

Supplemental Digital Content is available in the text.
Keywords: apolipoprotein, cell membrane, HDL cholesterol, lipid metabolism, low-density lipoprotein receptor–related protein 1, triglycerides
Abstract
Objective—
Studies into the role of LRP1 (low-density lipoprotein receptor–related protein 1) in human lipid metabolism are scarce. Although it is known that a common variant in LRP1 (rs116133520) is significantly associated with HDL-C (high-density lipoprotein cholesterol), the mechanism underlying this observation is unclear. In this study, we set out to study the functional effects of 2 rare LRP1 variants identified in subjects with extremely low HDL-C levels.
Approach and Results—
In 2 subjects with HDL-C below the first percentile for age and sex and moderately elevated triglycerides, we identified 2 rare variants in LRP1: p.Val3244Ile and p.Glu3983Asp. Both variants decrease LRP1 expression and stability. We show in a series of translational experiments that these variants culminate in reduced trafficking of ABCA1 (ATP-binding cassette A1) to the cell membrane. This is accompanied by an increase in cell surface expression of SR-B1 (scavenger receptor class B type 1). Combined these effects may contribute to low HDL-C levels in our study subjects. Supporting these findings, we provide epidemiological evidence that rs116133520 is associated with apo (apolipoprotein) A1 but not with apoB levels.
Conclusions—
This study provides the first evidence that rare variants in LRP1 are associated with changes in human lipid metabolism. Specifically, this study shows that LRP1 may affect HDL metabolism by virtue of its effect on both ABCA1 and SR-B1.
Since its discovery in 1988 through homology cloning with the LDL (low-density lipoprotein) receptor, the LRP1 (LDL receptor–related protein 1) has been considered an important regulator of plasma lipid concentrations by its effect on the clearance of atherogenic triglyceride-rich lipoproteins.1,2 LRP1 is also recognized for its role in other biological processes, including homeostasis of proteinases and proteinase inhibitors, activation of lysosomal enzymes, cellular signal transduction, and neurotransmission.3,4 Like other members of the LDLR family, LRP1 contains several ligand-interacting CCR (clusters of complement-like repeats) domains, cysteine-rich EGF (epidermal growth factor) repeats and a β-propeller domain. LRP1 is composed of a 515-kDa extracellular α-chain which contains binding sites for many unrelated ligands and an 85-kDa β-chain consisting of an extracellular domain, a transmembrane region and a cytoplasmic tail.4
A recent genome-wide association study in humans showed that common variants in LRP1 are associated with triglyceride but also with HDL-C (high-density lipoprotein cholesterol) levels.5 Because of the intricate relationship between triglyceride and HDL-C levels, it is not known whether LRP1 directly affects HDL metabolism. In mice, a clear role for LRP1 in HDL metabolism has, however, been established.6,7 Hepatic LRP1 deficiency was shown to result in 33% lower plasma HDL-C levels compared with wild-type (WT) mice, whereas no effect on triglyceride levels was observed.6 This was attributed to the observed negative effect of hepatic LRP1 deficiency on cell surface localization of ABCA1 (ATP-binding cassette transporter A1) which is essential for the transport of phospholipids and cholesterol across the cellular membrane to lipid-free apo (apolipoprotein) A1.8 It was proposed that LRP1 acts as an endocytic receptor for the binding and internalization of CTSD (cathepsin D), which is involved in the processing of PSAP (prosaposin), the precursor of the glycosphingolipid-hydrolyzing saposins.9 The latter plays a crucial role in regulating transport of glycosphingolipids and cholesterol through the late endosomes, which in turn regulates ABCA1 expression and activity. Accordingly, Lrp1 loss of function resulted in reduced intracellular levels of CTSD and impairment of PSAP activation and a corroborated trafficking of ABCA1 toward the plasma membrane. Other insights into the role of LRP1 in cholesterol metabolism were provided by Zhou et al,10 who elucidated a role of LRP1 in regulating LXR (liver X receptor)-mediated gene transcription and participation in reverse cholesterol transport by controlling cytosolic phospholipase A2 activation and ABCA1 expression. More recently, additional convergent LRP1-mediated signaling pathways were found to be crucial for cellular cholesterol homeostasis in mouse embryonic fibroblasts and HEK293 cells. In particular, the extracellular α-chain of LRP1 was reported to mediate a TGF (transforming growth factor) β-induced increase of WNT-5a (Wnt family member 5A), which reduced intracellular cholesterol accumulation via inhibition of cholesterol biosynthesis and stimulation of ABCG1 (ATP-binding cassette transporter G1)-mediated cholesterol efflux. In the absence of LRP1, WNT-5a is downregulated and cells accumulate cholesterol. Another pathway has been shown to be mediated through the cytoplasmic β-chain of LRP1 which is sufficient to limit cholesterol accumulation in LRP1 knockout cells by increasing the expression of ABCA1 and NCEH1 (neutral cholesterol ester hydrolase 1).7 In addition, the intracellular domain of LRP1 has been recently found to interact with the nuclear receptor Pparγ (peroxisome proliferator-activated receptor gamma), a central regulator of lipid and glucose metabolism, acting as its transcriptional coactivator in endothelial cells. This study showed that LRP1 mediates metabolic responses not only by acting as an endocytic receptor but also by directly participating in gene transcription.11
The studies performed to date clearly indicate that LRP1 has a large impact on cellular lipid homeostasis, which could directly affect HDL metabolism. However, this evidence has been obtained from studies performed in mice and cell culture. Confirmation of a role of LRP1 in human cholesterol homeostasis is apart from genome-wide association study5 largely lacking.12–16 The importance of a clear understanding of LRP1 in human lipid metabolism and associated pathophysiology is illustrated by the recently published association of a common variant in LRP1 (rs11172113) with incidence of coronary artery disease.17
In the current study, we investigated 2 extremely rare naturally occurring variants in LRP1 in individuals with plasma HDL-C levels below the first percentile. Despite the general concept that LRP1 affects TRL metabolism, we provide evidence that LRP1 may directly affect human HDL metabolism through effects on ABCA1 as previously observed in mice6 but also through effects on SR-B1 (scavenger receptor class B type 1).
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplementary file in the online-only Data Supplement.
Subjects and Mutation Analysis
A cohort of individuals with very high (n=40) and very low (n=40) plasma HDL-C levels (<1st and >99th percentile for age and sex) from the general population was studied to identify the genetic background underlying the HDL-C phenotype as described.18 Coding sequence and exon-intron boundaries of 195 lipid-related genes and 78 lipid-unrelated genes were sequenced using Agilent Sure select custom capture library on the Illumina HiSeq 2000 platform. LRP1 (the gene encoding LRP1; National Center for Biotechnology Information reference sequence NM_002332) was sequenced, and LRP1 variants (c. 9730G>A p.Val3244Ile, and c. 11949 G>T; p.Glu3983Asp) were identified in 2 individuals. Written informed consent was obtained from all individuals, and the study was approved by the Medical Ethical Committee of the Amsterdam Medical Center, Amsterdam, The Netherlands.
Population Data
The CCHS (Copenhagen City Heart Study) is a prospective study from the general population started between 1976 and 1978 with 3 follow-up examinations between 1981 and 1983, 1991 and 1994, and 2001 and 2003. Data were obtained from a questionnaire, a physical examination, and from blood samples including DNA extraction. Follow-up was 100% complete and ended on April 10, 2013. DNA extraction from the blood samples was performed at the 1991 to 1994 and 2001 to 2003 examination. Nonfasting total cholesterol, HDL-C, and triglycerides were measured by colorimetric assays (Boehringer Mannheim and Konelab). LDL cholesterol was calculated using the Friedewald equation when plasma triglycerides were ≤4.0 mmol/L (≤352 mg/dL), otherwise, LDL cholesterol was measured directly (Thermo Fisher Scientific and Konelab). TaqMan-based assays and ABI PRISM 7900HT Sequence Detection System (Applied Biosystems Inc, Foster City, CA) were used to genotype the genetic variant.
Antibodies
In the experimental procedures described, the following antibodies were used: rabbit monoclonal anti-LRP1 (Ab92544; Abcam), mouse monoclonal anti-ABCA1 (Ab18180; Abcam), rabbit monoclonal anti-PSAP (ab166910; Abcam), mouse monoclonal anti-CTSD (NBP1-04278 Novus Biologicals), goat anti–SR-B1 (NB400-131; Novus Biologicals), rabbit anti-ABCG1 (ab36969; Abcam), rat anti–Wnt-5a (MAB645; R&D Systems), mouse monoclonal anti–β-actin (A5441, Sigma-Aldrich, 1:5,000), anti–α-tubulin (Ab4047, Abcam), anti-GAPDH (Ab8245), rabbit anti-GST (sc-459), goat anti-rabbit IgG-horseradish peroxidase conjugate (170–6515: Bio-Rad Laboratories,), goat anti-mouse IgG-horseradish peroxidase conjugate (170-6516; Bio-Rad Laboratories), and goat anti-Rat IgG-horseradish peroxidase conjugated (HAF005; R&D Systems). For immunofluorescence, Alexa Fluor–conjugated secondary antibodies and Alexa Fluor 488–conjugated and 568–conjugated antibodies were purchased from Jackson ImmunoResearch.
