compared to CLM. We have noted, however, that the inhibitory properties of the semi-synthetic CLM-analogues do not correlate with their ribosome binding affinities and the compounds with high affinities, such as His-CAM, inhibit translation less efficiently than CLM, while compounds with low affinities, such as β-Ala-CAM, demonstrated inhibitory properties similar to those of the parental drug. Our crystal structures show that amino-acid analogues of CLM establish compound-specific interactions with the nucleotides of the 23S rRNA at the PTC and orient the amino acid moiety in the direction of the upper part of the peptide exit tunnel. The idiosyncratic interactions with the ribosome of the amino acid residue attached to the CLM core open the possibility of increasing the selectivity of this class of antibiotics and possibly diminish their side effects mediated by the action upon mitochondrial translation. Moreover, the possibility that some of the amino-acid derivatives of CLM might act upon drug-resistant ribosome makes them attractive compounds for further exploration by medicinal chemists, and we expect that our findings, including the structure of the AA-CAM-ribosome complexes described here, will serve as a starting point for such studies.
MATERIALS AND METHODS
Reagents
Various amino acid derivatives used in chemical synthesis (Supplementary Methods) were from Fluka or Reanal; CLM was from Sigma; Succinimide ester of BODIPY was from Invitrogen; N-hydroxysuccinimide, N,N’-dicyclohexylcarbodiimide (DCC) and DMAP were from Merck. BODIPY-ERY was synthesized as described previously [37].
Chemical synthesis of AA-CAM derivatives
The general scheme for the synthesis of AA-CAM derivatives is shown in Figure S1 and the details of chemical synthesis are provided in the Supplementary Methods section. CAM [(1R,2R)-2-amino-1-(4-nitrophenyl)propane-1,3-diol)] was prepared as described previously [23]. Amino acids with protected α- and side-chain amino groups were activated by reaction with N-hydroxysuccinimide in the presence of DCC at 0°C. The resulting succinimide-reactive esters were used for the acylation of CAM in the presence of diisopropylethylamine as a base at room temperature. Subsequent deprotection was achieved by treatment of the obtained amino-acid CAM derivatives with trifluoroacetic acid and appropriate scavengers. Synthesized AA-CAM derivatives were purified by column chromatography on silica gel using suitable systems of solvents. For generating N-acetylated variants of AA-CAM, additional acetylation was performed by reacting the unprotected AA-CAM derivatives with the N-acetylsuccinimide. Purity and chemical structures of obtained compounds were confirmed by HPLC, LC-MS and NMR spectroscopy (see Supplementary Methods).
In vitro binding assay
Binding affinities of CAM-derivatives to E. coli ribosomes was analyzed by competition binding assay using fluorescently-labeled BODIPY-ERY as described before [26, 27, 37]. BODIPY-ERY (4 nM) was incubated with ribosomes (25 nM) for 30 min at 25°C in the buffer containing 20 mM HEPES-KOH (pH 7.5), 50 mM NH4Cl, 10 mM Mg(CH3COO)2, and 0.05% Tween-20. The solution of CAM derivatives in a range of concentrations from 0.1 μM to 1 mM was added to the formed complex. The mixture was incubated for 2 hours until equilibrium was reached and the values of fluorescence polarization were measured.
In vitro translation
Inhibition of firefly luciferase synthesis in cell-free translation systems by AA-CAMs was tested as described previously [38]. Briefly, the in vitro transcribed firefly luciferase mRNA was translated using either PURExpress system (New England Biolabs) or E. coli S30 extract prepared according to [39]. Reactions were programmed with 100 ng mRNA and were carried out in 5 μL aliquots at 37°C for 30 minutes. The AA-CAMs were added to 30 μM final concentration. The activity of in vitro synthesized luciferase was assessed using 5 μL of the substrate from the Steady-Glo Luciferase Assay System (Promega).
