

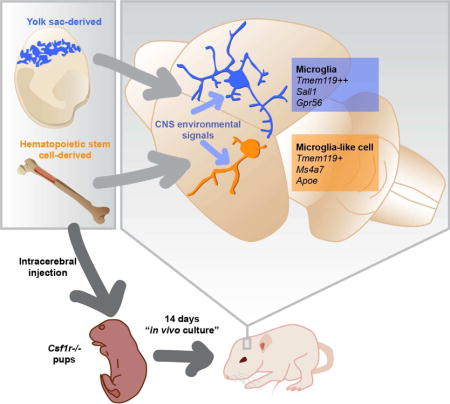
Summary
Microglia, the brain’s resident macrophages, are dynamic CNS custodians with surprising origins in the extra-embryonic yolk sac. The consequences of their distinct ontogeny are unknown but critical to understanding and treating brain diseases. We created a brain macrophage transplantation system to disentangle how environment and ontogeny specify microglial identity. We find that donor cells extensively engraft in the CNS of microglia-deficient mice, and even after exposure to a cell culture environment, microglia fully regain their identity when returned to the CNS. Though transplanted macrophages from multiple tissues can express microglial genes in the brain, only those of yolk-sac origin fully attain microglial identity. Transplanted macrophages of inappropriate origin, including primary human cells in a humanized host, express disease-associated genes and specific ontogeny markers. Through brain macrophage transplantation, we discover new principles of microglial identity, which have broad applications to the study of disease and development of myeloid cell therapies.
Graphical abstract
Introduction
Microglia are vital residents of the brain parenchyma, where they play important but poorly understood roles in development, injury and disease (Li and Barres, 2017; Salter and Stevens, 2017). Arising from yolk sac erythro-myeloid progenitors (YS EMPs) that colonize neural tissue during embryogenesis and undergo a distinct maturational process, microglia are unique among tissue macrophages in that they are thought to remain YS-derived throughout life, without contribution from the fetal liver or definitive hematopoiesis (Ginhoux et al., 2010; Hagemeyer et al., 2016; Hoeffel et al., 2015; Kierdorf et al., 2013; Perdiguero et al., 2014; Schulz et al., 2012). After insults such as experimental autoimmune encephalomyelitis, stroke, malignancy, or genetic depletion, however, microglia dramatically change their phenotype and are joined by infiltrating monocytes/macrophages from the circulation (Ajami et al., 2011; Varvel et al., 2012). These CNS-alien, hematopoietic stem cell (HSC)-derived occupants can resemble microglia in morphology and surface marker expression but appear to participate differently in disease pathogenesis, making it essential to further clarify their functions. Identification of a microglia-specific gene cassette has improved resolution of resident from infiltrating cells (Bennett et al., 2016; Hickman et al., 2013), but does not address whether infiltrating cells inherently lack the capacity to become microglia under appropriate conditions, nor whether microglia can irreversibly lose their identity in abnormal environments.
Although circulating monocytes can effectively differentiate into tissue macrophages in the lung and liver (Scott et al., 2016; van de Laar et al., 2016), the CNS environment is highly distinct, and protected from exposure to circulating factors and cells by the blood-brain barrier (Obermeier et al., 2013). As such, the precise contributions of ontogeny and CNS environment, which have been highly implicated in microglial identity (Gosselin et al., 2014; Lavin et al., 2014; Mass et al., 2016), remain incompletely understood. Do microglia permanently change their identity following disease, or can they return to a homeostatic state? Like YS-derived cells, can macrophages derived from HSCs become microglia? The answers are critical not only to how microglia and infiltrating myeloid cells affect the brain, but the growing use of myeloid cell therapies such as HSC transplantation to treat brain disease in humans (Biffi et al., 2013).
Since our goal was to precisely measure how ontogeny and environment affect microglial identity, we aimed to create a system for transplantation of myeloid cells across development into the brain. We took advantage of Csf1r−/− mice, which lack microglia (Dai et al., 2002; Ginhoux et al., 2010; Liddelow et al., 2017), and found that directly injected myeloid cells extensively engraft in the brain parenchyma, allowing study of donor populations with varied ontogeny. Transplantation into Csf1r−/− hosts offers several advantages. It can be used to study donor cells of diverse origin and developmental stage, and does not require conditioning irradiation or chemotherapy. It yields large numbers of donor derived microglia-like cells (MLCs) that have been conditioned by the brain parenchyma to express microglial genes in the absence of potentially confounding host macrophages, overcoming limitations of prior foundational approaches to understanding microglial identity (Bennett et al., 2016; Bruttger et al., 2015; Mildner et al., 2007).
By comparing multiple engrafted microglia types to MLCs from YS- and HSC-lineages, we found that microglial identity remains intact ex vivo, even following cell culture. We noted general similarity between MLCs derived from all donor lineages, but found striking ontogeny-dependent differences between HSC- and YS-derived populations, leading to discovery of durable markers of parenchymal macrophage ontogeny. We extended this approach to a humanized transplantation system and verified fundamental conclusions in human microglia and MLCs. In sum, we devised an experimental system to unravel the contributions of brain environment and ontogeny to macrophage identity in mouse and human.
Results
Directly transplanted microglia engraft and ramify in the Csf1r−/− CNS
We recently demonstrated that cultured microglia have the capacity to engraft in the Csf1r−/− brain parenchyma, which otherwise lacks microglia, after intracerebral transplantation (ICT) (Bohlen et al., 2017). To further study intrinsic versus acquired properties of microglial identity, we compared three distinct microglia populations after ICT into the CNS between postnatal day 0 to 4 (P0-4): 1) acutely isolated mature microglia (P21, “ICT MG”) 2) developmentally immature microglia (P5, “ICT P5 MG”), which lack expression of the full microglial gene cassette (Bennett et al., 2016), and 3) cultured microglia (P18-35, “ICT Cultured MG”) that undergo dramatic transcriptional changes in vitro including loss of expression of the microglial signature cassette (Bohlen et al., 2017; Gosselin et al., 2017) (Figure 1A). By 14 days after intracerebral injection, all donor microglia types extensively engrafted and ramified in the brain parenchyma, often filling entire sagittal sections (Figure 1B, Figure S1A). When normalized to area of engraftment, transplanted microglia reached a similar density to endogenous microglia in a wildtype Csf1r+/+ (WT) host (Figure S1B). By flow cytometric analysis, engrafted cells were CD45+CD11B+ and expressed WT levels of Tmem119 (Figure S1C-E). By immunostaining, 100% were Tmem119+ in sections from 4-7 biological and at least 5 technical replicates each across the brain. As in WT mice, we found no Tmem119 staining in the meninges and choroid plexus of ICT mice (not shown). Extent of donor cell engraftment varied - by FACS, we retrieved fewer microglia from transplanted hosts than WT controls, and occasionally observed minimal to no engraftment. Because the host strain for Csf1r−/− transplant experiments was FVB, for which no robustly expressed fluorescent reporters exist, we also verified that sorted engrafted microglia were WT at the Csf1r locus (Figure S1F). These data show that microglia from multiple developmental stages can occupy the postnatal brain, ramify, and express Tmem119 only when engrafted in the parenchyma.
Figure 1. Transplanted microglia engraft in the Csf1r−/− brain and express Tmem119.
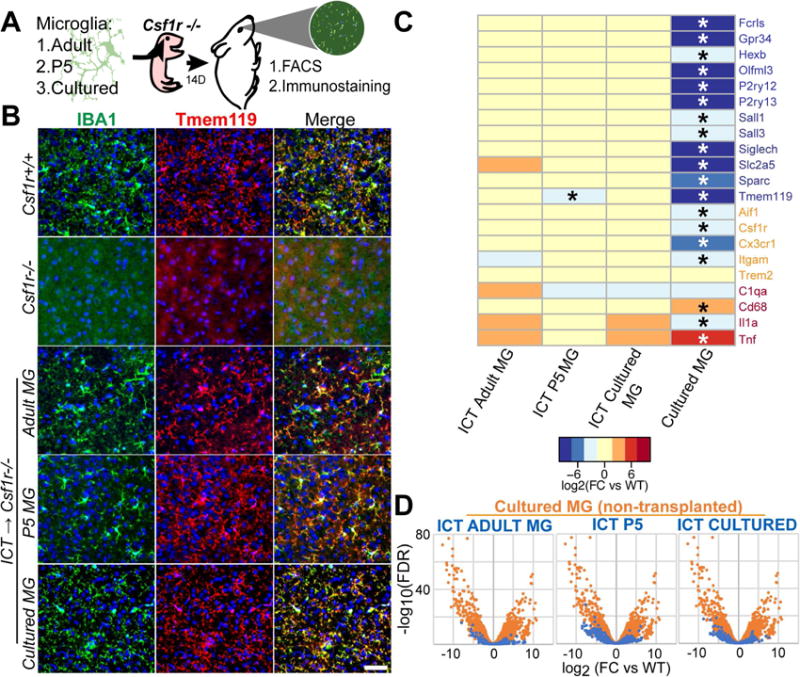
A) Schematic of microglial transplantation system. B) Immunostaining showing ramified IBA1+(green)/TMEM119+(red) microglia in Csf1r+/+ (WT) and microglia-transplanted Csf1r−/−hosts, and untransplanted Csf1r−/− control. Scale bar = 50μm. C) Expression heat map (log2(FC/WT)) of microglia (blue), myeloid (orange) and reactivity (red) genes by microglia after culture or transplantation into Csf1r−/− CNS. Detailed inventory of experimental replicates is listed in methods. * = FDR <0.05 compared with WT D) Overlaid volcano plots show reduced differential gene expression of ICT-cells (blue) compared to non-transplanted cultured MGs (orange), represented by reduced spread of volcano. See also Figure S1.
