

Supplemental Digital Content is available in the text
Keywords: metastasis, osteosarcoma, weighted gene co-expression network analysis
Abstract
Osteosarcoma (OS), the most common malignant bone tumor, accounts for the heavy healthy threat in the period of children and adolescents. OS occurrence usually correlates with early metastasis and high death rate. This study aimed to better understand the mechanism of OS metastasis.
Based on Gene Expression Omnibus (GEO) database, we downloaded 4 expression profile data sets associated with OS metastasis, and selected differential expressed genes. Weighted gene co-expression network analysis (WGCNA) approach allowed us to investigate the most OS metastasis-correlated module. Gene Ontology functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were used to give annotation of selected OS metastasis-associated genes.
We select 897 differential expressed genes from OS metastasis and OS non-metastasis groups. Based on these selected genes, WGCNA further explored 142 genes included in the most OS metastasis-correlated module. Gene Ontology functional and KEGG pathway enrichment analyses showed that significantly OS metastasis-associated genes were involved in pathway correlated with insulin-like growth factor binding.
Our research figured out several potential molecules participating in metastasis process and factors acting as biomarker. With this study, we could better explore the mechanism of OS metastasis and further discover more therapy targets.
1. Introduction
Osteosarcoma (OS) is the most common primary malignant bone tumor in childhood and adolescence. OS exhibits highly aggressive and early systemic metastasis.[1,2] About 20% of OS patients appear focus metastasis when they receive the first diagnosis. With the great development of effective chemotherapy combinations, the survival rates of nonmetastatic OS have significantly increased from <20% before 1970s to present rates of 65% to 75%.[3,4] However, OS systemic metastasis, especially the occurrence of lung metastasis is still the most prominent reason for OS-caused death. Only 11% to 30% of patients suffering from OS metastasis can survive after the combination of surgery resection and chemotherapy.[5,6] In this way, OS metastasis has become the obstacle for successful OS treatments. OS metastasis is a multistep dynamic progression facilitated by pro-metastasis genes such as myc,[7]ras,[8] and inhibited by metastasis-resistant genes including nm23,[9]p16,[10] kangai1 (KAI-1),[11] KiSS-1 metastasis-suppressor (KISS-1),[12] mitogen-activated protein kinase kinase 4 (MKK4). Although the biological characteristic of OS has been understood a lot, a large scale of regions in molecular mechanisms involved in OS metastasis are still uncovered. Therefore, in order to profoundly improve early diagnostic efficacy and clinical outcomes for patients with OS, it is not only urgent to find out the metastasis mechanism but also to identify molecular that can be used as candidate diagnostic biomarkers or therapeutic targets in OS.
Bioinformatic approaches are increasingly being used in target genes or proteins exploration and analysis. Weighted gene co-expression network analysis (WGCNA) is a novel gene co-expression network-based approach. WGCNA is a systems biology method for molecular interaction mechanism analysis and correlation networks resolving.[13] In accordance with high throughput microarray data, WGCNA can be used to seek synergetic expressed modules and explore the relationship between gene networks and clinical phenotype at transcriptome level.[13–15] As an effective and accurate bioinformatics method, WGCNA has been widely applied in identifying susceptibility genes, screening candidate targets for disease and cancer fields.[16,17] In 2017, Liu et al[18] predict gene clusters correlated with the mechanisms of OS using WGCNA, finding that the genes in module 5 are involved in OS. However, the pathogenesis of OS has not been fully revealed.
Previous studies offered a series of Gene Expression Omnibus (GEO) datasets presenting OS related genes expression profile. These analyses comprise bioinformatics data derived from tissue sample of OS patients. In this study, we selected some of these dataset with specific limitation for our bioinformatics analysis.[19–21] We conducted WGCNA and the following support vector machines (SVM) to analyze OS metastasis related genes and identified key genes potentially participated in OS metastasis progression. With the usage of effective bioinformatic analysis, we can figure out specific key genes that have important clinical implications in disease basic researches and clinical therapy.
2. Methods
2.1. Data collection and preprocessing
Four expression profile data sets of OS metastasis were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/gds). Datasets GSE14359, GSE21257, GSE32981, and GSE14827 were chosen for further study in accordance with the following rules: datasets belong to OS gene expression profiles; datasets contain OS metastasis and nonmetastasis classification information. GSE14359 dataset was used for OS significantly correlated modules and genes selection, and SVM establishment. The other three datasets were used for independent verification of SVM. Detail information of datasets was shown in Table 1. GSE14359 and GSE14827 data sets in GEL format were annotated according to the Affymetrix Human Genome U133A and U133 Plus 2.0 Array platforms respectively. The background correction and normalization of these raw data sets were performed by the Affy[22] package in R. GSE32981 was derived from ABI Human Genome Survey Microarray v2.0 platform. GSE21257 was obtained from Illumina[23] human-6 v2.0 expression beadchip (using nuIDs as identifier). GSE32981 and GSE21257 datasets were downloaded in original txt format.
Table 1.
Datasets of gene expression profiles.