Toe-printing analysis of compound-induced ribosome stalling
Toe-printing experiments were performed in the PURExpress cell-free translation system (New England Biolabs) following our published protocols [40, 41]. The synthetic template encoding the amino acid sequence MFKAFKNIIRTRTL was initially generated by PCR reaction using a combination of five primers: 2 μM T7 (ATTAATACGACTCACTATAGGG), 2 μM NV1 (GGTTATAATGAATTTTGCTTATTAAC) and 0.2 μM of each of the following: Fv (AATACGACTCACTATAGGGCAACCTAAAACTTACACACGCCCCGGTAAGGAAATAAAAAT), inner (GCCCCGGTAAGGAAATAAAAATGTTCAAAGCATTCAAAAACATCATACGTACTCGTACTC) and Rev (GGTTATAATGAATTTTGCTTATTAACCTTGCCTGCGCTTAAAGAGTACGAGTACGTATGATGT) as described in [42]. The product was cloned into the pUC18 plasmid cut with the SmaI restriction enzyme and the resulting pUCMFKAFK plasmid was verified by capillary sequencing. For the toe-printing reaction, the template was PCR-amplified from the pUCMFKAFK plasmid using T7 and NV1 primers. When needed, the other antibiotics (retapamulin, CLM, and AA-CAM analogues) were present in the reaction at the final concentration of 100 μM. In addition, all the toe-printing reactions were supplemented with the Ile-RS inhibitor mupirocin (final concentration 50 μM), which caused translating ribosomes to pause when an isoleucine codon was encountered.
Crystallographic structure determination
Ribosome complexes with mRNA and tRNAs or with protein Y were formed as described previously [35]. CAM-derivatives were added to the pre-formed ribosome complexes to a final concentration of 250 μM prior to crystallization. All Tth 70S ribosome complexes were formed in the buffer containing 5 mM HEPES-KOH (pH 7.6), 50 mM KCl, 10 mM NH4Cl, and 10 mM Mg(CH3COO)2, and then crystallized in the buffer containing 100 mM Tris-HCl (pH 7.6), 2.9% (w/v) PEG-20K, 7–12% (v/v) MPD, 100–200 mM arginine, 0.5 mM β-mercaptoethanol. Crystals were grown by the vapor diffusion method in sitting drops at 19°C and stabilized as described previously [35] with the corresponding CAM-derivatives being added to the stabilization buffers (100 μM each). Diffraction data were collected using beamlines 24ID-C and 24ID-E at the Advanced Photon Source (Argonne, IL). All crystals belonged to the primitive orthorhombic space group P212121 with approximate unit cell dimensions of 210Å × 450Å × 620Å and contained two copies of the 70S ribosome per asymmetric unit. Each structure was solved by molecular replacement using PHASER from the CCP4 program suite [43]. The search model was generated from the previously published structures of T. thermophilus 70S ribosome with bound mRNA and tRNAs (PDB entry 4Y4P from [35]) or with protein Y (PDB entry 4Y4O from [35]). The initial molecular replacement solutions were refined by rigid body refinement with the ribosome split into multiple domains, followed by positional and individual B-factor refinement. The final models of the 70S ribosome in complex with His-CAM and mRNA/tRNAs, or in complex with D-His-CAM/Lys-CAM and protein Y were generated by multiple rounds of model building in COOT [44], followed by refinement in PHENIX [45]. The statistics of data collection and refinement are compiled in Table S1.
Supplementary Material
RESEARCH HIGHLIGHTS.
Aminoacyl derivatives of the antibiotic chloramphenicol (AA-CAMs) can bind to the peptidyl transferase center of the bacterial ribosome with better affinities than the parent compound;
AA-CAM derivatives show different site-specificity of action compared to chloramphenicol and establish compound-specific interactions with the nucleotides of the 23S rRNA;
The inhibitory properties of the semi-synthetic CLM-analogues do not correlate with their ribosome binding affinities;
AA-CAMs represent promising chemical scaffolds that target the peptidyl transferase center and the nascent peptide exit tunnel.
Acknowledgments
We thank V. N. Tashlitsky for help with LCMS analysis, Y. K. Grishin and I. A. Godovikov for help with interpreting NMR spectra, E. N. Shapovalova for help with chiral chromatography analysis, M. S. Svetlov for help with translation inhibition assays, G. A. Korshunova for providing Boc derivatives of amino acids, and А. А. Bogdanov Jr. for providing Ac derivatives of amino acids. We are also thankful to the members of the A.A.B, P.V.S., A.S.M., and Y.S.P. laboratories for discussions and critical feedback.