Cell Culture and Transfections
Human embryonic kidney 293 cells (American Type Culture Collection), liver hepatocellular cell lines Hep3B (ATCC [American Type Culture Collection], HB-8064), and human skin fibroblasts were cultured in high-glucose Dulbecco modified Eagle medium GlutaMAXTM (4.5 g/L D-Glucose and Pyruvate; Invitrogen Life Technologies Corporation) supplemented with 10% fetal bovine serum, 1% penicillin, and streptomycin at 37°C in 5% CO2. In the recovery of LRP1 mutant expression, experiments transfected cells were either incubated at 37°C or 30°C for 48 hours. Subsequently, cells were lysed in RIPA (radioimmunoprecipitation assay buffer), and protein expression was determined by Western blot analysis. Experiments were performed in triplicate.
Generation of LRP1 Constructs
WT LRP1-GFP (green fluorescence protein) pFastBac1 vector was kindly provided by Joerg Heeren (University Medical Center Hamburg-Eppendorf). This vector carries the human cDNA of LRP1 fused in frame with a GFP cDNA. Such a vector was digested with KpnI and HindIII restriction enzymes purchased from New England Biolabs (Beverly, MA). The resulting fragment (2.7 kb) was used for subcloning into pCR-Blunt vector (Thermo Fisher Scientific). Both missense variants c.9730G > A and c.11949G > T were introduced via site-directed mutagenesis using QuikChange Lightning Site-Directed Mutagenesis kit (Stratagene). DNA sequences were checked out by Sanger sequencing.
Quantitative Reverse Transcriptase Polymerase Chain Reaction
Total RNA was isolated from cells by means of TRIZOL (Invitrogen Life Technologies Corporation). cDNA synthesis was performed using Transcription Universal cDNA Master (Roche). mRNA expression was assayed by quantitative polymerase chain reaction using Sybrgreen (Applied Biosystem) and the real-time polymerase chain reaction cycler (7900HT Applied Biosystem) using primers listed in Table II in the online-only Data Supplement. Expression data were analyzed using SDS 2.3 software (Applied Biosystems) and the standard curve method of calculation. Results were presented as fold induction and normalized to the expression of β-actin and GAPDH, which were selected as the most stable reference genes.
Western Blot Analysis
Protein concentrations were determined by Bradford Protein Assay. Transiently transfected cells and human skin fibroblasts were rinsed twice in ice-cold PBS before lysis in lysis buffer (5 mol/L NaCl, NP-40, 1 mol/L Tris; pH 8.0, 0.5 mol/L EDTA, H2O) supplemented with 1 mmol/L Na3VO4, 1 mmol/L PMSF (phenylmethylsulfonyl fluoride), 10 mmol/L dithiothreitol, and protease inhibitors (Complete; Roche, Basle, Switzerland). Protein lysates were boiled at 95°C for 5 minutes before gel loading. In all experiments, SDS-PAGE (6% and 8%) was followed by protein transfer onto polyvinylidene fluoride (Amersham) or nitrocellulose membranes (Bio-Rad Laboratories) for immunoblot analysis. Mouse monoclonal anti–β-actin, anti–α-tubulin, or anti-GAPDH was used as internal controls. Proteins were detected with either ECL detection system or SuperSignal West Femto ECL kit (Thermo Scientific). Densitometry analysis of Western blot bands was performed using Image Laboratory 5.0 software (Bio-Rad Laboratories).
Immunofluorescence Staining
Staining was performed as previously described.19 Skin-derived fibroblasts were grown on coverslips. Forty-eight hours after seeding, cells were washed twice in PBS and fixed using 4% (w/v) paraformaldehyde for 10 minutes at room temperature. Cells were permeabilized and blocked using, respectively, cold 0.3% Triton X-100 for 5 minutes and blocking buffer (0.2%; w/v) BSA (Sigma-Aldrich) in PBS for 30 minutes at room temperature in the dark. Immunolabeling was performed in blocking buffer with the indicated primary antibodies for 1 hour. After washing 3×, secondary labeling was performed with Alexa Fluor–conjugated secondary antibodies for 1 hour. DAPI (4’,6-diamidino-2-phenylindole) staining was performed in the final washing step preceding the mounting of coverslips (Vector Laboratories). Images were acquired using an LSM-710 laser scanning confocal microscope and analyzed by ZEN software (Carl Zeiss).
Protein Stability Assay
To measure protein stability, cycloheximide, a translation inhibitor in eukaryotic cells, was used. HEK293T cells were transfected with either WT or mutant constructs before addition of cycloheximide (Sigma, 100 μmol/L). Cells were harvested at various time points after cycloheximide treatment (0, 3, 6, and 24 hours), collected in lysis buffer, and protein processed for Western blot analysis. LRP1 protein stability was assessed as the proportion of the initial protein remaining after cycloheximide treatment.
Targeted Quantitative Proteomics on LRP1
WT and p.Glu3983Asp-mutant LRP1 were quantified using synthetic peptides carrying a 13C15N-isotope label on the C-terminal lysine (PEPotec synthetic peptides-grade 2; Thermo Scientific) in human fibroblasts of an LRP1 variant carrier and 3 controls. In brief, lysed fibroblasts (starting with 1×106 cells) were subjected to in-gel tryptic digestion (1:100 g/g) after reduction with 10 mmol/L dithiothreitol and alkylation with 55 mmol/L iodoacetamide. On the basis of the total protein concentration (determined by a BCA [bicinchoninic acid] assay), 20 µg of total protein was mixed with 45 fmol of the synthetic LRP1 synthetic isotopically labeled peptides DVIEVAQMK for the quantification of WT LRP1 and DVIDVAQMK for the GluE3983Asp-mutant LRP1. Both peptides were targeted and analyzed by a triple quadrupole mass spectrometer equipped with a nano-electrospray ion source (TSQ Vantage; Thermo Scientific). The chromatographic separation (gradient 110 minutes) of the peptides was performed by liquid chromatography on a nano-UHPLC system (Ultimate UHPLC focused; Dionex). The mass spectrometer traces were manually curated using the Skyline software before integration of the peak areas for quantification.20 The sum of all transition peak areas for the endogenous peptides and isotopically labeled synthetic peptide standards was used to calculate the ratio between the endogenous and standard peptides. The concentrations of the endogenous peptides were calculated from the known concentrations of the standards and expressed in fmol/μg of total protein.
CRISPR/Cas9 Genome Editing
The LRP1 knockout cell line was generated by using the CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/clustered regularly interspaced short palindromic repeat–associated 9) system as previously described.21 Candidate guide RNAs were designed using the Zhang Laboratory CRISPR Design website (http://crispr.mit.edu) to target LRP1 (Table IVA in the online-only Data Supplement). A plasmid based on gRNA_Cloning Vector (https://www.addgene.org/41824/) was used to express the guide RNA with the chosen protospacer sequence from the U6 promoter. pSpCas9(BB)-2A-Puro (PX459; https://www.addgene.org/62988/) was used to coexpress a human codon-optimized Cas9 gene with a C-terminal nuclear localization signal. HEK293T cell line was grown on 10 cm dish and assessed for efficacy with Surveyor assay. Single colonies were manually picked, dispersed, and replated individually to wells of 96-well plates. Colonies were subsequently screened by polymerase chain reaction and Sanger sequencing to identify insertion-deletion (Indels) at the target site. Several clones had compound heterozygote deletions flanking the target site (Table IVB in the online-only Data Supplement), and 3 were chosen for expansion and differentiation along with WT clones from the same 96-well plate. Absence of LPR1 protein expression was assessed by Western blot.
Knockdown in Hepatic Cells
RNAi silencing of LRP1 was performed by transfection of double-stranded siRNA oligonucleotides (Trilencer-27) designed and synthesized by Origene. Control transfections included both a proven nontargeting siRNA provided by Origene and transfection reagent only without oligonucleotides according to the manufacturers protocol.