Probes were mapped to gene symbols. Empty probes were discarded according to the annotation platform of each expression profiles. If there were multiple probes that mapped to the same gene symbol, their mean value was considered as the gene expression value. Data were normalized by the limma (linear models for microarray data) package in R.
2.2. Screening for differentially expressed genes
The differentially expressed genes (DEGs) between nonmetastasis and metastasis group in GSE14359 were screened by the limma package in R. The adjusted P-value (false discovery rate, FDR) <.05 and |logFC| > 1 were set as the cut-off criteria.
2.3. Screening for OS associated genes and modules by WGCNA
Module identification was accomplished with the dynamic tree cut method. Highly similar modules were identified by cluster analysis and then merged together with a height cut-off of 0.95. For further quantification of OS related genes and modules, the p-value of gene expression difference between normal group and disease group were evaluated with T-test, and log P was considered as gene significance. The mean value of gene significance (GS) derived from modules comprising gene was defined as module significance.
2.4. Establishment of SVM
GSE14539 was set as training dataset. The genes feature combination was obtained through recursive feature elimination (RFE) arithmetic. SVM was established with the selected optimized gene feature combination. To test the stability and transferability of SVM, the generated SVM was verified by using independent test datasets: GSE14827, GSE21257, and GSE32981. The classification effect was determined according to sensitivity (Se), specificity (Sp), positive predictive value (PPV), negative predictive value (NPV), and proportion under Receiver operating characteristic curve (ROC) curve.
3. Results
3.1. Datasets preprocessing and DEGs selection
In order to select OS metastasis associated genes, datasets about OS associated gene expression profiles were downloaded from the GEO Database (see Methods and Fig. 1 for details on the experimental design).
Figure 1.
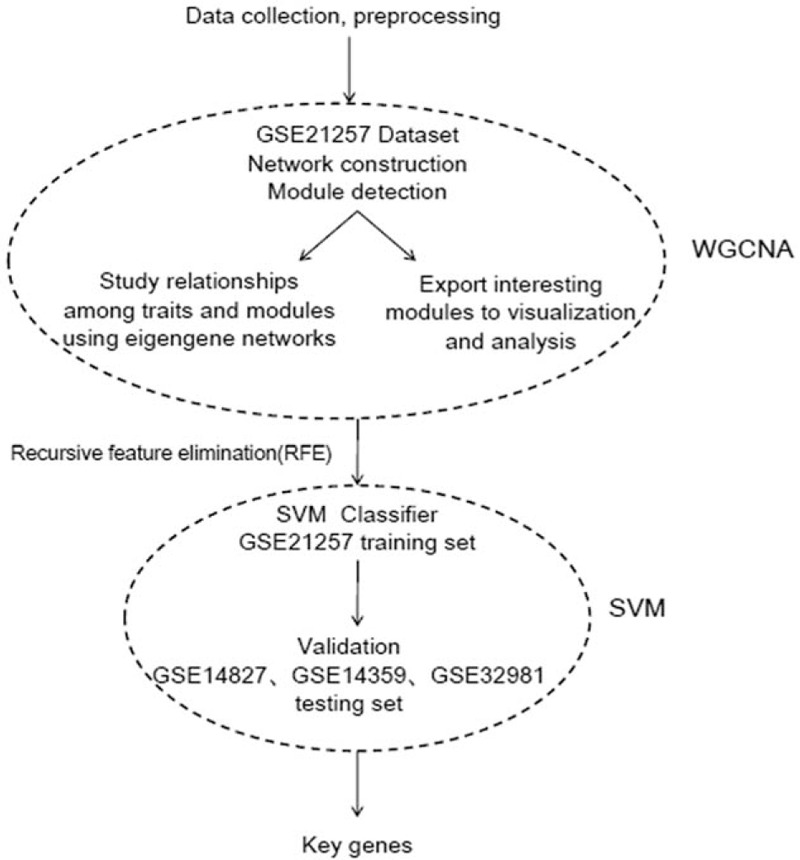
The workflow of data preparation, processing, analysis, and validation in this study.
Under the thresholds of |log FC| > 1 and FDR < 0.05, total 897 DEGs were obtained from GSE14359 dataset. Bidirectional hierarchical clustering was carried out according to the genes expression level and the results were presented by the heat map exhibiting genes mRNA expression level (Fig. 2).
Figure 2.