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on 24ID-C beamline is funded by a NIH-ORIP HEI grant (S10 RR029205). The Eiger 16M detector on 24ID-E beamline is funded by a NIH-ORIP HEI grant (S10 OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
This work was supported by Illinois State startup funds (to Y.S.P.), Russian Science Foundation grant 14-24-00061-P (to Tatyana S. Oretskaya, “Study of antibiotics binding to bacterial ribosomes”), and Russian Foundation for Basic Research grants 16-04-00709 (to N.V.S., “Synthesis of CLM analogues”) and 15-34-20139 (to I.A.O., “Translation inhibition analysis”), and the National Institutes of Health grants R01 AI125518 (to A.S.M.) and R01 GM106386 (to A.S.M.).
Footnotes
AUTHOR CONTRIBUTIONS
A.G.T. and V.A.S. performed chemical synthesis of CAM-derivatives; N.V.S. purified CAM-derivatives; A.G.T. performed 70S ribosome binding assay; A.A.S. and I.A.R. performed mass spectrometry analysis; I.A.O., E.S.K., and P.V.S. designed and performed in vitro translation inhibition assays; P.S.K. and A.L.K. prepared E. coli 70S ribosomes; J.M. and A.S.M designed and performed toe-printing experiments; M.D.B. and Y.S.P. designed and performed X-ray crystallographic experiments. N.V.S., A.S.M., A.A.B., and Y.S.P. supervised the experiments. All authors interpreted the results. A.G.T., N.V.S., A.S.M., A.A.B., and Y.S.P. wrote the manuscript.
ACCESSION NUMBERS
Coordinates and structure factors were deposited in the RCSB Protein Data Bank with accession code 5XXX for the T. thermophilus 70S ribosome in complex with His-CAM, mRNA, A-, P- and E-site tRNAs; 5XXX for the T. thermophilus 70S ribosome in complex with D-His-CAM and protein Y; and 5XXX for the T. thermophilus 70S ribosome in complex with Lys-CAM and protein Y.
References
- 1.Polacek N, Mankin AS. The ribosomal peptidyl transferase center: structure, function, evolution, inhibition. Crit Rev Biochem Mol Biol. 2005;40:285–311. doi: 10.1080/10409230500326334. [DOI] [PubMed] [Google Scholar]
- 2.Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol. 2014;12:35–48. doi: 10.1038/nrmicro3155. [DOI] [PubMed] [Google Scholar]
- 3.Schwarz S, Shen J, Kadlec K, Wang Y, Brenner Michael G, Fessler AT, et al. Lincosamides, Streptogramins, Phenicols, and Pleuromutilins: Mode of Action and Mechanisms of Resistance. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a027037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKie SA. Antibiotics: where to throw the spanner in the ribosomal machinery? Future Med Chem. 2016;8:1981–2002. doi: 10.4155/fmc-2016-0014. [DOI] [PubMed] [Google Scholar]
- 5.Dinos GP, Athanassopoulos CM, Missiri DA, Giannopoulou PC, Vlachogiannis IA, Papadopoulos GE, et al. Chloramphenicol derivatives as antibacterial and anticancer agents: Historic problems and current solutions. Antibiotics. 2016;5 doi: 10.3390/antibiotics5020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J Mol Biol. 2003;330:1061–75. doi: 10.1016/s0022-2836(03)00668-5. [DOI] [PubMed] [Google Scholar]
- 7.Bulkley D, Innis CA, Blaha G, Steitz TA. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc Natl Acad Sci USA. 2010;107:17158–63. doi: 10.1073/pnas.1008685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pestka S. Chloramphenicol. In: Corcoran JW, Hahn FE, Snell JF, Arora KL, editors. Antibitoics: Mechanism of action of antimicrobial and antitumor agents. Springer-Verlag; Berlin, Heidelberg, New York: 1975. pp. 370–95. [Google Scholar]