Cell Surface Biotinylation Assay
Cells were washed (3×) with ice-cold PBS-CM (phosphate-buffered saline plus CaCl2 and MgCl2) buffer (PBS, 1 mmol/L MgCl2 and 0.1 mmol/L CaCl2). EZ-link-sulfo-NHS-LC biotin reagent solution (0.5 mg/mL) and biotinylation buffer (10 mmol/L triethanolamine, pH 8.0, 150 mmol/L NaCl, and 2 mmol/L CaCl2) were added for 30 minutes at 4°C. Biotin reagent was removed, and cells were washed (2×) with quenching buffer (PBS-CM, 0.1% BSA) followed by 1× washing step with PBS-CM buffer. Cells were then incubated for 5 minutes at 4°C with lysis buffer (1.0% NP-40, 150 mmol/L NaCl, 5 mmol/L EDTA, 50 mmol/L Tris pH 7.5, and fresh protein inhibitors) collected by scraping and centrifuged (14 000g, 10 minutes, 4°C). Protein concentration was determined; 0.2 mg was used as input; 2 mg was used and diluted in lysis buffer (0.8 mL); and 80 μL Neutravidin beads (Neutravidin Agarose Resin, Thermo Scientific) were added and incubated for 3 hours at 4°C. Beads were collected by centrifugation (500g, 2 minutes, 4°C) and washed 3× with lysis buffer containing 1 mmol/L Biotin at 4°C. Finally, the beads were eluted in 50 μL 2× loading buffer (2 volume 4× laemli, 1 volume milli-Q-H2O, and 1 volume 1 mol/L dithiothreitol).
Statistical Analysis
Statistical analyses were performed using GraphPad software (La Jolla, CA). The quantitative data in this article are represented as mean±SEM. Statistical evaluation was performed using Student unpaired t test (normality and equal variances were visually checked and tested using F test), Kruskal-Wallis ANOVA followed by a Dunn test for multiple group comparisons, and 2-way ANOVA with Tukey test for multiple comparisons. The population data were analyzed using Stata/SE version 13.0 (Stata Corp, College Station, TX). To assess whether common variants were associated with levels of triglyceride, HDL-C, apoA1, apoB, and total cholesterol, a linear regression adjusted for age and sex was used and Cuzick extension of a Wilcoxon rank-sum test as a test for trend was applied. Differences were considered to be significant at P<0.05.
Results
Rare LRP1 Variants in Subjects With HDL-C Below the First Percentile
We previously identified 2 heterozygous carriers of 2 missense variants in LRP1 c.9730G>A (p.Val3244Ile; V3244I) and c.11949G>T (p.Glu3983Asp; E3983D) in a cohort of individuals with very low HDL-C levels and without any rare variant in 195 lipid-related genes.18 In the same cohort, we identified numerous mutations known to cause familial HDL deficiency (APOA1, ABCA1, and LCAT) illustrating the validity of this cohort to find novel regulators of HDL-C.22,23 Table 1 shows the HDL-C and triglyceride levels of the carriers of the rare LRP1 variants. Both variants have a high CADD (combined annotation-dependent depletion) score24 (Table 1) indicating that they are likely pathogenic. The c.9730G>A has not been reported elsewhere, whereas the second variant c.11949G>T was also identified in the Exome Sequencing Project (annotated as rs146923187) with a very low frequency of 0.07% and in the ExAC database with a minor allele frequency of 9×10−5 (Table 1). More detailed information on the variants (using ANNOVAR [annotate variation]25) is given in the Table I in the online-only Data Supplement. The amino acid sequences in the regions where the variants are located are highly conserved across mammalian species (Figure 1A) and are localized in the LDL receptor class B 30 and 32 domains of the α-chain (515 kDa) and β-chain (85 kDa) of LRP1, respectively (Figure 1B). Taken together, these in silico analyses suggest that both LRP1 variants identified in our study subjects may impact the function of the protein.
Table 1.
Rare LRP1 Variants Identified in Individuals With Low HDL-C Levels
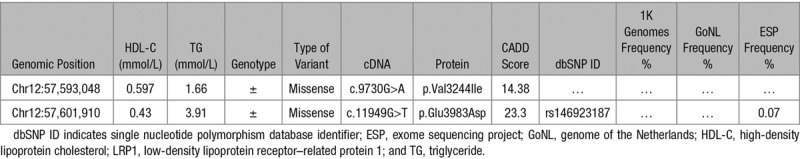
Figure 1.
Effect of LRP1 (low-density lipoprotein receptor–related protein 1) variants on protein expression and stability in vitro. A, Orthologue conservation of amino acids residues at positions 3244 and 3983 in LRP1 (p.Val3244Ile and p.Glu3983Asp identified in individuals with HDL-C [high-density lipoproteins cholesterol] levels <0.6 mmol/L). B, Scheme of the LRP1 encoded by LRP1 with the α-chain (light gray) and β-chain (dark gray) and the location of the newly identified variants. C, Real-time polymerase chain reaction quantification of LRP1 mRNA expression in HEK293T cells. β-actin and GAPDH were used as housekeeping genes. D, Representative Western blot analysis showing wild-type (WT) and LRP1 mutant expression in HEK293T cells. Either WT or mutant hLRP1 constructs (1 μg) were transiently expressed in HEK293T cell line along with GST (glutathione S-transferase) construct to control for transfection efficiency. Bar graphs show the quantification of LRP1 bands, black bar (WT), dark gray (p.Val3244Ile, V3244I) white (p.Glu3983Asp, E3983D), and light gray (nontransfected). E, Incubation of transfected cell with cycloheximide and LRP1 expression measured at 0, 3, 6, and 24 hours, respectively. Lines represent the percentage of the value before the addition of cycloheximide. Note that SEM was too small to visualize in some cases. Representative Western blot analysis showing the time course of LRP1 protein levels. F, Recovery of LRP1 mutants expression by incubating transfected cells at either 37°C or 30°C for 48 hours. Bars represent fold induction. Unchanged levels of α-tubulin or β-actin are shown as loading controls. The WT form of LRP1 was used as reference. Groups were compared using a Kruskal-Wallis ANOVA followed by a Dunn test for multiple group comparisons (C, D) and unpaired t tests (E, F). Data are expressed as mean±SEM of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.005 vs control cells.
Heterologous Expression of LRP1 Loss-of-Function Variants Affect Protein Expression and Stability
In light of the above in silico analysis, both variants were expressed in HEK293T cells to compare their properties to WT LRP1. Without a significant effect on the expression of the messenger RNA (Figure 1C), mutant LRP1 protein levels were significantly reduced (>50%; P<0.01; Figure 1D) suggesting that both LRP1 variants might influence protein stability. This was evaluated by measuring protein degradation over time in a cellular system where we overexpressed both WT and mutant LRP1 in the presence of the translation inhibiting agent cycloheximide. As expected, LRP1 WT levels decreased progressively with a half-life of >24 hours.26 In contrast, the half-life of p.Val3244Ile mutant was ≈50% and of the p.Glu3983Asp ≈85% shorter compared with WT (Figure 1E). In an attempt to restore the mutant LRP1 expression, transiently transfected cells were cultured at lower temperature (30°C), which has been previously shown to improve protein folding and subsequently stability.27 Notably, after 48-hour incubation, LRP1 protein levels were in both cases recovered close to WT levels (Figure 1F). These data suggest that the respective amino acid substitutions adversely affect LRP1 protein expression and stability.