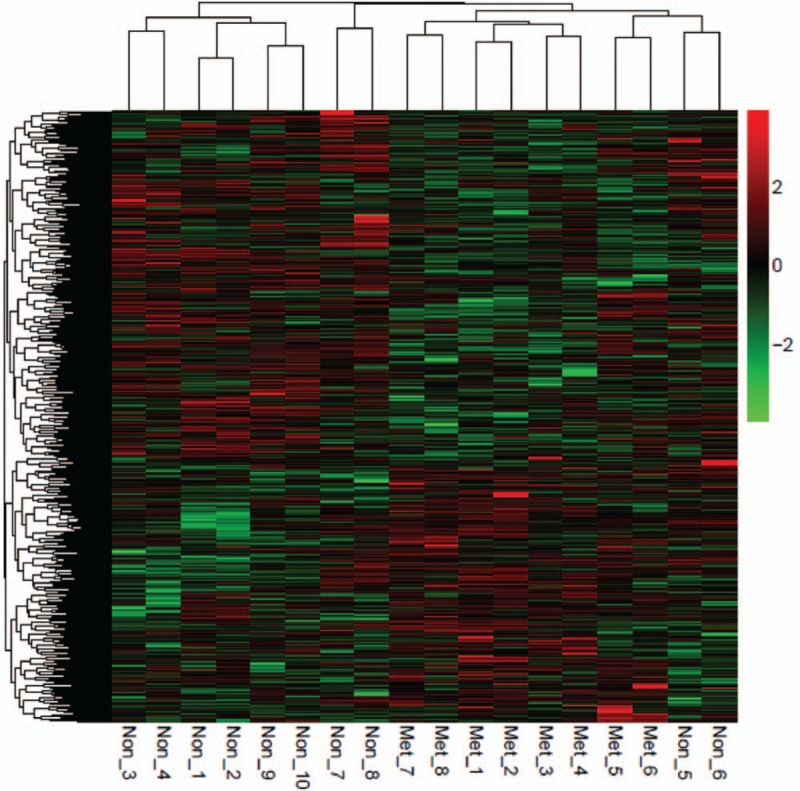
Heat map of the difference of genes mRNA expression in GSE14359. Red color exhibits upregulated genes while the green means the downregulated genes. The correlation between color and the fold change of genes mRNA level is displayed in the upper right.
3.2. Dataset network construction and modules detection
In order to identify OS-metastasis associated modules and genes, WGCNA was performed on the obtained 897 DEGs. Firstly, we explored the value of adjacency matrix weighting, power. As shown in Figure 3, X-axis represents matrix weighting power while the Y-axis represents quadratic correlation index derived from log (k) and log (P(k)) of the corresponding network. From Figure 3, we took power as 12 when correlation index reached 0.9 the first time.
Figure 3.
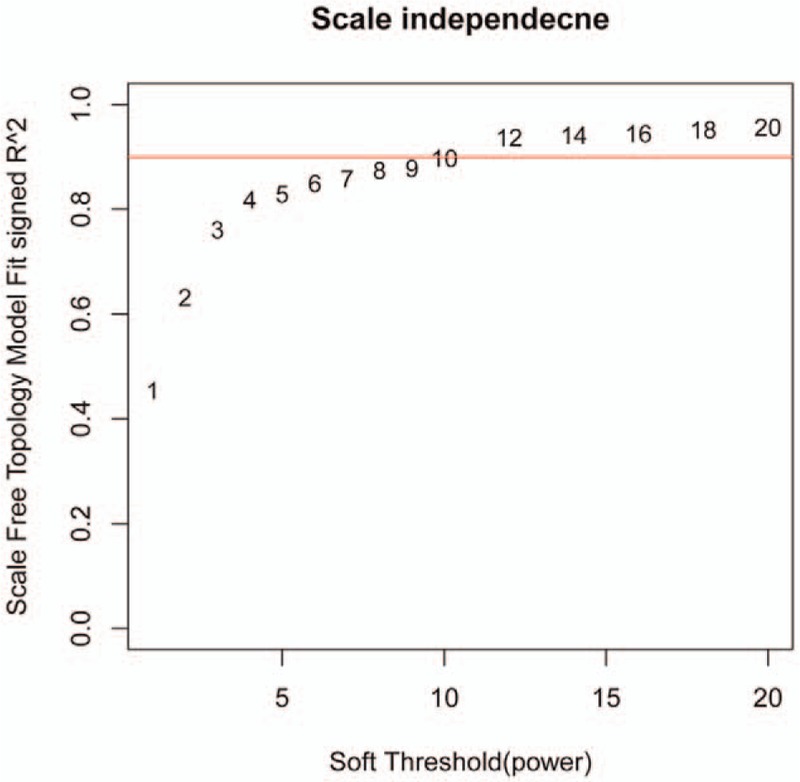
Calculation chart of adjacency matrix weighting parameters (power). The X-axis represents weighting parameters (power). The Y-axis represents quadratic of correlation index from log (k) and log (P(K)).
In the next step, we calculated community dissimilarity index of DEGs and obtained system clustering tree. According to the standard of dynamic cut tree, we set 30 as the least gene number of each gene network and 0.95 as the cut-height, respectively. After determination of gene modules by using dynamic tree cut method, we computed the eigenvector value of each module. Based on cluster analysis of modules, the closer ones were merged together for new modules’ acquisition. Modules partition was shown in Figure 4, we totally obtained 10 osteosarcoma-metastasis associated modules.
Figure 4.