CNS signals are sufficient to induce, sustain, and re-induce microglial identity
To better understand relationships between microglial ontogeny, environment, and transcriptional phenotype, we used optimized techniques to isolate RNA from highly pure parenchymal microglia after ICT into Csf1r−/− hosts based on Tmem119 immunoreactivity (Bennett et al., 2016). Transcriptomic profiling by RNAseq showed that by 14 days in vivo, microglia which had either lost expression of signature genes in vitro (ICT Cultured MG) or had not attained full maturity (ICT P5 MG), expressed mature microglial signature genes at nearly normal levels, including Tmem119, P2ry12, Olfml3, and Sall1 (Figure 1C). More broadly, ICT cultured, P5, and adult microglia were highly similar to each other and to their WT counterparts. Of 1827 differentially expressed genes in in vitro microglia, all but 16 returned to within 2-fold of WT levels after re-engraftment of cultured microglia in the CNS (Figure S1G). Volcano plot overlays demonstrate that differences between cultured WT microglia are largely restored after re-engraftment in the brain (Figure 1D). While transplanted microglia have statistically meaningful differences in gene expression compared to untransplanted WT microglia, these changes likely represent an “engraftment signature” from donor cell isolation, culture, and the Csf1r−/− host environment. Gene expression changes were generally of small magnitude, included several chemokine genes, tetraspanins and G-protein coupled receptors but not a signature of reactivity or specific functional process (Table S1). These experiments show that the Csf1r−/− CNS is sufficient to sustain, induce, and re-induce microglial identity, and that microglial identity potential persists despite dramatic transcriptional perturbations induced ex vivo.
Transplanted cells of diverse ontogeny engraft and ramify in the Csf1r−/− CNS
Given stable microglial identity despite highly plastic gene expression between adult, P5 and cultured microglia, we appreciated that ICT could clarify relationships between brain macrophage ontogeny and environment. In particular, we wondered whether HSC- or YS-derived macrophages originating outside the developed brain could become microglia in a permissive CNS environment capable of supporting homeostatic microglia. Therefore, we individually transplanted whole tissues and sorted myeloid cells into the Csf1r−/− CNS at P0-P4 including YS-derived cells from yolk sac and fetal brain, HSC-derived cells from blood and bone marrow (BM), and monocytes from the fetal liver which, at E13-14, contains a mix of HSC- and YS-derived cells (Hoeffel et al., 2015; Perdiguero et al., 2014). We observed extensive engraftment of ramified Iba1+/Tmem119+ microglia-like cells (MLCs) using all tissue types tested across both embryonic and postnatal lineages (Figure 2A-B; S2A-C,F), though YS-derived MLCs (YS-MLCs) had a consistently more ramified morphology than HSC-derived (HSC-MLCs). We verified donor origin of MLCs using a GFP reporter after back crossing the Csf1r−/− allele to C57Bl/6 (Figure 2B), and additionally noted extensive coverage of the spinal cord by donor cells delivered by supratentorial injection (Figure S2D)
Figure 2. Diverse myeloid populations engraft in the Csf1r−/− brain, ramify, and are Tmem119+.
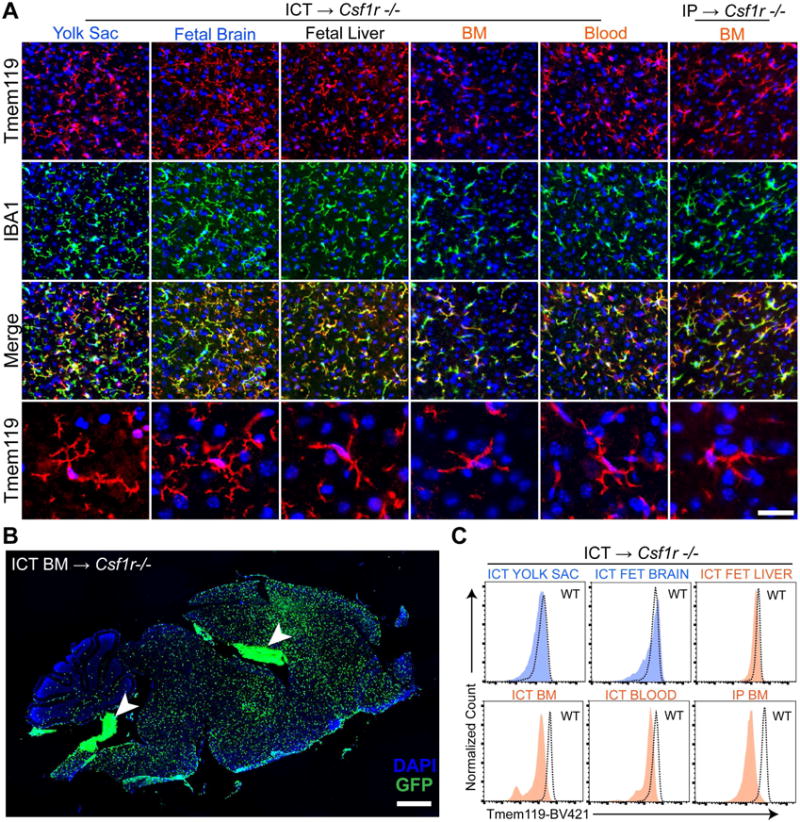
A) IBA1+(green) MLCs ramify and are TMEM119+ (red) in the brain parenchyma 14 days after ICT into Csf1r−/− hosts. Bottom row depicts TMEM119+ MLCs at high magnification. Scale bar = 36.5μm (top 3 rows), 25μm (bottom row). B) Engrafted GFP+ MLCs 14 days after bone marrow ICT into P1 Csf1r−/− host. Arrowheads indicate non-parenchymal donor cells in the ventricles and choroid plexus, of which virtually all are IBA1 positive (see Figure S2F). Scale bar = 900μm. C) Histograms show Tmem119 expression in MLCs 14 days after transplantation by flow cytometry, including reduced staining in HSC-derived MLCs. See also Figure S2.
By flow cytometry, nearly all CD45+CD11B+ cells were Tmem119 immunoreactive, although HSC donor tissues consistently showed lower intensity staining than WT (Figure 2C). Since we saw a small Tmem119- population in some cases, we again confirmed by immunostaining that, as with transplanted microglia, all parenchymal but no other Iba1+ MLCs were Tmem119+ (Figure S2G). All donor tissues engrafted to similar densities as microglia in Csf1r−/− brains, except for fetal liver, which reached a significantly higher density (Figure S2E). FACS plots, engraftment levels and percent Tmem119 positive values are further detailed in Figure S2 to provide potential users a realistic assessment of the robustness of this system.
An inherent limitation of the Csf1r−/− model is poor host viability, which required us to measure the effects of CNS residence in ICT experiments after 14 days. To better study the trajectory of effects of longer incubation, we also created a chemotherapy- and irradiation-free peripheral bone marrow transplantation system that allows study of long term MLC engraftment. Whereas Csf1r−/− mice do not typically survive past weaning age (Dai et al., 2002; Li et al., 2006), simple intraperitoneal injection (IP) of WT bone marrow “rescued” approximately 50% of pups, leading to prolonged survival, tooth eruption, occasional fertility, and engraftment of donor-derived myeloid cells in multiple tissues including the brain parenchyma and liver (Figures 3A-D, S3A-B). By 1 month, the brain parenchyma of rescued mice showed complete, uniform coverage by donor-derived cells (Figure 3A). We harvested well-appearing rescued mice up to 1 year after transplantation and observed stable occupancy of the brain parenchyma by MLCs (Figure 3B). Taken together, these studies show that the CNS niche readily hosts macrophages from multiple donor tissues, including for long periods using bone marrow.