ABCA1 and SR-B1 Cell Surface Localization Are Significantly Affected in Fibroblasts From the LRP1 p.Glu3983Asp Carrier
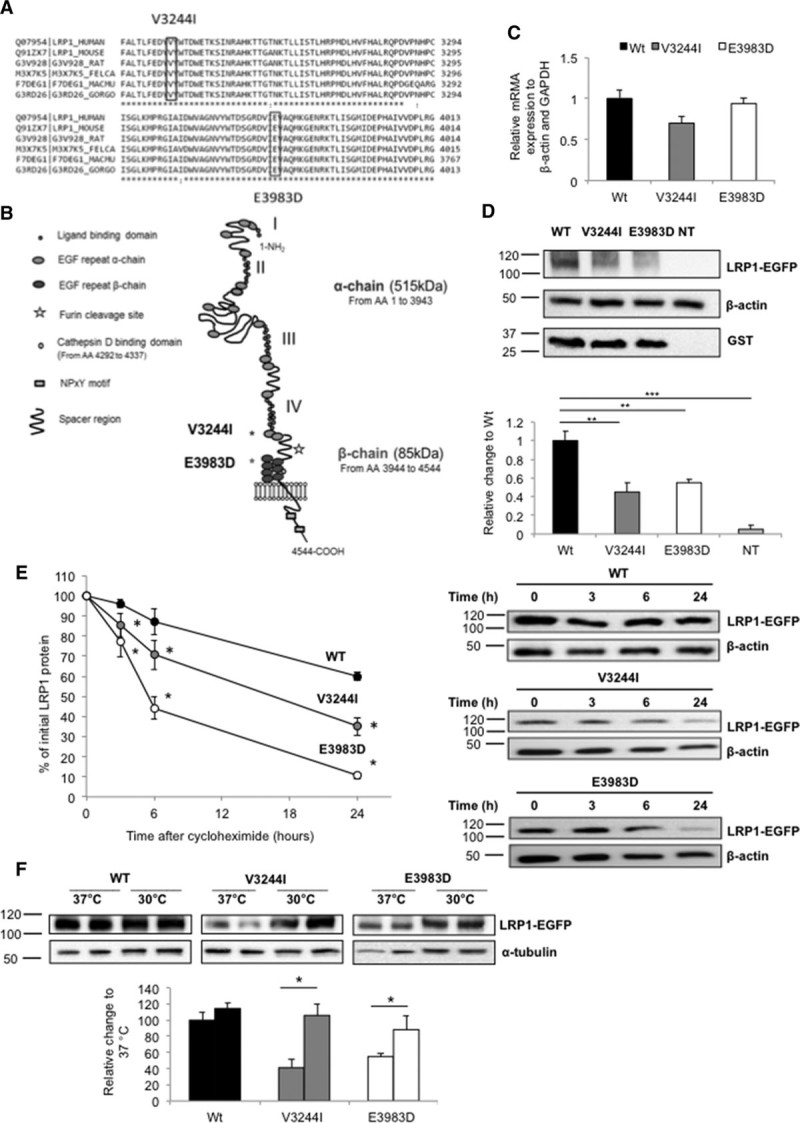
Skin-derived fibroblasts were collected from the index case carrying the p.Glu3983Asp (E3983D) variant. In line with our expression studies in HEK293T cells, this variant was found to have no effect on mRNA abundance (Figure 2A), but total LRP1 protein expression as assessed by Western Blotting was reduced by ≈35% (Figure 2B). LRP1 expression was again restored to control levels when fibroblasts were cultured at 30°C (Figure 2C).
Figure 2.
ABCA1 (ATP-binding cassette A1) and SR-B1 (scavenger receptor class B type 1) protein levels in the LRP1 (low-density lipoprotein receptor–related protein 1) variant carrier and control fibroblasts. A, Real-time polymerase chain reaction quantification of LRP1 expression in total cell lysates. β-actin and GAPDH were used as housekeeping genes. B, Representative Western blot showing LRP1 expression in 3 control individuals and the variant carrier. Quantification of LRP1 expression in controls (black bar) and variant carrier (white bar). C, Recovery of LRP1 mutant expression by culturing fibroblasts at both 37°C and 30°C for 48 hours. Bars represent fold induction. Quantification of LRP1 expression. D, Wild-type and E3983D-mutant LRP1 quantification using mass spectrometry. Endogenous signal is shown in light gray, standard in dark gray. Carrier (left) and 3 controls (right) are shown. E, PSAP (prosaposin; upper) and CTSD (cathepsin D; lower) expression levels in controls and variant carrier. Quantification of PSAP and CTSD expression. F, ABCA1 protein expression in total lysates (left) and on the cell surface (right). G, ABCA1 and SR-B1 mRNA expression levels. H, SR-B1 protein expression in total lysates (left) and on the cell surface (right). I, Immunofluorescence studies. Control (left) and carrier (right) fibroblasts were assessed for LRP1 (green), ABCA1 (red), and SR-B1 (purple) expression using confocal microscopy. In the above experiments, unchanged levels of β-actin or α-tubulin are shown as loading controls. Fibroblasts from controls were averaged and used as reference. Groups were compared using unpaired t tests. Data are expressed as mean±SEM of 3 independent experiments. *P<0.05, **P<0.01 vs control cells.
WT and mutant LRP1 protein levels were also specifically quantified by mass spectroscopy in the fibroblasts from the carrier and 3 controls. The WT protein in the fibroblasts from the carrier was present at 40% lower levels compared with the WT protein levels in controls. As expected the mutant protein was only detected in the carrier (Figure 2D; for details, see Table II in the online-only Data Supplement). These quantitative data indicate that both the mutant and WT LRP1 are present at approximate similar levels.
To acquire more insights into the possible molecular mechanism(s) underlying the low HDL-C phenotype, CTSD and PSAP expression levels were measured in fibroblast lysates. CTSD levels were reduced by 50% and PSAP levels were increased by 60% in the carrier compared with controls, respectively (Figure 2E). Next, ABCA1 levels were measured in total cell lysates and on the cellular surface in the presence or absence of an LXR agonist. A significant 50% reduction (P<0.05) was observed in both cellular compartments (Figure 2F and 2G). To validate the immunoblot data, ABCA1 protein expression was also studied by confocal microscopy. ABCA1 protein expressed at the plasma membrane and in the cytoplasm was decreased in fibroblasts derived from the heterozygote for the LRP1 variant (Figure 2I, middle).
In addition, we studied expression of SR-B1 as a major player in HDL metabolism. Interestingly, SR-B1 protein was increased by >2-fold in total cell lysate and in the cell membrane fraction (Figure 2G and 2H). It was recently observed that ABCG1 levels and WNT5a signaling pathways are controlled by LRP1 in mouse embryonic fibroblasts and HEK293.7 In contrast to these observations, both ABCG1 and WNT5a mRNA and protein levels were marginally decreased in fibroblasts of the E3983D carrier (Figure II in the online-only Data Supplement).
HEK293T LRP1 Knockout Cells Mimic the Phenotype Observed in Fibroblasts From the Carrier of the LRP1 Variant
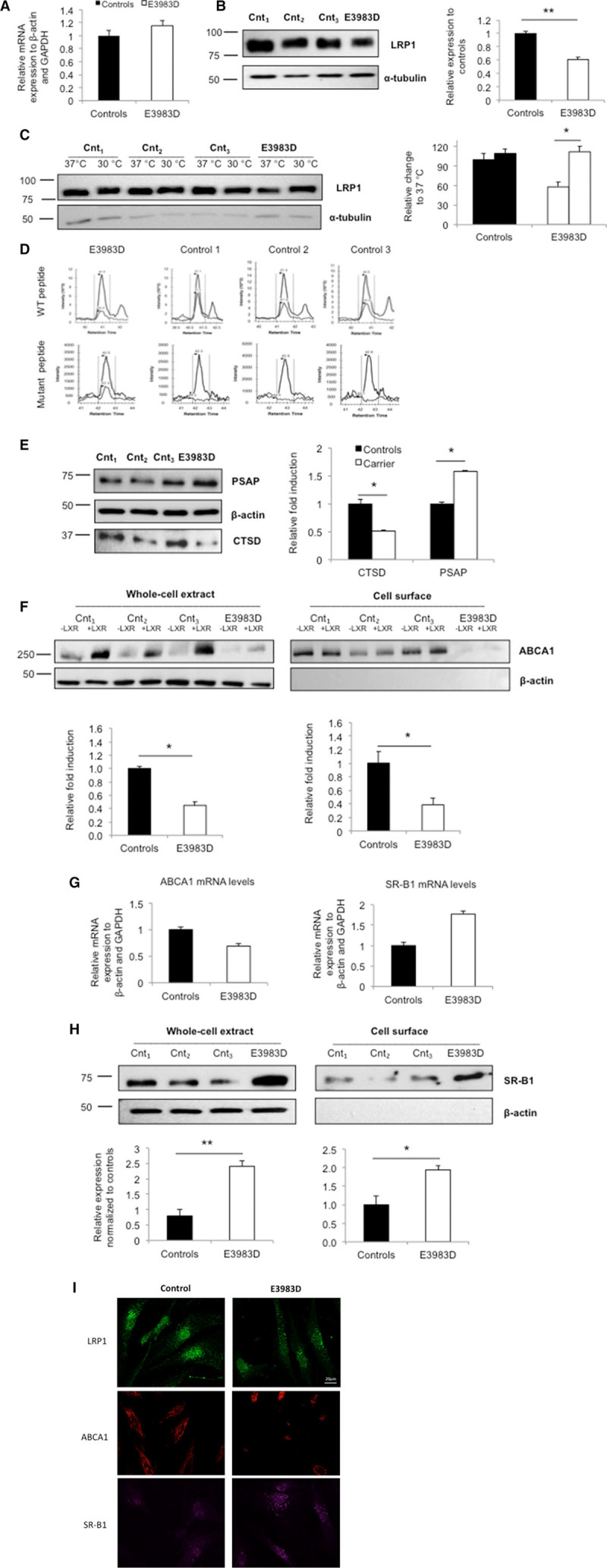
As the individuals are heterozygous carriers of the LRP1 variant, it means that both the WT protein and the mutated protein are present (Figure 2D). To study the sole effect of WT LRP1 and the LRP1 variants, we generated HEK293T LRP1 knockout cells using CRISPR/Cas9 technology.21 Specific sgRNA(s) were designed to target LRP1 (Table IVA in the online-only Data Supplement). Mutation efficiency was evaluated by the surveyor nuclease assay (Figure III in the online-only Data Supplement), followed by direct DNA sequencing and immunoblotting. Several heterozygous compound deletions were identified (Table IVB in the online-only Data Supplement). Upon confirmation that LRP1 expression was completely lost (Figure 3A), 3 clones were taken for further studies. CTSD and PSAP protein levels were measured in cell lysates. CTSD levels were 40% lower, whereas PSAP levels were 35% higher in the knockout compared with WT clones (Figure 3B). In line with previous experiments, ABCA1 expression was lower, whereas SR-B1 expression was higher at the cell surface of LRP1-deficient clones (Figure 3C).