- 9.Marks J, Kannan K, Roncase EJ, Klepacki D, Kefi A, Orelle C, et al. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc Natl Acad Sci USA. 2016;113:12150–5. doi: 10.1073/pnas.1613055113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnhill AE, Brewer MT, Carlson SA. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial components. Antimicrob Agents Chemother. 2012;56:4046–51. doi: 10.1128/AAC.00678-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh R, Sripada L, Singh R. Side effects of antibiotics during bacterial infection: mitochondria, the main target in host cell. Mitochondrion. 2014;16:50–4. doi: 10.1016/j.mito.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 12.Jones CN, Miller C, Tenenbaum A, Spremulli LL, Saada A. Antibiotic effects on mitochondrial translation and in patients with mitochondrial translational defects. Mitochondrion. 2009;9:429–37. doi: 10.1016/j.mito.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Ibrahim NG, Burke JP, Beattie DS. The sensitivity of rat liver and yeast mitochondrial ribosomes to inhibitors of protein synthesis. J Biol Chem. 1974;249:6806–11. [PubMed] [Google Scholar]
- 14.Lamb AJ, Clark-Walker GD, Linnane AW. The biogenesis of mitochondria. 4. The differentiation of mitochondrial and cytoplasmic protein synthesizing systems in vitro by antibiotics. Biochimica et biophysica acta. 1968;161:415–27. [PubMed] [Google Scholar]
- 15.Hanekamp JC, Bast A. Antibiotics exposure and health risks: chloramphenicol. Environ Toxicol Pharmacol. 2015;39:213–20. doi: 10.1016/j.etap.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 16.Cohen BH, Saneto RP. Mitochondrial translational inhibitors in the pharmacopeia. Biochimica et biophysica acta. 2012;1819:1067–74. doi: 10.1016/j.bbagrm.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 17.Smith LK, Mankin AS. Transcriptional and translational control of the mlr operon, which confers resistance to seven classes of protein synthesis inhibitors. Antimicrob Agents Chemother. 2008;52:1703–12. doi: 10.1128/AAC.01583-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long KS, Poehlsgaard J, Kehrenberg C, Schwarz S, Vester B. The Cfr rRNA methyltransferase confers resistance to Phenicols, Lincosamides, Oxazolidinones, Pleuromutilins, and Streptogramin A antibiotics. Antimicrob Agents Chemother. 2006;50:2500–5. doi: 10.1128/AAC.00131-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Locke JB, Zurenko GE, Shaw KJ, Bartizal K. Tedizolid for the management of human infections: in vitro characteristics. Clin Infect Dis. 2014;58(Suppl 1):S35–S42. doi: 10.1093/cid/cit616. [DOI] [PubMed] [Google Scholar]
- 20.Mamos P, Krokidis MG, Papadas A, Karahalios P, Starosta AL, Wilson DN, et al. On the use of the antibiotic chloramphenicol to target polypeptide chain mimics to the ribosomal exit tunnel. Biochimie. 2013;95:1765–72. doi: 10.1016/j.biochi.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Bougas A, Vlachogiannis IA, Gatos D, Arenz S, Dinos GP. Dual effect of chloramphenicol peptides on ribosome inhibition. Amino acids. 2017;49:995–1004. doi: 10.1007/s00726-017-2406-5. [DOI] [PubMed] [Google Scholar]
- 22.Vince R, Almquist RG, Ritter CL, Daluge S. Chloramphenicol binding site with analogues of chloramphenicol and puromycin. Antimicrob Agents Chemother. 1975;8:439–43. doi: 10.1128/aac.8.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rebstock MC, Crooks HM, Controulis J, Bartz QR. Chloramphenicol (chloromycetin). IV. Chemical studies J Am Chem Soc. 1949;71:2458–62. [Google Scholar]
- 24.Drainas D, Mamos P, Coutsogeorgopoulos C. Aminoacyl analogs of chloramphenicol: examination of the kinetics of inhibition of peptide bond formation. J Med Chem. 1993;36:3542–5. doi: 10.1021/jm00075a008. [DOI] [PubMed] [Google Scholar]
- 25.Michelinaki M, Mamos P, Coutsogeorgopoulos C, Kalpaxis DL. Aminoacyl and peptidyl analogs of chloramphenicol as slow-binding inhibitors of ribosomal peptidyltransferase: a new approach for evaluating their potency. Mol Pharmacol. 1997;51:139–46. doi: 10.1124/mol.51.1.139. [DOI] [PubMed] [Google Scholar]