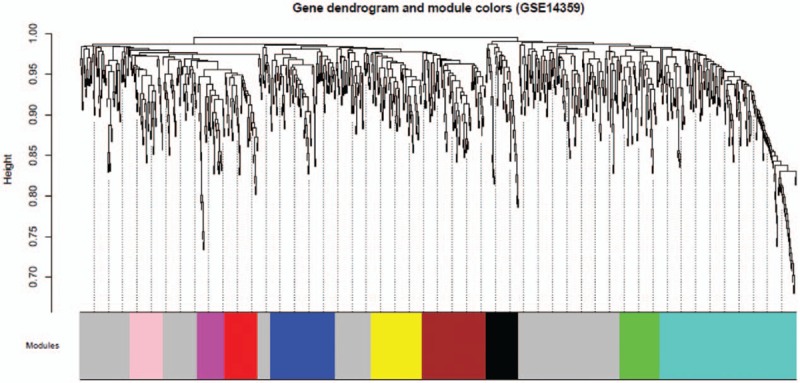
Clustering dendrograms of GSE14359. System clustering tree was built based on GSE14359 dataset. Ten kinds of color present 10 modules.
In further, we qualified the relevance between eigenvalue of network modules and osteosarcoma-metastasis condition (metastasis and nonmetastasis). As shown in Figure 5, gene significance of each modules were over 0.8 while modules significance P-value was only .0072, far lower than 0.05, indicating significant difference among modules. The specific DEGs in obtained 10 modules were recorded in Table S1. The turquoise pattern contained the largest number of DEGs among these modules. So we took DEGs in this module for further study.
Figure 5.
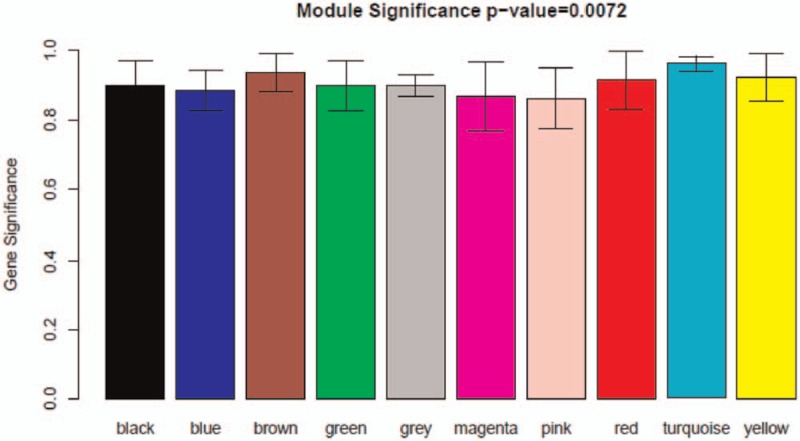
Diagram of correlation of module's color and osteosarcoma. The relevance between eigenvalue of network modules and osteosarcoma-metastasis condition was qualified. The colored row indicates modules and the Y-axis represents gene significance.
3.3. Generation and annotation of OS associated module network
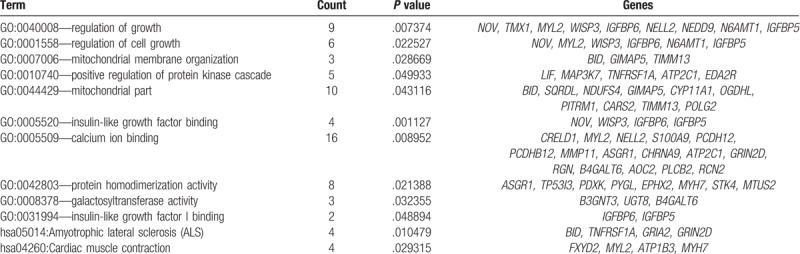
We established genes expression network by using OS associated gene expression value from turquoise panel. As shown in Figure 6, the network contains 231 connection sides and 142 nodes comprising 59 downregulated genes and 83 upregulated genes. All these 142 genes were subjected to Gene Ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses and the annotations were presented in Figure 7 and Table 2. As shown in Table 2, nephroblastoma overexpressed (NOV), insulin-like growth factor binding protein 5 (IGFBP5), insulin-like growth factor binding protein 6 (IGFBP6), WNT1 inducible signaling pathway protein 3 (WISP3) and myosin light chain 2 (MYL2) were found in both cell growth regulation and insulin-like growth factor binding groups. These data indicated that these genes and associated functional pathway may contribute to the progression of tumor cell migration.
Figure 6.