Figure 3.
ABCA1 (ATP-binding cassette A1) and SR-B1 (scavenger receptor class B type 1) levels in LRP1 (low-density lipoprotein receptor–related protein 1) CRISPR/Cas9 knockout cells. A, Representative Western blot showing LRP1 expression in 3 wild-type (WT) and knockout (KO) cell lines, respectively. Quantification of LRP1 expression in WT (black bar) and KO (white bar). B, Quantification of CTSD (cathepsin D) and PSAP (prosaposin) expression in controls and KO cells. C, ABCA1 and SR-B1 protein expression in total lysates and on the cell surface and quantification of ABCA1 and SR-B1 expression. Unchanged levels of β-actin, α-tubulin are shown as loading controls. WT cell lines were used as reference. Groups were compared using unpaired t tests. Data are expressed as mean±SEM of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.005 vs control cells.
ABCA1 and SR-B1 Plasma Membrane Expression Are Not Rescued by Introduction of LRP1 Mutants in LRP1 Knockout Cells
Next, we investigated whether reduced cell surface expression of ABCA1 and increased expression of SR-B1 in LRP1 knockout cells could be rescued by reintroducing WT LRP1 or the 2 LRP1 variants (Figure 4A). After transient transfection, the levels of LRP1 protein were significantly lower in the case of LRP1 mutants when compared with WT (Figure 4B gray and white bars). ABCA1 membrane expression was restored after transfection with WT-LRP1, but for the 2 LRP1 mutants, no rescue of ABCA1 was observed (Figure 4C gray and white bars, respectively). Unexpectedly, SR-B1 expression at the cell membrane was increased after the introduction of the WT-LRP1 and mutant LRP1. This latter result is not agreement with all other experiments in which attenuated or reduced LRP1 was found to also increase SR-B1.
Figure 4.
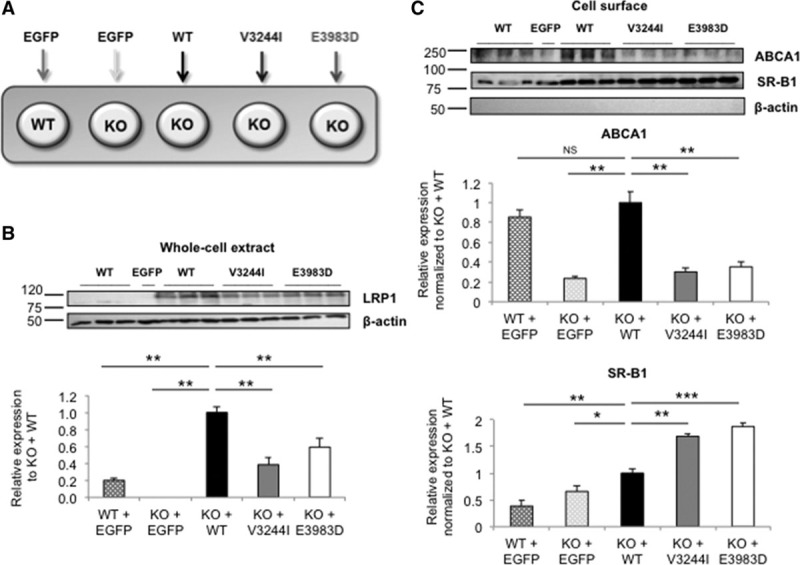
ABCA1 (ATP-binding cassette A1) and SR-B1 (scavenger receptor class B type 1) phenotype rescue studies in CRISPR/Cas9 knockout cells. A, LRP1 (low-density lipoprotein receptor–related protein 1)-deficient HEK293T cells were transfected with either the wild-type (WT) or mutant forms of LRP1. ABCA1 and SR-B1 (cell surface) and LRP1 (total lysates) expression was measured. B, Representative Western blot of LRP1 expression. Upper band (exogenous LRP1), lower band (endogenous LRP1). C, ABCA1 and SR-B1 cell surface expression. Legend: WT+empty construct (dark gray pattern), knockout (KO)+empty construct (light gray pattern), KO+WT construct (black), KO+V3244I construct (gray), and KO+E3983D construct (white). Unchanged levels of β-actin is shown as loading control. The KO+WT group (black) was used as reference. Groups were compared using 2-way ANOVA with Tukey test for multiple group comparisons. Data are expressed as mean±SEM of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.005 vs control cells.
LRP1 Knockdown in a Liver Cell Line Shows Similar Effects on ABCA1 and SR-BI
Next, we assessed whether LRP1 also affects ABCA1 and SR-B1 as major players in HDL metabolism in a human hepatoma cell line. To this purpose, we silenced LRP1 using siRNA in Hep3B cells. The 2 specific siRNA duplexes used resulted in 40% reduction of the expression of LRP1 protein (Figure 5A), which mimics the LRP1 expression in fibroblasts of the heterozygous carrier (Figure 2B). CTSD levels were significantly reduced by 35%, and PSAP levels were increased by 50%, respectively (Figure 5B). In line with previous data, ABCA1 expression at the cell surface was reduced, whereas SR-B1 was significantly increased (Figure 5C).
Figure 5.
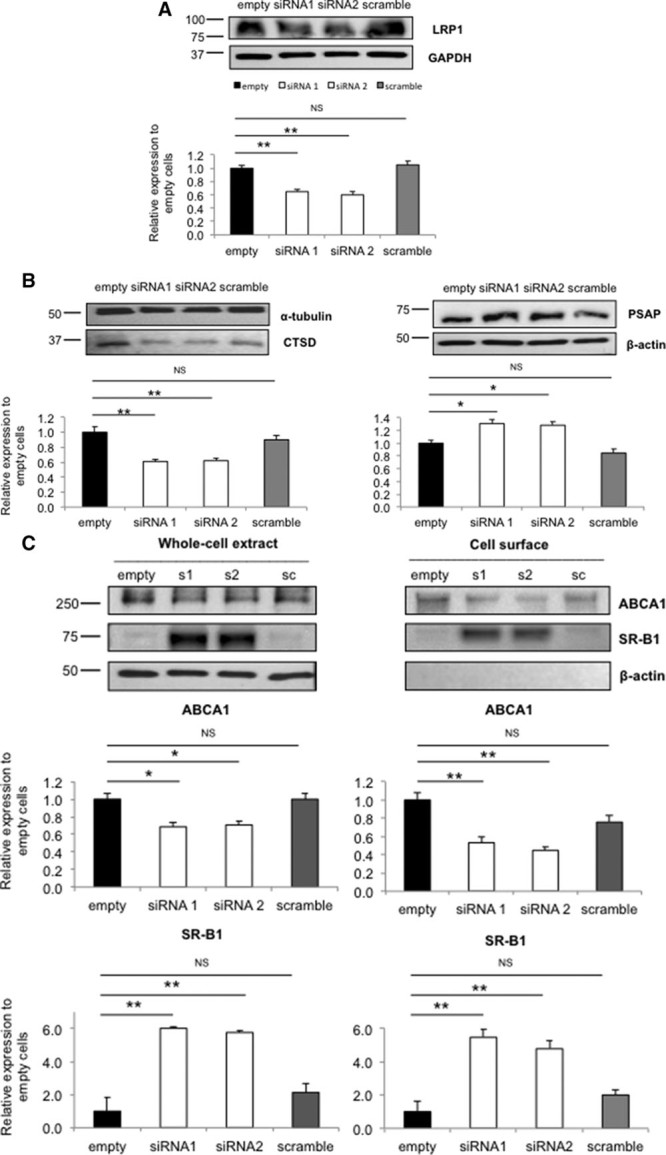
ABCA1 (ATP-binding cassette A1) and SR-B1 (scavenger receptor class B type 1) levels in LRP1 (low-density lipoprotein receptor–related protein 1) siRNA-mediated knockdown cells. A, Representative Western blot showing knockdown of LRP1 in Hep3B cell line. Untreated cells (empty, black bar), siRNA1 (s1), siRNA2 (s2; white bars), and scrambled (sc, gray bar) are shown, respectively. B, CTSD (cathepsin D) expression and PSAP (prosaposin) expression. C, ABCA1 and SR-B1 protein expression in total cell lysates and on the cell surface. Quantification of ABCA1 (upper) and SR-B1 expression (lower). Unchanged levels of β-actin, α-tubulin, and GAPDH are shown as loading controls. The empty group (black) was used as reference. Groups were compared using a Kruskal-Wallis ANOVA followed by a Dunn test for multiple group comparisons. Data are expressed as mean±SEM of 3 independent experiments. *P<0.05, **P<0.01 vs control cells.