Figure 3. Peripheral bone marrow injection leads to widespread engraftment of donor-derived cells and results in partial rescue of the Csf1r−/− phenotype; both purified monocytes and Ccr2 −/− bone marrow cells engraft in the Csf1r −/− brain and express Tmem119.
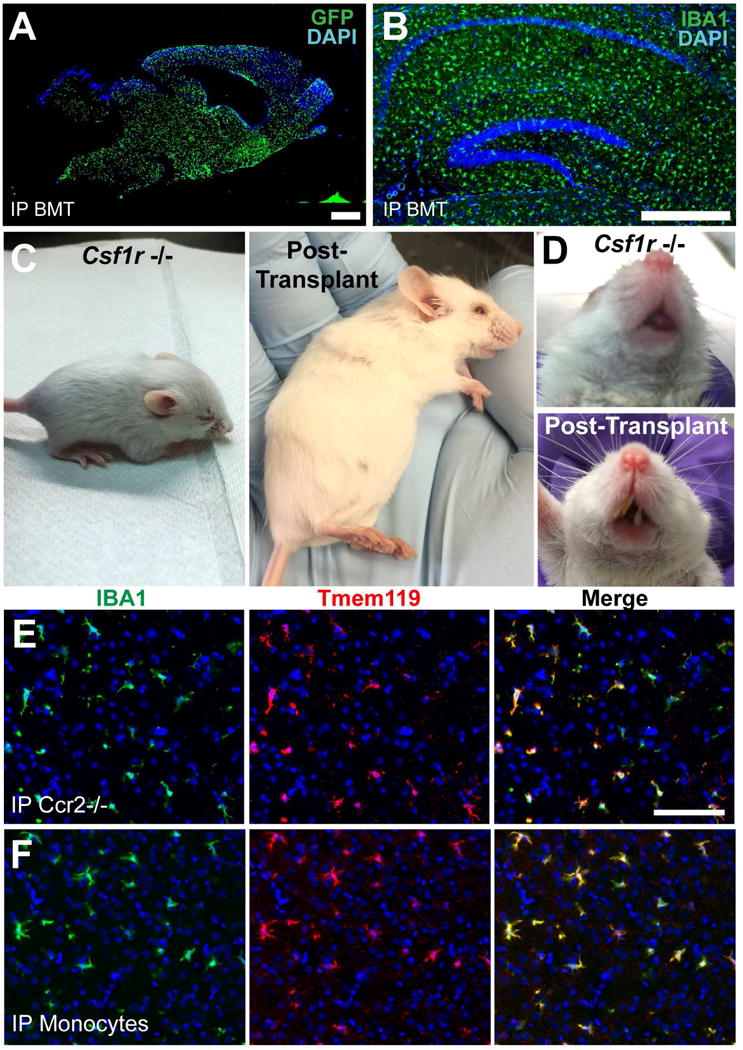
A) Engrafted MLCs 1 month after intraperitoneal (IP) bone marrow injection into P1 host. Scale bar = 900μm. B) Hippocampal section of Csf1r−/− brain stained for IBA1 8 months after IP BMT. Scale bar = 400μm. C) Typical Csf1r−/− mouse showing abnormal head shape, small size (left), compared to 3 months after intraperitoneal bone marrow injection at P2 (right). D) Untransplanted Csf1r−/− mouse lacks teeth (top), while transplanted Csf1r−/− mouse shows tooth growth (bottom). E) Both Ccr2 Rfp/Rfp BM and F) purified BM monocytes engraft in the Csf1r −/− brain and express TMEM119 at T=21 days. Scale bar = 100μm. See also Figure S3.
The surprising observation that IP injected bone marrow populated the Csf1r−/− brain without preconditioning led us to further characterize how donor cells might enter the brain. Since a prior study of macrophage repopulation by peripheral cells found evidence for increased blood brain barrier (BBB) permeability (Varvel et al., 2012), we tested for the presence of increased levels of IgG and albumin in the brain, which are largely excluded from the parenchyma under homeostatic conditions. Quantitative ROI analysis of immunostained histological sections showed no evidence for increased albumin or IgG extravasation, and we did not observe focal areas of increased staining, suggesting “normal” BBB permeability (Figure S3C). Since multiple studies suggest that monocyte infiltration into the diseased or injured CNS is facilitated by CCR2 (Ajami et al., 2011; Dzenko et al., 2001), we wondered whether engraftment of MLCs was similarly CCR2-dependent in Csf1r−/− hosts. We found that, as with WT bone marrow, IP injection of CCR2 Rfp/Rfp (Ccr2 knockout) bone marrow (Saederup et al., 2010) into Csf1r−/−mice leads to robust Tmem119+ MLC engraftment at 2 weeks, meaning that CCR2 is dispensable for Csf1r−/− IP BMT (Figure 3E). Interestingly, we observed a range of RFP fluorescence levels in engrafted cells (Figure S3D), suggesting that either brain signals suppress CCR2 expression, or that more than one population of bone marrow cells (as distinguished by CCR2 reporter expression) are capable of brain engraftment. We also noted a preponderance of RFP+ cells in a periventricular distribution (Figure S3E). Finally, to determine whether myeloid progenitors or HSCs were strictly required to create MLCs, we also transplanted BM-monocytes that were stringently depleted of progenitor populations (Figure S3F), and observed abundant Tmem119+ parenchymal MLCs by 2 weeks (Figure 3F).
The CNS environment strongly and rapidly induces microglial gene expression in CNS-naive cells
The ability of both HSC- and YS-derived donor cells to engraft in the brain parenchyma and express Tmem119 only when engrafted attests to the potency of programming signals from the brain parenchyma. To comprehensively measure the ability of diverse transplanted cells to adopt a microglial transcriptional program, we purified parenchymal Tmem119+ MLCs using identical methods to transplanted microglia, allowing highly specific isolation of parenchymal macrophages. We transcriptionally profiled MLCs derived from E8 yolk sac, E12-13 fetal brain, E13-14 fetal liver, adult blood, and BM, comparing them to each other and to transplanted microglia. Among these highly purified transcriptomes, we observed a striking degree of similarity between engrafted cell types (Figure 4A, S4A-C). MLCs, irrespective of ontogeny, expressed many microglia signature genes, including Tmem119, Fcrls, Hexb, and Olfml3, at near-microglial levels (Figure 4A). Gene expression in all MLC types was well-correlated (Spearman coefficients >0.6-0.8), and in exploratory analyses combining published datasets, MLCs were more closely related to microglia than to other tissue macrophages, monocytes, or neutrophils (Figure S4C,D). These data confirm the strong programming effects of the brain parenchyma on macrophages, and the intrinsic ability of even CNS-alien macrophages to respond to programming signals by expressing microglial genes.
Figure 4. Ontogeny shapes adoption of microglial transcriptional identity; BM-derived cells show highly similar transcriptomes at 2 weeks compared to 2-3 months of brain residence.