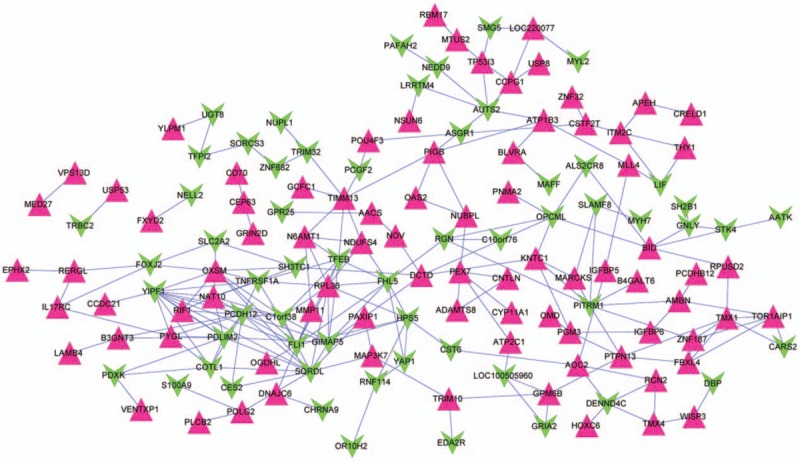
Gene expression network derived from turquoise module. Equilateral triangle and inverted triangle represent downregulation and upregulation, respectively. The red point meant significantly upregulated expression genes while the green point meant down-regulated expression genes.
Figure 7.
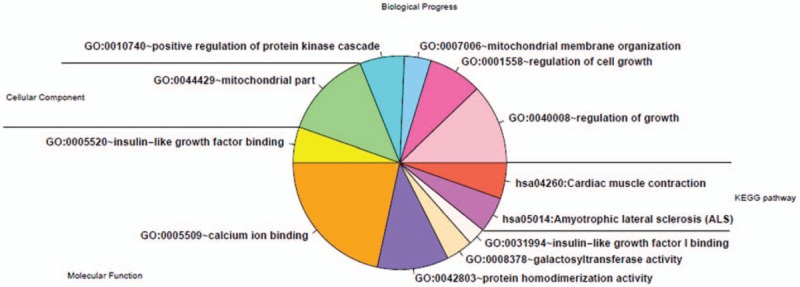
Annotation of KEGG pathway enrichment analysis and gene ontology function of differentially expressed genes. KEGG = Kyoto Encyclopedia of Genes and Genomes.
Table 2.
Annotation of KEGG pathway enrichment analysis and gene ontology function of differentially expressed genes list.
3.4. Optimal characterized genes selection
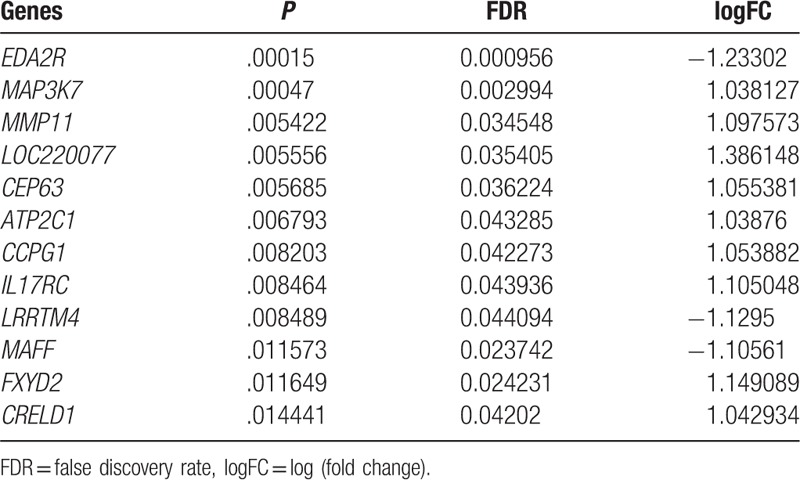
To confirm typical characterized genes involved in these 142 genes, we chose GSE14359 as training dataset and conducted feature selection with recursive feature elimination (RFE). If there were 12 genes combination, 17 samples were accurately classified with accuracy rate of 94.4% (Fig. 8). All these 12 genes, especial tumor-associated gene matrix metalloproteinase11 (MMP11), FXYD domain containing ion transport regulator 2 (FXYD2) were presented in Table 3.
Figure 8.
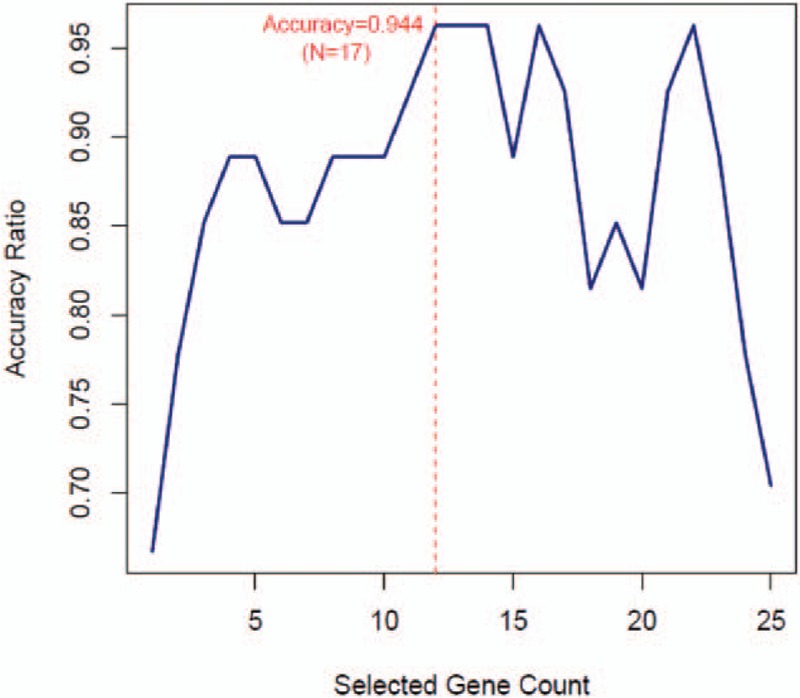
The optimal characterized genes combination selection by recursive feature elimination. The X-axis presents the number of selected characters. The Y-axis presents prediction accuracy of characters set.