A Common Variant Close to LRP1 Is Associated With HDL-C and ApoA1 Levels in the CCHS
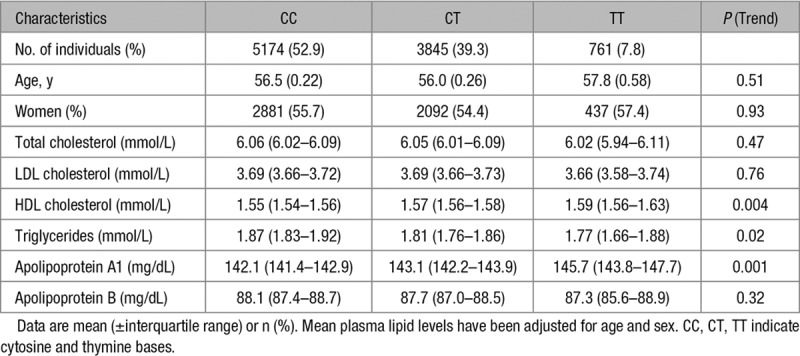
Genome-wide association studies have previously identified LRP1 as a locus that is associated with plasma triglycerides and HDL-C levels.5 Using the CCHS, we studied the association between the respective lead SNP (single nucleotide polymorphism; rs11613352) in 10 859 individuals and validated these associations with HDL-C (P=0.004) and triglyceride levels (P=0.02). We confirmed a per T-allele stepwise higher HDL-C (P for trend across genotypes=0.004) and a stepwise lower triglyceride level (P trend=0.02).
A step forward was taken by testing for association with plasma apolipoprotein levels: after adjustment for age and sex, we identified a highly significant association with apoA1 (P=0.001) but not with ApoB levels (P=0.32; Table 2). In agreement with lipid levels, plasma levels of apoA1 per T-allele were stepwise higher (P trend=0.001), whereas ApoB levels showed a weak, nonsignificant trend toward lower levels (P trend=0.32; Table 2).
Table 2.
Characteristics of Study Participants by LRP1 rs11613352 Genotype
Discussion
Despite the general concept that LRP1 affects triglyceride-rich lipoproteins, we provide evidence in humans that LRP1 may also directly affect HDL metabolism. This is shown through 2 very rare heterozygous loss-of-function variants in LRP1 identified in subjects with extremely low HDL-C who have no mutations in the canonical HDL genes. Our studies in various cell lines (including skin-derived fibroblasts and human hepatoma cells) indicate that these LRP1 variants cause lower cell membrane expression of ABCA1, which is in line with observations in liver-specific Lrp1 knockout mice.6 In addition, we observed higher levels of cell membrane SR-B1. Together, these findings may contribute to the extremely low HDL-C phenotype in carriers of these LRP1 variants. In support of our study, 1 of the 2 heterozygous loss of function variants (rs146923187) was also identified in the ExAC database (http://exac.broadinstitute.org/) and was found associated with a borderline reduction in plasma levels of HDL-C (P=0.1) in the Cardiovascular Disease Knowledge Portal despite the notion that it is exceedingly rare (http://broadcvdi.org). In addition, we validated earlier epidemiological evidence that common variation in LRP1 is associated with HDL-C5 but also show that this variant is associated with apoA1, but not with apoB levels in the CCHS. This finding could, however, not be replicated in publicly available data sets and awaits confirmation.
Consistent with a liver-specific Lrp1 knockout mouse that is characterized by low HDL-C levels, we showed that reduced LRP1 protein expression causes consistent downregulation of intracellular CTSD and upregulation of PSAP protein levels which resulted in impaired translocation of ABCA1 to the cell surface.6 Overexpression of the natural occurring human variants in LRP1 knockout cells failed to rescue the reduced ABCA1 protein content at the cell membrane. This low HDL-C phenotype is consistent with the notion that complete loss of ABCA1 function results in near absent plasma HDL-C in mice and men while heterozygotes for loss-of-function variants present with half normal HDL-C levels.28,29
A broad spectrum of unrelated extracellular ligands has been described that interact with LRP1 and may impact downstream physiological pathways.4 In this context, pro-CTSD, an aspartic protease, binds to the extracellular domain of the LRP1 β-chain.30 This enzyme has been implicated in several processes, such as intracellular digestion within lysosomal compartments, hormone and antigen processing,31,32 mitogenic activity in cancer,33,34 and apoptosis.35 CTSD may affect lipid metabolism by binding of ceramide to the prepro-CTSD resulting in activation of the enzymatically active isoforms36 and enabling PSAP conversion to saposin peptides involved in glycosphingolipids degradation, which in turn allows ABCA1 to be translocated to the cell surface where it mediates cholesterol efflux.37 Our study illustrates the complexity of ABCA1 expression at the cell membrane which was previously shown to be also modulated by syntaxin 13,38 ceramide,39 and cyclosporine A.40
Recently, El Asmar et al7 showed that WNT5a and ABCG1 levels are also controlled by LRP1 in mouse embryonic fibroblasts and HEK293. We found, however, no changes in WNT5a and ABCG1 protein in fibroblasts of the carrier compared with control subjects suggesting that this pathway might not be affected in our model.
In contrast, increased SR-B1 levels were consistently observed in our cellular models in which LRP1 function was attenuated or knocked down suggesting a potential involvement of this receptor in the low HDL-C phenotype of our study subjects. Overexpression of LRP1 in LRP1 knockout HEK293 cell line did not result in an opposite effect, which may be related to the overexpression conditions.
To date, little is known about the role of LRP1 in intracellular cholesterol homeostasis in humans, including the mechanisms involved in SR-B1 trafficking to the cell membrane. Our data suggest that the reduced HDL-C levels in our study subjects are the net result of attenuated LRP1-mediated CTSD uptake, which reduces the activation of saposins and subsequent ABCA1 translocation toward the plasma membrane (Figure 6) in combination with increased SR-B1 levels through a yet unidentified mechanism.
Figure 6.
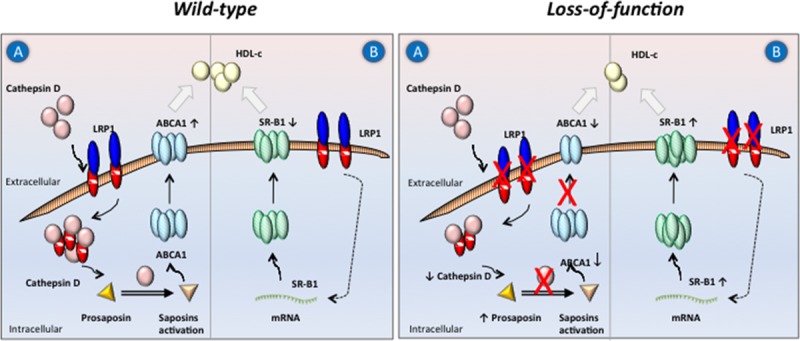
Proposed molecular mechanism(s) leading to decreased HDL-C (high-density lipoprotein cholesterol) as a result of LPR1 (low-density lipoprotein receptor–related protein 1) loss-of-function in humans. Two potential mechanisms affecting HDL-C levels are illustrated. In the presence of LRP1, cellular pro-CTSD (cathepsin D) recruitment leads to activating sphingolipid proteins SAP-A, -B, -C and -D (also called saposins). This allows ABCA1 (ATP-binding cassette A1) to be translocated from the late endosomes toward the plasma membrane (A). Another contributing player is SR-B1 (scavenger receptor class B type 1), which is known to mediate HDL-C uptake (B). Absence or reduction of LRP1 in humans affects downstream pathways reducing ABCA1 and increasing SR-B1 cell surface localization, respectively.