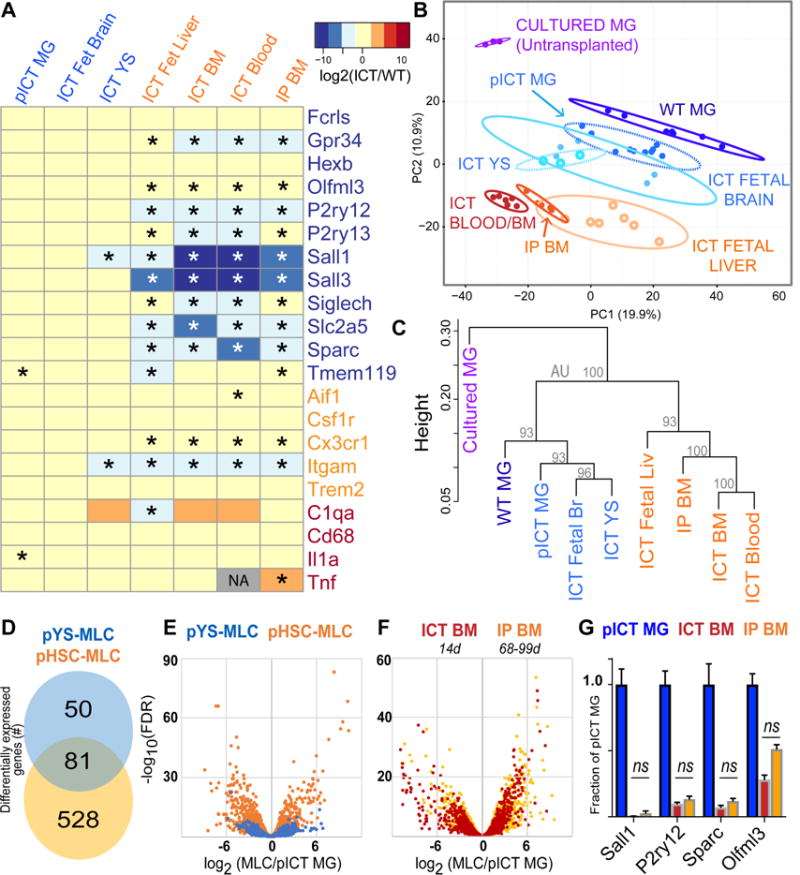
A) Heatmap showing log2(FC versus WT) expression of microglial (blue), myeloid (orange) and reactivity (red) genes across MLC types. * = FDR<0.05 compared to WT. Grey box indicates that edgeR algorithm could not compute log2(FC) due to low read abundance. B) Plot of largest principal components for cultured microglia (purple), WT microglia (dark blue), pooled ICT MG (blue), YS-MLCs (ICT yolk sac, fetal brain; lighter shades of blue), fetal liver MLCs (light orange), and HSC-MLCs (ICT Blood, BM, and BMT; orange/red), using top 2500 most variant genes. Ellipses demarcate 95% confidence interval for assigned clusters. C) Unsupervised hierarchical clustering of microglia, pooled ICT microglia and MLCs by Spearman coefficients using 1000 most variant genes, AU = approximately unbiased p-value using PVclust package, bootstrap n=10000 D) Venn diagram showing differential gene expression between pooled YS- and HSC-MLCs, both compared to pooled ICT-MGs (2-fold cutoff, FPKM>20, FDR <0.05). See also Figure S5A. E) Volcano plot overlay showing differential gene expression of YS- and HSC-MLCs types compared to ICT-MGs, measured as log2(pMLC/pMG). F) Volcano plot overlay comparing MLCs derived from ICT BM at 14 days (red), to MLCs from IP BM at 2-3 months (orange) showing no gross shift in transcriptome difference from MGs. G) MG identity genes do not change between ICT BM (red) and IP BM (orange), ns, FDR >0.05 by edgeR comparison. See also Figure S5C. MG= microglia, MLC = microglia-like cell, ICT = intracerebral transplant, HSC = hematopoietic stem cells, YS = Yolk Sac, pYS= pooled YS, Fetal Br = fetal brain, Fetal Liv = fetal liver, IP BM = intraperitoneal bone marrow transplant. See also Figure S4, S5.
HSC ontogeny prevents full adoption of microglial identity
Although grossly similar, we found major ontogeny-dependent differences between HSC- and YS-MLCs. Principal component analysis showed that the transcriptomes of transplanted microglia, yolk sac MLCs, and fetal brain MLCs overlap with each other, distinct from blood, BM and fetal liver MLCs (Figure 4B). Unsupervised hierarchical clustering similarly showed that YS-derived (YS, fetal brain) MLC gene expression is more closely related to transplanted microglia than HSC-derived (blood, BM) and mixed-origin MLCs (fetal liver) (Figure 4C).
To focus on ontogeny specific gene expression patterns, we pooled gene expression data from YS- and HSC-MLCs donor groups, excluding fetal liver-MLCs since E13-14 liver contains a mix of YS- and HSC-derived cells (Hoeffel et al., 2015; Perdiguero et al., 2014). As a group, YS-lineage MLCs had 131 differentially regulated genes compared to transplanted microglia, while HSC-derived MLCs had 609 (Figure 4D). Volcano plot overlay further depicts the higher similarity of YS-MLCs to transplanted microglia at a whole-transcriptome level (Figure 4E). At the gene level, YS-MLCs expressed microglia signature genes more faithfully than their HSC-derived counterparts, including Slc2a5, Olfml3, Gpr34, Sparc, and P2ry12 (Figure S5A). Among microglia-enriched genes in Tmem119+ cells from a prior study (Bennett et al., 2016), YS-MLCs were significantly closer to MG expression levels in 30 of 32 measured genes (Table S2). Similarly, among 31 genes enriched in non-microglia CNS myeloid cells, YS-MLCs were closer to microglial levels for 29 (Table S3). Of particular significance, HSC-MLCs did not express Sall1, a transcription factor recently implicated in microglial identity (Buttgereit et al., 2016). Interestingly, the pattern of Sall1 expression by YS-MLCs correlated with its expression during microglial development, from low levels in yolk sac MLCs, intermediate levels in fetal brain MLCs, and highest in transplanted microglia (Figure S5B).
While YS-MLCs were able to express all microglia signature genes by 2 weeks, HSC-MLCs were not. Based on prior studies using microglial repopulation systems (Bruttger et al., 2015; Mildner et al., 2007; Priller et al., 2001; Varvel et al., 2012), along with observations in other tissues (Scott et al., 2016; van de Laar et al., 2016) we felt it was critical to address whether longer CNS engraftment was sufficient to better reprogram HSC-MLCs as microglia. If so, one would expect interval increases in the expression of microglial signature genes with prolonged brain engraftment, akin to observations in the liver between 15 and 30 days after engraftment (Scott et al., 2016). We therefore analyzed MLCs that were engrafted in the brain parenchyma of rescued mice for 2-3 months (mean 81 days) following peripheral bone marrow transplantation, and assessed for a trajectory of higher expression of microglial identity genes. We compared gene expression patterns to directly injected BM-MLCs harvested after 14 days and found almost no interval induction of microglia signature genes after prolonged CNS incubation. In particular, longer-term engrafted cells showed persistently low expression of Sall1, Sall3, Sparc, P2ry12, Gpr34 and Olfml3, none of which were statistically different from expression in short term engrafted cells (Figures 4A,G, S5C). By unbiased clustering, long term engrafted BM-MLCs were highly similar to short term engrafted BM and blood-MLCs (Figure 4B,C). Volcano plot overlay does not show increased similarity of longer term engrafted cells to microglia (Figure 4F). Longer-term engraftment was associated with differential gene expression (Tables S4,5), though with no clear evidence of further reprogramming based on microglia signature genes (Figure S5C). Taken together, these data show that when macrophages derived from HSCs engraft in the brain, they become similar to microglia by 14 days, but do not further increase expression of microglial identity genes when incubated in the brain at least four times longer. In contrast, YS-derived macrophages, which share a common ancestor with microglia, have the intrinsic potential to become highly similar to transplanted microglia by 14 days.
HSC-MLCs share transcriptional signatures with disease-associated microglia, including highly elevated Apoe expression
To better understand transcriptomic differences between HSC- and YS-MLCs, we assessed transcriptional networks by multiple approaches. Pathway analysis (Ingenuity) suggested HSC-MLCs were enriched in pathways associated with CNS perturbation (neuroinflammatory signaling, NOS/ROS production, and TLR signaling, Figure S5D). Recent studies of mouse and human microglia found significant overlap between transcriptional changes in cultured, Alzheimer’s disease (AD), Amyotrophic Lateral Sclerosis (ALS), LPS exposed, and immature microglia, suggesting common downstream reactivity pathways (Bohlen et al., 2017; Gosselin et al., 2017). We wondered if the observed gene expression differences between HSC- and YS-MLCs overlapped with differences between brain myeloid cells in disease compared to health. Indeed, HSC- compared to YS-MLCs were significantly enriched in gene sets associated with ALS, AD, LPS treatment, immaturity, and in vitro culture from prior studies (Bennett et al., 2016; Chiu et al., 2013; Matcovitch-Natan et al., 2016; Wang et al., 2015), along with major histocompatibility complex class II genes (Figure 5A). YS-MLCs were relatively enriched in gene sets associated with homeostasis but these did not reach significance cutoff (FDR<0.05).
Figure 5. HSC-MLCs resemble microglia lacking identity genes and in disease states.