Table 3.
The optimal characterized genes combination list.
3.5. Verification and assessment of SVM
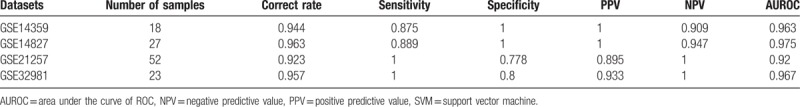
GSE14827, GSE21257, and GSE32981 datasets were utilized to verify the SVM generated by the selected optimized gene feature combination. In GSE14827, SVM can classify 26 samples with accuracy rate of 96.3%. In GSE32981 and GSE21257, the classified samples were 22 and 48 with accuracy rate of 95.7% and 92.3%, respectively. The sample recognition and classification effect was synthetically determined according to Se, Sp, PPV, NPV, and areas under ROC curve (Fig. 9). The analysis results of SVM evaluation index were shown in Table 4.
Figure 9.
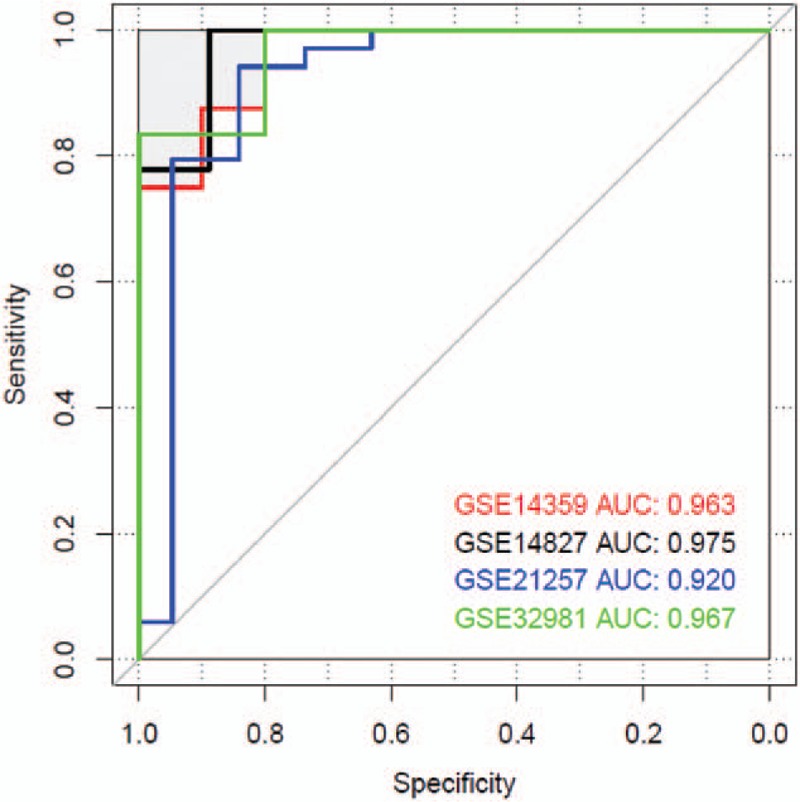
Area under the curve of ROC diagram of support vector machine.
Table 4.
List of support vector machines (SVM) evaluation index in training datasets and test datasets.
4. Discussion
OS is the most common primary malignant bone tumor, which especially displays highly aggressive and early systemic metastasis in people at young age.[1,2] More knowledge about the mechanism of OS metastasis is required for OS early diagnosis and treatment. In this study, we identified 897 DEGs. With the usage of WGCNA approach, 142 genes highly related with OS metastasis were obtained. These genes were enriched in cell growth regulation and insulin-like growth factor binding associated GO functions and KEGG pathways. Finally, considered GSE14359 as training dataset and conducted following RFE, we obtained 12 optimal characterized genes including FXYD2 and MMP11. SVM was further verified with the usage of the other 3 GSE14827, GSE21257, and GSE32981 datasets.