Both study subjects were heterozygotes for LRP1 variants. The use of antibodies in our Western Blotting and immune-fluorescence studies show that total LRP1 levels are significantly reduced in carriers compared with controls. These results, however, do not allow conclusions whether this is because of reductions in mutant or WT LRP1 or both. To study this, we used mass spectrometry to quantify WT and mutant LRP1 peptides in fibroblasts of one of the study subjects. Interestingly, both forms of LRP1 were present at similar levels indicating that the mutant does not have dominant negative effects (downregulation of the WT protein). The reductions of total LRP1 levels as observed with antibodies can, however, not be reconciled with these mass spectroscopy data. These discrepancies may be related to the specific LRP1 peptides that were chosen to quantify WT and mutant LRP1 and the epitope that is recognized by the monoclonal LRP1 antibody that was used. In addition, it is possible that the mass spectrometry analysis still detects the mutant LRP1 peptide, although the actual full protein is degraded because of lower stability. This may not be the case for the monoclonal LRP1 antibody.
Our study supports a direct role for LRP1 in HDL metabolism through CTSD, PSAP, ABCA1, and SR-B1. Additional studies are, however, required to pinpoint how changes in lipid species of the late endosomal compartment can result in the observed changes in the cellular trafficking and localization of proteins. Substantial support for this working model could come from the identification of additional natural functional variants in LRP1 in subjects with very low HDL-C levels. Such studies may benefit from a focus on those exons encoding for the domains involved in the uptake of CTSD (ie, exons 83–84).26 Further support may come from in vitro studies showing that decreased ABCA1 at the cell membrane because of LRP1 downregulation is associated with reduced cholesterol efflux to apoA1. In addition, downregulation of LRP1 in human hepatic cell lines may help to study the anticipated increased SR-B1–mediated uptake of cholesteryl ester from HDL. In vivo studies, however, are in our opinion much preferred to study the actual effects of LRP1 on plasma levels of HDL-C. Unfortunately, such studies in mice are hampered by species-specific effects that we observed although our findings support a role of SR-B1 in human cells, it has already been shown that a lack of hepatic LRP1 in mice does not affect SR-B1.6 To truly bolster our hypothesis, we, therefore, deem segregation studies in larger families with (additional) rare LRP1 defects to be pivotal.
In summary, we provide evidence that common and rare LRP1 gene variants are associated with effects on HDL metabolism. Our in vitro studies show that attenuated LRP1 function decreases ABCA1 and increases SR-B1 plasma membrane expression, which combined may explain reduced HDL-C levels in individuals with loss-of-function LRP1 variants.
Acknowledgments
F. Oldoni, B. van de Sluis, and J.A. Kuivenhoven designed the experiments; F. Oldoni performed the experiments; J.C. van Capelleveen contributed to the patient-derived fibroblasts study; J.C. Wolters performed and analyzed proteomics data; K.G. Hovingh and G.M. Dallinga-Thie provided clinical data; N. Dalila, R. Frikke-Schmidt, and A. Tybjærg-Hansen provided epidemiological data; and F. Oldoni and J.A. Kuivenhoven wrote the article with input from B. van de Sluis, K.G. Hovingh, G.M. Dallinga-Thie, and D.Y. Hui. All authors critically read the article.
We thank all participants for taking part in the study and Nicolette Huijkman and Alinda Schimmel for technical assistance.
Sources of Funding
J.A. Kuivenhoven and K.G. Hovingh are supported through funding from the Netherlands CardioVascular Research Initiative (CVON2011-2016, Acronym Genius; CVON2017-2020, Acronym Genius2), the European Union (FP7-603091; Acronym TransCard). J.A. Kuivenhoven is an established investigator of the Netherlands Heart Foundation (2015T068). K.G. Hovingh is holder of a VIDI grant (016.156.445) from the Netherlands Organisation for Scientific Research (NWO). D.Y. Hui was supported by research grant RO1 DK074932 from the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases.
Disclosures
K.G. Hovingh or his institution received honoraria for consultancy, advisory boards and conduct of clinical trials from Amgen, Aegerion, Pfizer, Astra Zeneca, Sanofi, Regeneron, KOWA, IOnis, and Cerenis. The other authors report no conflicts.
Nonstandard Abbreviations and Acronyms
- ABCA1
- ATP-binding cassette transporter A1
- ABCG1
- ATP-binding cassette transporter G1
- APOA1
- apolipoprotein A1
- CCHS
- Copenhagen City Heart Study
- CTSD
- cathepsin D
- GFP
- green fluorescence protein
- HDL
- high-density lipoprotein
- LRP1/Lrp1
- low-density lipoprotein receptor–related protein 1
- NCEH
- neutral cholesterol ester hydrolase
- PSAP
- prosaposin
- SR-B1
- scavenger receptor class B type 1
- TGFβ
- transforming growth factor β
- TRL
- triglyceride-rich lipoproteins
- WNT5a
- Wnt family member 5A
- WT
- wild type
These authors contributed equally to the article.
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.117.310309/-/DC1.
Highlights
The LRP1 (low-density lipoprotein receptor–related protein 1) affects the clearance of triglyceride-rich lipoproteins, but its role in cholesterol homeostasis in humans is not well understood.
We provide the first evidence in humans that variants in LRP1 have a large functional impact on lipid metabolism by virtue of their effect on 2 main players in HDL (high-density lipoprotein) metabolism, that is, ABCA1 (ATP-binding cassette A1) and SR-B1 (scavenger receptor B1 class B type 1).
We report that the lead LRP1 SNP (single nucleotide polymorphism) locus is associated with HDL cholesterol/apo (apolipoprotein) A1 levels in the CCHS (Copenhagen City Heart Study).
References
- 1.Herz J, Hamann U, Rogne S, Myklebost O, Gausepohl H, Stanley KK. Surface location and high affinity for calcium of a 500-kd liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. EMBO J. 1988;7:4119–4127. doi: 10.1002/j.1460-2075.1988.tb03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dieckmann M, Dietrich MF, Herz J. Lipoprotein receptors–an evolutionarily ancient multifunctional receptor family. Biol Chem. 2010;391:1341–1363. doi: 10.1515/BC.2010.129. doi: 10.1515/BC.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–784. doi: 10.1172/JCI13992. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willer CJ, Schmidt EM, Sengupta S, et al. Global Lipids Genetics Consortium. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basford JE, Wancata L, Hofmann SM, Silva RA, Davidson WS, Howles PN, Hui DY. Hepatic deficiency of low density lipoprotein receptor-related protein-1 reduces high density lipoprotein secretion and plasma levels in mice. J Biol Chem. 2011;286:13079–13087. doi: 10.1074/jbc.M111.229369. doi: 10.1074/jbc.M111.229369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El Asmar Z, Terrand J, Jenty M, et al. Convergent signaling pathways controlled by LRP1 (receptor-related protein 1) cytoplasmic and extracellular domains limit cellular cholesterol accumulation. J Biol Chem. 2016;291:5116–5127. doi: 10.1074/jbc.M116.714485. doi: 10.1074/jbc.M116.714485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singaraja RR, Kang MH, Vaid K, Sanders SS, Vilas GL, Arstikaitis P, Coutinho J, Drisdel RC, El-Husseini Ael D, Green WN, Berthiaume L, Hayden MR. Palmitoylation of ATP-binding cassette transporter A1 is essential for its trafficking and function. Circ Res. 2009;105:138–147. doi: 10.1161/CIRCRESAHA.108.193011. doi: 10.1161/CIRCRESAHA.108.193011. [DOI] [PubMed] [Google Scholar]