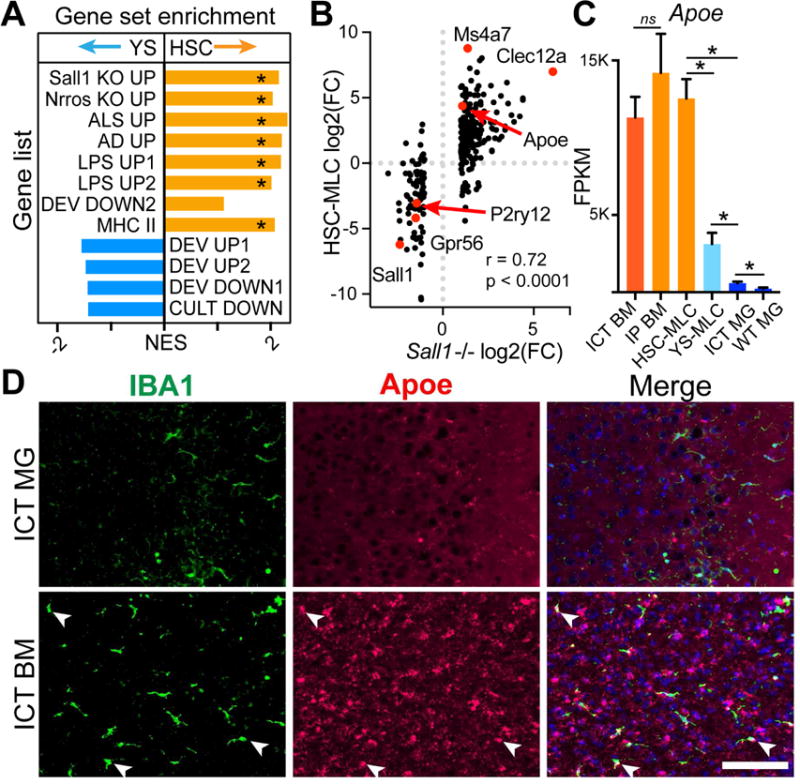
A) Normalized Enrichment Scores (NES) from GSEA comparing YS- to HSC-MLCs for enrichment in genes upregulated in Sall1−/− (Sall1 KO UP), Nrros−/− (Nrros KO UP), Amyotrophic Lateral Sclerosis (ALS UP from), AD (AD UP), after LPS treatment (LPS UP1, 2), changed in during development (DEV UP 1, 2 or DEV DOWN1,2), in culture (CULT DOWN) and MHCII genes. * = FDR <0.05. B) Expression plot comparing HSC-MLCs and Sall1−/− microglia, both expressed as log2(FC/WT). Red dots highlight genes of interest. r = correlation coefficient, p = p-value for linear regression analysis. C) Apoe gene expression in MGs/MLCs. * = FDR < 0.05. D) RNA in situ hybridization for Apoe (red) with IBA1 counterstain in Csf1r−/− brains 14D post-transplantation with BM or MGs. Arrows indicate Apoe+ MLCs. Scale bar = 100μm. See also Figure S5.
Since HSC-MLCs lack Sall1, we also tested for enrichment of genes that change with loss of microglial Sall1 and Nrros, which were recently found to cause similar shifts in transcriptional identity (Buttgereit et al., 2016; Wong et al., 2017). We compared our datasets to published gene expression profiles of Sall1 and Nrros deficient microglia, and indeed HSC-MLCs showed significant enrichment for genes upregulated in both Sall1 and Nrros −/− microglia (Figure 5A). In fact, gene expression in HSC-MLCs and Sall1 −/− microglia were highly correlated (Figure 5B), suggesting that lack of Sall1 could contribute to differential gene expression in HSC-MLCs.
Among these dysregulated genes, Apoe stands out as the most highly expressed gene in HSC-MLCs, and does not decrease with longer engraftment (Figure 5C). To validate high Apoe expression observed in HSC-MLCs, we performed RNA in situ hybridization. As anticipated, we found high intensity signal in many brain macrophages after BM but not MG ICT. Surprisingly, we also found increased probe signal throughout the brain parenchyma in Iba1 negative cells of BM-ICT animals, which may reflect increased expression by astrocytes in the presence of HSC-MLCs (Figure 5D). Taken together, these results show that HSC- but not YS-MLCs nor transplanted microglia share gene expression signatures with microglia in disease states or after loss of identity genes.
HSC-MLCs express markers distinct from YS-MLCs and microglia
The Csf1r−/− transplantation approach demonstrates that many macrophage types have the intrinsic potential to masquerade as microglia by expressing signature genes and TMEM119 protein in the brain. This does not occur following conventional BMT (Bennett et al., 2016) and complicates the use of microglia signature genes to distinguish microglia from HSC-MLCs in complex disease models, which now requires creation of new tools. To identify stable markers of brain parenchymal macrophage ontogeny, we screened our dataset for genes 1) highly expressed across HSC-MLC types, but lowly expressed in YS-MLCs and microglia or 2) highly expressed in YS-MLCs and microglia but not HSC-MLCs. We then eliminated genes dysregulated in microglia in AD and after LPS stimulation, yielding five candidate ontogeny markers: Clec12a, Ms4a7, Lilra5, Klra2, and Gpr56 (Figure 6A). We performed RNA in situ hybridization to validate the localization of Ms4a7, Clec12a, and Gpr56 in engrafted brain tissues (Figure 6B-D; S6A). As predicted by transcriptomic data, virtually all HSC-MLCs but no microglia nor YS-MLCs were Ms4a7+ and Clec12a+. Meanwhile, no HSC lineage cells were Gpr56+, a gene recently associated with microglial ontogeny in transcriptomic studies (Gosselin et al., 2014) and also expressed by other glia, such as astrocytes (Zhang et al., 2014). Importantly, we probed healthy postnatal CNS tissue and did not find Ms4a7 expression by microglia across the lifespan (Figure S6C). We also tested whether these HSC ontogeny markers were unique to the Csf1r−/− system. We transplanted GFP+ BM directly into the CNS of neonatal and adult Cx3cr1-CreER+/−; Csf1r fl/fl mice, treated with tamoxifen, to partially and transiently deplete microglia. At 2 months, we observed abundant GFP+Tmem119+ cells in the brain parenchyma, and concordant clusters of Ms4a7+ cells by RNA in situ (Figure S6D-G), suggesting that HSC ontogeny marker induction is neither unique to the Csf1r−/− mouse system nor the neonatal CNS.
Figure 6. Ontogeny markers discriminate HSC- from YS-MLCs and microglia.
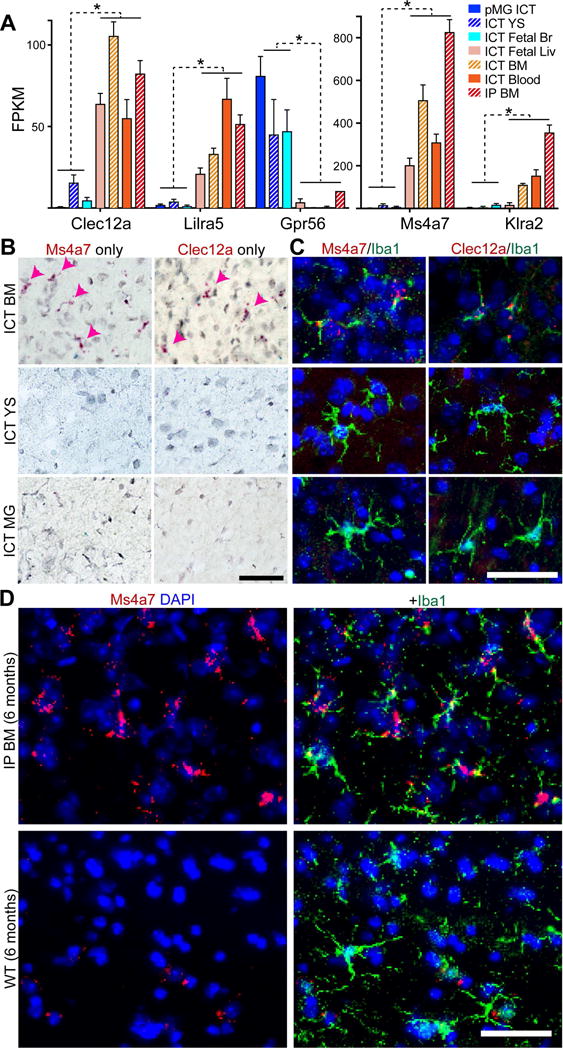
A) FPKM values for HSC- and YS-MLC/MG enriched genes (red-orange and blue bars, respectively). Error bars represent SEM, FDR < 0.005 for all genes between YS/MG and HSC groups. B) RNA in situ hybridization showing expression of Ms4a7 or Clec12a (red) in ICT BM- but not YS-MLCs nor ICT MGs. Arrowheads highlight BM-MLCs. Scale bar = 50μm. C) Fluorescent RNA in situ hybridization showing expression of Ms4a7 or Clec12a (red) in ICT BM- but not YS-MLCs nor ICT MGs, co-stained for IBA1 (green). Scale bar = 50μm. D) RNA in situs show persistent expression of Ms4a7 (red) in IBA1+ cells (green) six months after transplantation in HSC-MLCs, but no expression in WT microglia. Scale bar = 50μm. See also Figure S5, 6.