In this study, the selected 142 DEGs had been classified into several classical pathways (Table 2). Insulin-like growth factor binding proteins (IGFBPs) members can both positively or negatively regulate IGFs function.[24,25] Among these IGFBPs members, IGFBP6 expression level was increased in OS metastasis group (Fig. 6). IGFBP-6 was able to inhibit actions of IGF-II, including IGF-II induced cell proliferation and differentiation by typical IGF-dependent pathway.[26] However, recently studies showed that IGFP6 can also promote cancer cell migration through IGF-independent manner, which may be transduced by mitogen-activated protein kinase (MAPK).[27]WISP3, also named as CCN6, was another increased genes involved in insulin-like growth factor binding. WISP3 was regarded as one potential anti-cancer therapy target because it elicited OS cells metastasis by suppressing activation of TAK1 and p38.[28] To our surprise, IGFBP5, one protein that inhibit tumorigenicity and metastasis of human osteosarcoma, also increased significantly in OS metastasis panel,[29] which indicated the complexity of metastasis process.
In the 12 optimal characterized genes, MMP11 has been detected in many invasive human carcinomas. In oral squamous cell carcinoma, MMP11 expression level was associated with lymph node metastasis. Overexpression of MMP11 in oral squamous cell carcinoma cells also promoted cell migration in vitro.[30] In addition, another report indicated the potential role of MMP11 existing in plasma in disease progression assessment, which suggested MMP11 can be used as biomarker for diagnosis.[31] FXYD domain Containing Ion Transport Regulator 2 (FXYD2) was the modulating subunit of Na+/K+-ATPases. In OCCC tissues, FXYD2 significantly expressed. Deficiency of FXYD2 led to tumor growth suppression, which indicated that FXYD2 had the possibility to be antitumor target.[32]
Although we identified DEGs between OS metastasis and non-metastasis through bioinformatics methods, we did not conduct experimental test for any of these selected genes, which was a limitation of this study. Functional analysis is necessary to further study the functions of these genes in OS metastasis progress regulation.
In conclusion, IGFBP5, IGFBP6, WISP3, and MYL2 were involved in Insulin-like growth factor binding. This correlation gave us hypothesis that insulin-like growth factor binding associated genes may play important role in deciding OS metastasis progression. More basic functional experiments need to be conducted to investigate these involved genes function to explore more anti-tumor and metastasis inhibition targets.
Author contributions
Conceptualization: Honglai Tian, Donghui Guan, Jianmin Li.
Data curation: Honglai Tian, Donghui Guan.
Formal analysis: Jianmin Li.
Writing – original draft: Honglai Tian, Donghui Guan.
Writing – review & editing: Honglai Tian, Jianmin Li.
Supplementary Material
Footnotes
Abbreviations: DEGs = differentially expressed genes, FDR = false discovery rate, GEO = Gene Expression Omnibus, GO = gene ontology, GS = gene significance, IGFBP5 = insulin-like growth factor binding protein 5, IGFBP6 = insulin-like growth factor binding protein 6, IGFBPs = insulin-like growth factor binding proteins, KAI-1 = kangai1, KEGG = Kyoto Encyclopedia of Genes and Genomes, MAPK = mitogen-activated protein kinase, MKK4 = mitogen-activated protein kinase kinase 4, MMP11 = matrix metalloproteinase11, MYL2 = myosin light chain 2, NOV = nephroblastoma overexpressed, NPV = negative predictive value, OS = osteosarcoma, PPV = positive predictive value, RFE = recursive feature elimination, ROC = receiver operating characteristic curve, Se = sensitivity, Sp = specificity, SVM = support vector machines, WISP3 = WNT1 inducible signaling pathway protein 3.
HT and DG should be regarded as co-first authors.
Ethics committee: This is a meta-analysis manuscript, there is no ethics committee or institutional review board approved by the study.
Competing interests: The authors declare that they have no competing interests.
Supplemental Digital Content is available for this article.
References
- [1].Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer 2009;125:229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].PJ M, RM G, FW A-K, et al. Osteosarcoma. J Am Acad Orthop Surgeons 2009;17:515. [DOI] [PubMed] [Google Scholar]
- [3].Eilber F, Giuliano A, Eckardt J, et al. Adjuvant chemotherapy for osteosarcoma: a randomized prospective trial. J Clin Oncol 1987;5:21–6. [DOI] [PubMed] [Google Scholar]
- [4].Allison DC, Carney SC, Ahlmann ER, et al. A meta-analysis of osteosarcoma outcomes in the modern medical era. Sarcoma 2012;2012:704872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Meyers PA, Heller G, Healey JH, et al. Osteogenic sarcoma with clinically detectable metastasis at initial presentation. J Clin Oncol 1993;11:449–53. [DOI] [PubMed] [Google Scholar]
- [6].Chou A, Merola P, Lh, et al. Treatment of osteosarcoma at first recurrence after contemporary therapy: the Memorial Sloan-Kettering Cancer Center experience. Cancer 2005;104:2214–21. [DOI] [PubMed] [Google Scholar]