- 9.Hiesberger T, Hüttler S, Rohlmann A, Schneider W, Sandhoff K, Herz J. Cellular uptake of saposin (SAP) precursor and lysosomal delivery by the low density lipoprotein receptor-related protein (LRP). EMBO J. 1998;17:4617–4625. doi: 10.1093/emboj/17.16.4617. doi: 10.1093/emboj/17.16.4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou L, Choi HY, Li WP, Xu F, Herz J. LRP1 controls cPLA2 phosphorylation, ABCA1 expression and cellular cholesterol export. PLoS One. 2009;4:e6853. doi: 10.1371/journal.pone.0006853. doi: 10.1371/journal.pone.0006853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao H, Lockyer P, Li L, Ballantyne CM, Patterson C, Xie L, Pi X. Endothelial LRP1 regulates metabolic responses by acting as a co-activator of PPARγ. Nat Commun. 2017;8:14960. doi: 10.1038/ncomms14960. doi: 10.1038/ncomms14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohlmann A, Gotthardt M, Hammer RE, Herz J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. 1998;101:689–695. doi: 10.1172/JCI1240. doi: 10.1172/JCI1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willnow TE, Armstrong SA, Hammer RE, Herz J. Functional expression of low density lipoprotein receptor-related protein is controlled by receptor-associated protein in vivo. Proc Natl Acad Sci USA. 1995;92:4537–4541. doi: 10.1073/pnas.92.10.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Espirito Santo SM, Pires NM, Boesten LS, Gerritsen G, Bovenschen N, van Dijk KW, Jukema JW, Princen HM, Bensadoun A, Li WP, Herz J, Havekes LM, van Vlijmen BJ. Hepatic low-density lipoprotein receptor-related protein deficiency in mice increases atherosclerosis independent of plasma cholesterol. Blood. 2004;103:3777–3782. doi: 10.1182/blood-2003-11-4051. doi: 10.1182/blood-2003-11-4051. [DOI] [PubMed] [Google Scholar]
- 15.Yu KC, Chen W, Cooper AD. LDL receptor-related protein mediates cell-surface clustering and hepatic sequestration of chylomicron remnants in LDLR-deficient mice. J Clin Invest. 2001;107:1387–1394. doi: 10.1172/JCI11750. doi: 10.1172/JCI11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laatsch A, Panteli M, Sornsakrin M, Hoffzimmer B, Grewal T, Heeren J. Low density lipoprotein receptor-related protein 1 dependent endosomal trapping and recycling of apolipoprotein E. PLoS One. 2012;7:e29385. doi: 10.1371/journal.pone.0029385. doi: 10.1371/journal.pone.0029385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webb TR, Erdmann J, Stirrups KE, et al. Wellcome Trust Case Control Consortium; MORGAM Investigators; Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69:823–836. doi: 10.1016/j.jacc.2016.11.056. doi: 10.1016/j.jacc.2016.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Motazacker MM, Peter J, Treskes M, Shoulders CC, Kuivenhoven JA, Hovingh GK. Evidence of a polygenic origin of extreme high-density lipoprotein cholesterol levels. Arterioscler Thromb Vasc Biol. 2013;33:1521–1528. doi: 10.1161/ATVBAHA.113.301505. doi: 10.1161/ATVBAHA.113.301505. [DOI] [PubMed] [Google Scholar]
- 19.Lin YC, Lin CH, Kuo CY, Yang VC. ABCA1 modulates the oligomerization and Golgi exit of caveolin-1 during HDL-mediated cholesterol efflux in aortic endothelial cells. Biochem Biophys Res Commun. 2009;382:189–195. doi: 10.1016/j.bbrc.2009.03.005. doi: 10.1016/j.bbrc.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 20.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holleboom AG, Kuivenhoven JA, Peelman F, Schimmel AW, Peter J, Defesche JC, Kastelein JJ, Hovingh GK, Stroes ES, Motazacker MM. High prevalence of mutations in LCAT in patients with low HDL cholesterol levels in The Netherlands: identification and characterization of eight novel mutations. Hum Mutat. 2011;32:1290–1298. doi: 10.1002/humu.21578. doi: 10.1002/humu.21578. [DOI] [PubMed] [Google Scholar]
- 23.Candini C, Schimmel AW, Peter J, Bochem AE, Holleboom AG, Vergeer M, Dullaart RP, Dallinga-Thie GM, Hovingh GK, Khoo KL, Fasano T, Bocchi L, Calandra S, Kuivenhoven JA, Motazacker MM. Identification and characterization of novel loss of function mutations in ATP-binding cassette transporter A1 in patients with low plasma high-density lipoprotein cholesterol. Atherosclerosis. 2010;213:492–498. doi: 10.1016/j.atherosclerosis.2010.08.062. doi: 10.1016/j.atherosclerosis.2010.08.062. [DOI] [PubMed] [Google Scholar]
- 24.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cal R, García-Arguinzonis M, Revuelta-López E, Castellano J, Padró T, Badimon L, Llorente-Cortés V. Aggregated low-density lipoprotein induces LRP1 stabilization through E3 ubiquitin ligase CHFR downregulation in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2013;33:369–377. doi: 10.1161/ATVBAHA.112.300748. doi: 10.1161/ATVBAHA.112.300748. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Koulov AV, Kellner WA, Riordan JR, Balch WE. Chemical and biological folding contribute to temperature-sensitive DeltaF508 CFTR trafficking. Traffic. 2008;9:1878–1893. doi: 10.1111/j.1600-0854.2008.00806.x. doi: 10.1111/j.1600-0854.2008.00806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bodzioch M, Orsó E, Klucken J, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 29.Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denèfle P, Assmann G. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 30.Beaujouin M, Prébois C, Derocq D, et al. Pro-cathepsin D interacts with the extracellular domain of the beta chain of LRP1 and promotes LRP1-dependent fibroblast outgrowth. J Cell Sci. 2010;123(pt 19):3336–3346. doi: 10.1242/jcs.070938. doi: 10.1242/jcs.070938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liaudet-Coopman E, Beaujouin M, Derocq D, Garcia M, Glondu-Lassis M, Laurent-Matha V, Prébois C, Rochefort H, Vignon F. Cathepsin D: newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Cancer Lett. 2006;237:167–179. doi: 10.1016/j.canlet.2005.06.007. doi: 10.1016/j.canlet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Moss CX, Villadangos JA, Watts C. Destructive potential of the aspartyl protease cathepsin D in MHC class II-restricted antigen processing. Eur J Immunol. 2005;35:3442–3451. doi: 10.1002/eji.200535320. doi: 10.1002/eji.200535320. [DOI] [PubMed] [Google Scholar]
- 33.Tsukamoto T, Iida J, Dobashi Y, Furukawa T, Konishi F. Overexpression in colorectal carcinoma of two lysosomal enzymes, CLN2 and CLN1, involved in neuronal ceroid lipofuscinosis. Cancer. 2006;106:1489–1497. doi: 10.1002/cncr.21764. doi: 10.1002/cncr.21764. [DOI] [PubMed] [Google Scholar]
- 34.Sebzda T, Saleh Y, Gburek J, Andrzejak R, Gnus J, Siewinski M, Grzebieniak Z. Cathepsin D expression in human colorectal cancer: relationship with tumour type and tissue differentiation grade. J Exp Ther Oncol. 2005;5:145–150. [PubMed] [Google Scholar]
- 35.Démoz M, Castino R, Cesaro P, Baccino FM, Bonelli G, Isidoro C. Endosomal-lysosomal proteolysis mediates death signalling by TNFalpha, not by etoposide, in L929 fibrosarcoma cells: evidence for an active role of cathepsin D. Biol Chem. 2002;383:1237–1248. doi: 10.1515/BC.2002.137. doi: 10.1515/BC.2002.137. [DOI] [PubMed] [Google Scholar]
- 36.Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, Weber T, Saftig P, Peters C, Brunner J, Krönke M, Schütze S. Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J. 1999;18:5252–5263. doi: 10.1093/emboj/18.19.5252. doi: 10.1093/emboj/18.19.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haidar B, Kiss RS, Sarov-Blat L, Brunet R, Harder C, McPherson R, Marcel YL. Cathepsin D, a lysosomal protease, regulates ABCA1-mediated lipid efflux. J Biol Chem. 2006;281:39971–39981. doi: 10.1074/jbc.M605095200. doi: 10.1074/jbc.M605095200. [DOI] [PubMed] [Google Scholar]
- 38.Bared SM, Buechler C, Boettcher A, Dayoub R, Sigruener A, Grandl M, Rudolph C, Dada A, Schmitz G. Association of ABCA1 with syntaxin 13 and flotillin-1 and enhanced phagocytosis in tangier cells. Mol Biol Cell. 2004;15:5399–5407. doi: 10.1091/mbc.E04-03-0182. doi: 10.1091/mbc.E04-03-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witting SR, Maiorano JN, Davidson WS. Ceramide enhances cholesterol efflux to apolipoprotein A-I by increasing the cell surface presence of ATP-binding cassette transporter A1. J Biol Chem. 2003;278:40121–40127. doi: 10.1074/jbc.M305193200. doi: 10.1074/jbc.M305193200. [DOI] [PubMed] [Google Scholar]
- 40.Le Goff W, Peng DQ, Settle M, Brubaker G, Morton RE, Smith JD. Cyclosporin A traps ABCA1 at the plasma membrane and inhibits ABCA1-mediated lipid efflux to apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2004;24:2155–2161. doi: 10.1161/01.ATV.0000144811.94581.52. doi: 10.1161/01.ATV.0000144811.94581.52. [DOI] [PubMed] [Google Scholar]