Since ICT transplanted Csf1r−/− animals do not survive long enough to test prolonged time points, we also tested whether HSC-MLCs in animals rescued by IP BMT were able to downregulate Ms4a7 six months after transplantation, as would be expected if environmental signals superseded the limits of ontogeny. We found that all HSC-MLCs remained TMEM119+ and Ms4a7+ after 6 month incubation (Figure 6D; S6B). This is consistent with our transcriptomic observations at 2-3 months, when HSC-MLCs sustained expression of ontogeny markers, and in fact increased expression of one such ontogeny marker, Klra2 (Figure S5C). In sum, we capitalized on transcriptomic data from transplanted brain macrophages to validate ontogeny markers that are stable despite the high plasticity of brain macrophage gene expression in response to the CNS environment.
Human primary cells engraft, ramify and express Tmem119 in humanized Csf1r−/− mice, but only HSC-MLCs are Ms4a7 immunoreactive
To explore the utility of this transplantation system for human brain macrophage study, we crossed Csf1r+/− mice into an immunodeficient strain expressing the human form of MCSF (Rathinam et al., 2011), based on our observations that murine MCSF does not promote human microglial survival (unpublished observations), creating a Rag2−/− IL2rg−/− hMCSF Csf1r−/− mouse. We transplanted human blood, fetal brain macrophages, and postnatal microglia from neurosurgical cases directly into the mouse CNS. We observed engraftment, survival and ramification of all cell types, often over large territories of the mouse brain parenchyma (Figure 7A-D). Most all transplanted human fetal and postnatal brain parenchymal macrophages were Tmem119 immunoreactive (Figure 7C-D), whereas engrafted peripheral blood cells showed variable staining: in some animals, nearly all engrafted blood-derived cells were Tmem119+ (Figure 7A), while in others, expression was restricted to a fraction (Figure 7B).
Figure 7. Macrophage transplantation, origin markers, and anti-TMEM119 monoclonal antibodies for the study of primary human macrophages in vivo.
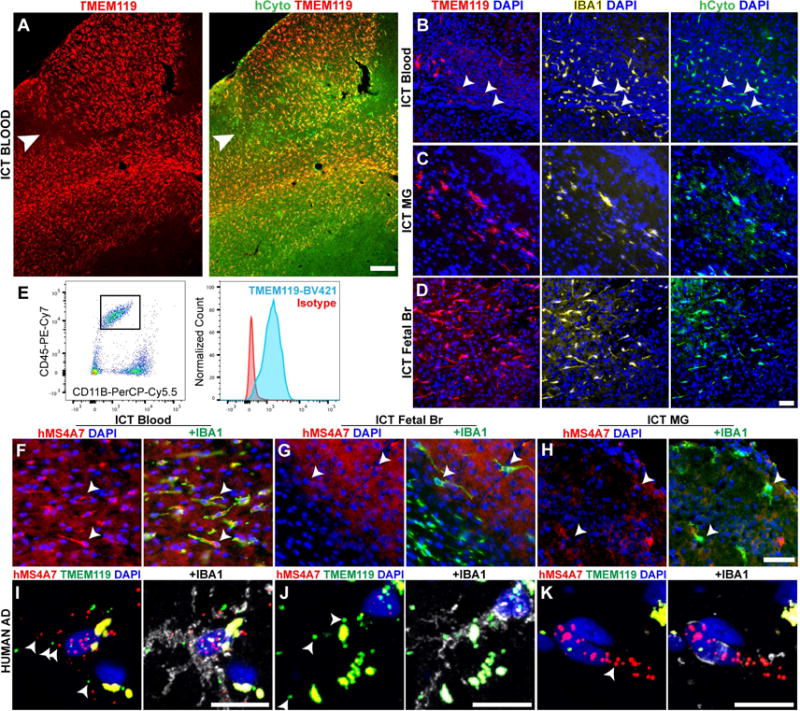
A) Pervasively engrafted TMEM119+ (red) cells from human blood in the Rag2−/−Il2rg−/−hMCSF+/+Csf1r−/− brain parenchyma, co-stained with human cytoplasm marker (hCyto, green). Arrowhead indicates TMEM119- cells at edge of engraftment territory. Scale bar = 200μm. (B-D) Representative images of MLCs from human blood (B), adult microglia (C), and cultured fetal brain (D), immunostained for human TMEM119 (red), IBA1 (yellow) and human cytoplasmic marker (green). Arrowheads in (B) identify human TMEM119 low/- cells. Scale bar = 50μm. (E) A custom anti-human TMEM119 antibody identifies CD45+/CD11B+ fetal human MLCs 14 days after transplantation into the mouse CNS. Tmem119 staining (blue) is shown compared to isotype control (red). (F-H) Representative images of MLCs from blood (F), fetal brain (G) and primary human microglia (H) immunostained for human MS4A7 (red) and IBA1 (green). Arrowheads (B,F-H) mark location IBA1+ cell bodies. Scale bar = 100μm. (I-K) Representative images from RNA in situ hybridization of post-mortem Alzheimer’s disease brain samples, showing (I) rare MS4A7+(red)/TMEM119+(green)/IBA1 protein+ (white) macrophages, (J) abundant MS4A7-/TMEM119+/IBA1+ macrophages, and (K) MS4A7+/TMEM119-/IBA1+ perivascular macrophages. Arrowheads show examples of positive puncta, given abundant autofluorescent signal (puncta in perfect registration) Scale bars = 12.5μm. See also Figure S7.
To better study human microglia, we generated a mouse monoclonal antibody against the extracellular domain of human TMEM119 using the same approach we recently used for mouse TMEM119 (Bennett et al., 2016). This custom antibody does not stain human blood (Figure S7A), but does identify a distinct subpopulation of CD45+/CD11B+ cells in human brain cell suspensions (Figure S7B), as well as human fetal brain macrophages engrafted in the mouse brain (Figure 7E, S7C). This tool will allow specific purification of human microglia in the future.
Finally, we wondered whether MS4A7 has potential for use as an ontogeny marker for human brain macrophages. We found that xenograft, blood-derived human HSC-MLCs, but not microglia were MS4A7 immunoreactive (Figure 7F-H, S7D-F). Having demonstrated that MS4A7 can identify human HSC-MLCs, we next tested if we could detect putative HSC-MLCs in a complex human disease. In post-mortem samples from cases of Alzheimer’s and severe cerebrovascular disease, we observed rare ramified TMEM119+/MS4A7+/IBA1+ cells, which we could not detect in healthy controls (Figure 7I-J S7G-H). Interestingly, we also found frequent MS4A7+/Iba1+ cells in a perivascular distribution that were not associated with disease (Figure 7K, S7I). Together, these findings demonstrate that MS4A7 is a conserved ontogeny marker, and that the presence of HSC-MLCs may be associated with human neurodegenerative disease.
Discussion
Direct CNS transplantation reveals fundamental principles of microglial identity and plasticity
Ex vivo manipulations cause dramatic shifts in microglial gene expression that resemble patterns found during disease and in immature embryonic microglia (Bohlen et al., 2017; Gosselin et al., 2014; 2017; Haynes et al., 2006). Using a cell transplantation system we found that upon return to the CNS, microglia readily inhabit the Csf1r −/− brain, adopt a ramified morphology, and revert to a normal transcriptional program despite profound ex vivo derangement. Our findings demonstrate that though remarkably sensitive to environment, microglia robustly retain the potential to return to a homeostatic state. Comparison between transplanted mature, young, and cultured microglia clarifies that the brain is replete with necessary factors to sustain, induce and re-induce homeostatic microglial gene expression. These findings have direct relevance to microglial reactivity, and development of myeloid cell therapies for brain disease. They suggest that microglia themselves could be cultured, modified, and used translationally for CNS cell therapies, and further, identify the parenchymal macrophage niche as a valid “in vivo culture system” for transplanted microglia. Our findings also encourage continued efforts to develop an in vitro culture system that better sustains microglial identity.
The CNS induces microglial gene expression in diverse myeloid populations
Cell depletion and bone marrow transplantation studies demonstrate the brain’s ability to host peripheral myeloid cells (Bruttger et al., 2015; Cronk et al., 2018; Mildner et al., 2007; Priller et al., 2001). Here, we show that the Csf1r−/− brain readily and durably hosts myeloid cells from blood, bone marrow, fetal liver, fetal brain, and yolk sac. We found strong induction of microglial signature genes and TMEM119 protein within 14 days, by as yet unknown CNS environmental cues. Based on observations using BMT and/or genetic microglial ablation with our study, macrophages likely also need access to an open parenchymal macrophage niche (Bruttger et al., 2015; Cronk et al., 2018). Here, without conditioning irradiation or chemotherapy, we were able to directly compare multiple engrafted TMEM119+ populations from the YS and HSC lineages at multiple stages of development, to clearly delineate the consequences of ontogeny on gene expression in brain resident macrophages.