- 26.Yan K, Hunt E, Berge J, May E, Copeland RA, Gontarek RR. Fluorescence polarization method to characterize macrolide-ribosome interactions. Antimicrob Agents Chemother. 2005;49:3367–72. doi: 10.1128/AAC.49.8.3367-3372.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tereshchenkov AG, Shishkina AV, Karpenko VV, Chertkov VA, Konevega AL, Kasatsky PS, et al. New fluorescent macrolide derivatives for studying interactions of antibiotics and their analogs with the ribosomal exit tunnel. Biochemistry (Mosc) 2016;81:1163–72. doi: 10.1134/S0006297916100138. [DOI] [PubMed] [Google Scholar]
- 28.Dunkle JA, Xiong L, Mankin AS, Cate JH. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc Natl Acad Sci USA. 2010;107:17152–7. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vazquez D. Binding of chloramphenicol to ribosomes. The effect of a number of antibiotics. Biochimica et biophysica acta. 1966;114:277–88. doi: 10.1016/0005-2787(66)90309-1. [DOI] [PubMed] [Google Scholar]
- 30.Lessard JL, Pestka S. Studies on the formation of transfer ribonucleic acid-ribosome complexes. 23. Chloramphenicol, aminoacyl-oligonucleotides, and Escherichia coli ribosomes. J Biol Chem. 1972;247:6909–12. [PubMed] [Google Scholar]
- 31.Tereshchenkov AG, Shishkina AV, Tashlitsky VN, Korshunova GA, Bogdanov AA, Sumbatyan NV. Interaction of chloramphenicol tripeptide analogs with ribosomes. Biochemistry (Moscow) 2016;81:392–400. doi: 10.1134/S000629791604009X. [DOI] [PubMed] [Google Scholar]
- 32.Shimizu Y, Kuruma Y, Kanamori T, Ueda T. The PURE system for protein production. Methods Mol Biol. 2014;1118:275–84. doi: 10.1007/978-1-62703-782-2_19. [DOI] [PubMed] [Google Scholar]
- 33.Vazquez-Laslop N, Klepacki D, Mulhearn DC, Ramu H, Krasnykh O, Franzblau S, et al. Role of antibiotic ligand in nascent peptide-dependent ribosome stalling. Proc Natl Acad Sci USA. 2011;108:10496–501. doi: 10.1073/pnas.1103474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polikanov YS, Blaha GM, Steitz TA. How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science. 2012;336:915–8. doi: 10.1126/science.1218538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polikanov YS, Melnikov SV, Soll D, Steitz TA. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat Struct Mol Biol. 2015;22:342–4. doi: 10.1038/nsmb.2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porse BT, Garrett RA. Sites of interaction of streptogramin A and B antibiotics in the peptidyl transferase loop of 23 S rRNA and the synergism of their inhibitory mechanisms. J Mol Biol. 1999;286:375–87. doi: 10.1006/jmbi.1998.2509. [DOI] [PubMed] [Google Scholar]
- 37.Shishkina A, Makarov G, Tereshchenkov A, Korshunova G, Sumbatyan N, Golovin A, et al. Conjugates of amino acids and peptides with 5-o-mycaminosyltylonolide and their interaction with the ribosomal exit tunnel. Bioconjug Chem. 2013;24:1861–9. doi: 10.1021/bc400236n. [DOI] [PubMed] [Google Scholar]
- 38.Polikanov YS, Osterman IA, Szal T, Tashlitsky VN, Serebryakova MV, Kusochek P, et al. Amicoumacin A inhibits translation by stabilizing mRNA interaction with the ribosome. Mol Cell. 2014;56:531–40. doi: 10.1016/j.molcel.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Svetlov MS, Kommer A, Kolb VA, Spirin AS. Effective cotranslational folding of firefly luciferase without chaperones of the Hsp70 family. Protein Sci. 2006;15:242–7. doi: 10.1110/ps.051752506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vazquez-Laslop N, Thum C, Mankin AS. Molecular mechanism of drug-dependent ribosome stalling. Mol Cell. 2008;30:190–202. doi: 10.1016/j.molcel.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 41.Orelle C, Szal T, Klepacki D, Shaw KJ, Vazquez-Laslop N, Mankin AS. Identifying the targets of aminoacyl-tRNA synthetase inhibitors by primer extension inhibition. Nucleic Acids Res. 2013;41:e144. doi: 10.1093/nar/gkt526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orelle C, Carlson S, Kaushal B, Almutairi MM, Liu H, Ochabowicz A, et al. Tools for characterizing bacterial protein synthesis inhibitors. Antimicrob Agents Chemother. 2013;57:5994–6004. doi: 10.1128/AAC.01673-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.