- [7].Han G, Wang Y, Bi W. C-Myc overexpression promotes osteosarcoma cell invasion via activation of MEK-ERK pathway. Oncol Res 2012;20:149–56. [DOI] [PubMed] [Google Scholar]
- [8].Muff R, Ram Kumar RM, Botter SM, et al. Genes regulated in metastatic osteosarcoma: evaluation by microarray analysis in four human and two mouse cell line systems. Sarcoma 2012;2012:937506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Steeg PS, Bevilacqua G, Kopper L, et al. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst 1988;80:200–4. [DOI] [PubMed] [Google Scholar]
- [10].Silva G. Aboussekhra A. p16 (INK4A) inhibits the pro-metastatic potentials of osteosarcoma cells through targeting the ERK pathway and TGF-beta1. Mol Carcinog 2016;55:525–36. [DOI] [PubMed] [Google Scholar]
- [11].Dong JT, Lamb PW, Rinker-Schaeffer CW, et al. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science 1995;268:884–6. [DOI] [PubMed] [Google Scholar]
- [12].Zhang Y, Tang YJ, Li ZH, et al. KiSS1 inhibits growth and invasion of osteosarcoma cells through inhibition of the MAPK pathway. Eur J Histochem 2013;57:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Presson AP, Sobel EM, Papp JC, et al. Integrated weighted gene co-expression network analysis with an application to chronic fatigue syndrome. BMC Syst Biol 2008;2:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhou Q, Su X, Jing G, et al. Meta-QC-chain: comprehensive and fast quality control method for metagenomic data. Genomics, Proteomics Bioinformatic 2014;12:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Malki K, Tosto MG, Jumabhoy I, et al. Integrative mouse and human mRNA studies using WGCNA nominates novel candidate genes involved in the pathogenesis of major depressive disorder. Pharmacogenomics 2013;14:1979–90. [DOI] [PubMed] [Google Scholar]
- [17].Udyavar AR, Hoeksema MD, Clark JE, et al. Co-expression network analysis identifies Spleen Tyrosine Kinase (SYK) as a candidate oncogenic driver in a subset of small-cell lung cancer. BMC Syst Biol 2013;7:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu X, Hu AX, Zhao JL, et al. Identification of Key Gene Modules for in Human Osteosarcoma by Co-expression Analysis weighted gene coexpression network analysis (WGCNA). J Cell Biochem 2017;118:3953. [DOI] [PubMed] [Google Scholar]
- [19].Namlos HM, Kresse SH, Muller CR, et al. Global gene expression profiling of human osteosarcomas reveals metastasis-associated chemokine pattern. Sarcoma 2012;2012:639038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Buddingh EP, Kuijjer ML, Duim RA, et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: a rationale for treatment with macrophage activating agents. Clin Cancer Res 2011;17:2110–9. [DOI] [PubMed] [Google Scholar]
- [21].Kobayashi E, Masuda M, Nakayama R, et al. Reduced argininosuccinate synthetase is a predictive biomarker for the development of pulmonary metastasis in patients with osteosarcoma. Mol Cancer Ther 2010;9:535. [DOI] [PubMed] [Google Scholar]
- [22].Liu WM, Mei R, Di X, et al. Analysis of high density expression microarrays with signed-rank call algorithms. Bioinformatics 2002;18:1593–9. [DOI] [PubMed] [Google Scholar]
- [23].Gentleman R, Carey V, Huber W, et al. Bioinformatics and Computational Biology Solutions Using R and Bioconductor (Statistics for Biology and Health). New York: Springer-Verlag; 2005. [Google Scholar]
- [24].Kelley KM, Oh Y, Gargosky SE, et al. Insulin-like growth factor-binding proteins (IGFBPs) and their regulatory dynamics. Int J Biochem Cell Biol 1996;28:619–37. [DOI] [PubMed] [Google Scholar]
- [25].Collett-Solberg PF, Cohen P. The role of the insulin-like growth factor binding proteins and the IGFBP proteases in modulating IGF action. Endocrinol Metab Clin North Am 1996;25:591. [DOI] [PubMed] [Google Scholar]
- [26].Bach LA. Insulin-like growth factor binding protein-6: The “forgotten” binding protein? Horm Metab Res 1999;31:226–34. [DOI] [PubMed] [Google Scholar]
- [27].Bach LA, Fu P, Yang Z. Insulin-like growth factor-binding protein-6 and cancer. Clin Sci 2013;124:215–29. [DOI] [PubMed] [Google Scholar]
- [28].Pal A, Huang W, Li X, et al. CCN6 modulates BMP signaling via the Smad-independent TAK1/p38 pathway, acting to suppress metastasis of breast cancer. Cancer Res 2012;72:4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Luther GA, Lamplot J, Chen X, et al. IGFBP5 domains exert distinct inhibitory effects on the tumorigenicity and metastasis of human osteosarcoma. Cancer Lett 2013;336:222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hsin CH, Chou YE, Yang SF, et al. MMP-11 promoted the oral cancer migration and Fak/Src activation. Oncotarget 2017;8:32783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hsin CH, Chen MK, Tang CH, et al. High level of plasma matrix metalloproteinase-11 is associated with clinicopathological characteristics in patients with oral squamous cell carcinoma 2014;9:e113129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hsu IL, Chou CY, Wu YY, et al. Targeting FXYD2 by cardiac glycosides potently blocks tumor growth in ovarian clear cell carcinoma. Oncotarget 2016;7:62925–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.