Peripheral bone marrow injection rescues Csf1r−/− animals and results in pervasive engraftment of brain macrophages without irradiation or chemotherapy
Intraperitoneal injection of whole BM with no preconditioning prolongs the survival of Csf1r −/− mice, which typically succumb around 2 weeks of life (Dai et al., 2002; Li et al., 2006). In rescued animals, we observed pervasive engraftment of donor cells in all tissues examined, including the brain and spinal cord. Surprisingly, although classic studies identify CCR2 as critical for CNS entry in the setting of inflammation, (Ajami et al., 2011; 2007), we find that MLC engraftment did not depend on CCR2, nor did the Csf1r−/− host show evidence for frank BBB breakdown. These data suggest the existence of an alternative mechanism for cell entry into the brain, which we hope to address in future studies. Furthermore, we found that isolated bone marrow monocytes themselves can become TMEM119+ MLCs, consistent with recent results using conditional Csf1r deletion to deplete microglia (Cronk et al., 2018). Though this does not exclude the possibility that myeloid progenitors or HSCs also contribute to MLC formation, it suggests monocytes as a candidate donor population for cell-based therapies. Provocatively, circulating cells do not contribute to repopulation following pharmacologic depletion of microglia (Huang et al., 2018), suggesting that surviving host microglia, which are absent in Csf1r −/− hosts, may be advantaged colonizers and/or limit parenchymal macrophage niche access.
Ontogeny regulates adoption of microglial identity, and is revealed by stable markers
Direct CNS injection allowed detailed and controlled exploration of fundamental differences between microglia and MLCs from multiple ontogenies across development. Although all MLC types shared similarities with microglia, we observed major effects of ontogeny on transcriptomic identity. YS-MLCs, which share a common progenitor with microglia, became more microglia-like than HSC-MLCs, notably in their expression of microglial signature genes. These findings contrast observations in lung, where engrafted yolk sac, fetal liver and bone marrow monocytes were reported to be near-identical to alveolar macrophages (van de Laar et al., 2016).
Since Tmem119 and other microglial markers may be expressed by HSC-MLCs, their presence does not assure microglial ontogeny. Here, we validated a panel of HSC ontogeny markers that are homogenously expressed among engrafted cells. Despite exposure to the same CNS signals, HSC-MLCs but not YS-MLCs/microglia express Ms4a7,Clec12a, Klra2, Lilra5. In contrast, HSC-MLCs do not express Gpr56, which is expressed by most if not all YS-MLCs/microglia. These ontogeny markers may augment current origin-mapping approaches such as parabiosis and genetic fate labeling. Interestingly, while Clec12a, Lilra5, and Klra2 are broadly expressed by circulating and tissue myeloid cells, Ms4a7 expression is restricted to macrophages (Heng et al., 2008). It is highly expressed by intestinal macrophages, which increasingly arise from HSCs in adulthood (Bain et al., 2014), raising the possibility that ontogeny markers may be valid outside of the CNS.
YS-MLCs were able to express the full complement of known microglial identity genes by 14 days, but HSC-MLCs were not. An important caveat to the Csf1r−/− system is the limited ability to study MLCs at long time points after transplantation due to host viability. Peripheral BM injection permitted comparison of long- to short-term resident HSC-MLCs to determine whether prolonged CNS residence further promoted microglial gene expression. We found surprisingly little difference between HSC-MLCs at 14 days versus 2-3 months using both unbiased and targeted analyses. In particular, HSC-MLCs remained unable to express Sall1/Sall3, did not further increase expression of signature genes such as Tmem119, P2ry12, Olfml3, and continued to express HSC ontogeny markers, some at increased levels. After 6 months of brain residence, HSC-MLCs continued to express Ms4a7, which also marked HSC-MLCs in both neonatal and adult tamoxifen-treated Cx3cr1-CreER;Csf1r fl/fl hosts after 2 months. Taken together, these observations suggest that prolonged CNS incubation is not sufficient to completely over-ride cell-intrinsic properties associated with ontogeny, an important consideration for the development of myeloid cell therapies for brain disease. The limitations of the Csf1r −/− model, or in fact any mouse model, call for future studies to determine if years-long incubation relevant for human lifespan is sufficient to fully induce microglial identity in HSC-derived cells.
HSC-MLCs resemble microglia in disease states
Sall1 and Nrros are central to microglial identity and when absent, microglia adopt an abnormal phenotype characterized by reduced expression of microglial signature genes and increased expression of macrophage markers associated with inflammation (Buttgereit et al., 2016; Wong et al., 2017). HSC-MLCs are unable to express Sall1 in Csf1r−/− hosts and resemble microglia from multiple disease states. Of particular interest, Apoe is the most highly expressed gene in HSC-MLCs – 4-20 times higher than in transplanted microglia or YS-MLCs. By in situ hybridization, HSC-MLC engrafted brains also showed increased Apoe levels in other brain cell types in addition to microglia. Apoe genotype remains one of the most important risk factors in Alzheimer disease (AD), and may directly drive microglial dyshomeostasis during neurodegeneration (Krasemann et al., 2017; Shi et al., 2017).
Intriguingly, HSC-MLC ontogeny markers from our study have previously been implicated in neurodegenerative processes. The Ms4a family of genes is associated with AD risk in large human studies (Hollingworth et al., 2011). In addition, Clec12a blockade attenuated a mouse model of multiple sclerosis (Sagar et al., 2017). Associations between HSC transcriptomes, ontogeny markers, and brain disease raise the tantalizing hypothesis that infiltrating cells could masquerade as microglia and contribute to brain malfunction. The future study of the functional consequences of HSC-MLCs and different ontogeny markers may be critical to understanding brain function in health and disease.
Transplantation of primary human cells and a custom anti-human Tmem119 antibody facilitate study of human microglia and MLCs
In vitro studies of human microglia are limited by loss of microglial identity in culture. Here, we found that cultured and acutely isolated primary cells from human blood and brain engraft, ramify, and express TMEM119 in humanized Csf1r −/− rodent brains. This shows that the murine brain is replete with factors to support survival of primary human macrophages and expression of a microglial protein not reliably detectable in culture (Abud et al., 2017; Gosselin et al., 2017). It also offers a new approach to studying human microglia in a living and highly controllable CNS environment, that we hope will facilitate future studies of disease. Since blood from patients with brain diseases is relatively accessible, direct CNS transplantation may be used to compare MLCs from healthy and disease states, complementing analogous approaches limited by lack of microglial gene expression in vitro (Gosselin et al., 2017; Ohgidani et al., 2014; Sellgren et al., 2016). We also validated a custom anti-human TMEM119 antibody for FACS sorting of pure human microglia from human and xenograft samples, for use by the microglia community.
Of equal importance, we confirmed that MS4A7 marks human HSC-MLCs but not microglia, suggesting that it is an ontogeny marker across species. The relevance of infiltrating myeloid cells to human brain disease is unresolved but of great consequence to the development of novel therapies. Since parabiosis and fate-labeling are not possible in human, MS4A7 may offer a feasible approach to detecting long term resident HSC-MLCs in the CNS. Importantly, we found MS4A7+/TMEM119+ cells in brains of neurodegenerative disease patients but not healthy controls, demonstrating the feasibility of this method and supporting the possibility that infiltrating myeloid cells may reside in the degenerating human brain.
Relevance of ontogeny and environment to microglia replacement therapy
Here, we provided new tools to study transplanted mouse and human brain macrophages, and applied them to relationships between environment, ontogeny, and tissue macrophage identity. In addition to the future studies discussed above, direct myeloid cell transplantation holds potential as a form of treatment for diverse neurological and psychiatric diseases. Our results reveal the importance of both ontogeny and CNS environmental cues to brain resident macrophage identity, as well as potential future avenues for understanding the CNS macrophage niche. These findings warrant future studies to reveal the functional and phenotypic consequences of these important differences, in order to make robust human microglial replacement therapies a reality.
STAR METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, F Chris Bennett (eph.bennett@gmail.com)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
All animal studies were performed with approval from the Stanford Administrative panel on Laboratory Animal Care in accordance with institutional and national regulations. With the exception of humanized mice, all mice were immunocompetent. All mice appeared healthy, received regular monitoring from veterinary and animal care staff, and were not involved in prior procedures or testing. No animals used in this study experienced health morbidities. Animals were housed in a non-barrier facility with 12 hour light/dark cycles at 23+/−2 degrees Celsius, in disposable ventilated cages with no more than 5 animals per cage. Animals were provided water and standard chow ad libitum. Cages and bedding were changed every 2 weeks. Csf1r −/− pups, which lack teeth, were supplemented with DietGel Boost soft food (ClearH20), sometimes diluted with water, starting at p12. Immune compromised Rag2−/− IL2rg−/−hMCSF+/+ Csf1r−/− mice were provided irradiated food, but otherwise treated identically. We observed no evidence of infection or illness in these mice. No sentinel animals tested positive for infectious agents on any rack used in this study.
Csf1r −/− (FVB.129X1-Csf1rtm1Ers) and Csf1r +/+ littermate animals on the FVB background were a generous gift from Dr. Richard Stanley, Albert Einstein College of Medicine, New York, USA. Adult WT FVB donor animals were bred from the identical starting strain. Timed FVB embryonic tissues were obtained from Charles River (Hollister, CA). For experiments using GFP-expressing donor cells, we back crossed the Csf1r KO allele onto the C57BL/6 strain using MaxBax speed congenics (Charles River) for 5 generations to > 99.7% C57BL/6, then crossed 1 further generation. We generated a Cx3Cr1CreER; Csf1rfl/fl line by intercrossing Jax 021212 and 021160. For experiments, we crossed Cx3Cr1CreER +/−; Csf1rfl/fl to Csf1rfl/fl animals. To test whether engraftment required CCR2, we use Ccr2 Rfp/Rfp homozygous donors (Jax 017586). We also used C57BL/6-Tg(CAG-EGFP)131Osb/LeySopJ (“Osb-GFP,” Jax 006567) as a source for GFP-tagged donor cells. To generate the Rag2−/− IL2rg−/− hMCSF+/+ Csf1r−/− mouse we crossed the Csf1rtm1Ers allele into C;129S4-Rag2tm1.1Flv Csf1tm1(CSF1)Flv Il2rgtm1.1Flv/J (Jax 017708). Animal lines were genotyped by Transnetyx (Cordova, TN) except pan-GFP animals which were genotyped by green florescence upon blue light exposure. We sometimes genotyped for Csf1r with in-house PCR (F1 5’-AGACTCATTCCAGAACCAGAGC-3’, F2 5’-CCGGTAGAATTCCTCGAGTCTA-3’, R1 5’-GAATTTGGAGTCCTCACCTTTG-3’). We verified Csf1r genotypes with second genotyping post-mortem. For all experiments, we used mixed sex donors and hosts at the specific fetal and adult developmental stages described in the method details below.
Human tissue samples
Human studies were approved by the Stanford Research Compliance Office and included IRB approval. Informed consent was obtained from all subjects. Postnatal brain macrophages used in transplantation experiments were obtained from n=5 pediatric and adult neurosurgical resections for epilepsy (7 mo female, 3 yo female, 14 yo male, 28 yo female, 42 yo male), via the Stanford Neurosurgery Tissue Bank. We used pieces of cortex outside the epileptic focus; these were described as normal on MRI and by the operating surgeon. Fetal brain tissue (n=2, 16-20 wk gestational age, biological sex could not be ascertained due to nature of clinical procedures preceding sample collection) was obtained from Stemexpress (Folsom, CA). Adult peripheral blood (n=6) was obtained from the Stanford Blood Center (Palo Alto, CA). To protect personal information from healthy blood donors, we did not request information on sex nor age. All donor information was anonymized prior to investigator acquisition of samples. For human in situ hybridizations, flash frozen post-mortem brain tissue was obtained from the Stanford Brain Bank and diagnosed by E.P., a board-certified neuropathologist, thawed and fixed overnight in 4% PFA, then cryoprotected in 30% sucrose prior to frozen sectioning. We assessed N=3 cases of severe AD pathology (80 yo male, 89 yo female also with diffuse Lewy Body disease, 79 yo female also with diffuse Lewy Body disease and cerebral amyloid angiopathy) and N=1 case of severe cerebrovascular disease (85 yo male), comparing to N=2 controls from temporal lobe epilepsy surgeries (49 yo male, 19 yo male).
METHOD DETAILS
General considerations for experimental design
Strategy for randomization and/or stratification
We performed no prospective treatment studies, and so did not have occasion to randomize a cohort to treatment arms. We performed all transplantation experiments identically, using all available Csf1r −/− hosts of age P0-4 on the day of donor tissue harvest. For BBB permeability stainings we included all animals of appropriate age on two separate days of harvest.
Blinding
Processing of RNAseq data, scoring of cell densities, percent TMEM119+ positive, and BBB integrity were performed blinded. RNAseq results were unblinded after mapping in order to group samples into donor cell types for subsequent analysis.
Sample-size estimations and method of computation
Sample-size estimations for RNAseq experiments were empirically based on past studies (Clarke et al., 2018; Zhang et al., 2014). For quantitative immunostaining experiments, we supplemented tissue from sequenced animals with littermates that we were not able to sort, or which did not yield adequate RNA for sequencing.
Inclusion and exclusion criteria of any data or subjects
For cell sorting, we included all surviving hosts for each transplantation experiment, and excluded animals that did not survive long enough to harvest. We excluded RNA samples from sorted cells when total yield was below the recommended input range for the Ovation V2 kit (Nugen), or when RIN was below 7 according to Bioanalyzer results (Agilent).
Microglia/MLC transplantation
For ICTs, P0-P4 Csf1r −/− pups and +/+ controls were injected as described previously (Bohlen et al., 2017) by hand using a pulled glass microcapillary tube in an electrode holder connected by silicon tubing to a syringe. One microliter containing a single cell suspension of donor cells in PBS was slowly injected bilaterally into cortex, 1-2 mm anterior and 2-3 mm lateral to lambda at a depth of 0.5-1 mm. Host animals were harvested after 14 days. Due to the constitutional fragility of Csf1r −/− animals, we harvested surviving animals of both sexes and pooled them for analyses. IP bone marrow injections were performed in P0-4 pups using an insulin syringe containing 20 μl of a single cell suspension in PBS. For adult CX3CR1-CreERT +/−;Csf1r fl/fl x Csf1r fl/fl ICTs, 5 month old mice were injected with 150mg/kg 4-hydroxy tamoxifen for two days. On day one, host animals were anesthetized with isofluorane, and five million bone marrow cells from Osb-GFP mouse donors were injected per hemisphere through two small burr holes bilaterally, approximately 1mm behind Bregma. The incision was closed using 4-0 vicryl sutures. Mice were treated with 5mg/kg subcutaneous carprofen analgesia and monitored closely post-operatively. Neonatal pups of CX3CR1-CreERT +/−; Csf1r fl/fl x Csf1r fl/fl mice were injected daily between P1-P4 with 200mg/kg 4-hydroxytamoxifen subcutaneously. These hosts were then transplanted via direct intracerebral injection on day 5.
Donor tissue preparation
Microglia
Microglia were isolated as previously described (Bennett et al., 2016) with the following substitutions: Mice were euthanized by CO2 asphyxiation and intravascularly perfused with 10 mL cold PBS, except for P5 animal brains which were carefully rinsed in PBS after dissection of the meninges. After myelin depletion using MACS beads, cell suspensions were positively selected for CD11b expression by magnetic bead separation using the MACS system (Miltenyi). Where applicable, mouse microglia were then cultured in TIC medium (described in (Bohlen et al., 2017)) supplemented with 10% heat inactivated FCS for 16-20 hours at 37 °C and 10%
CO2, and subsequently harvested for RNA isolation or intracranial transplantation. Cells were cultured on tissue culture plastic, and harvested by incubation on ice for 5-10 minutes, followed by 3-5 washes with ice cold FACS buffer (PBS, 25 mM HEPES, 2mM EDTA, and 2% FCS) and repeated pipetting. 20-100 x 103 cells were injected per host based on cell yields. For injection of human brain macrophages, we used identical methods, except that for 2 of the adult brain samples we omitted CD11B+ selection due to limited sample size. For postnatal samples, we injected 2-40x103 cells, and for fetal, 5 x 103. Where applicable, we cultured human fetal brain macrophages identically to mouse, with the exception of using human MCSF (Peprotech) instead of mouse, at identical concentration.
Yolk sac
We manually dissected the yolk sac from 4-6 pregnant females at E8 (Charles River) into cold PBS (counting plug date as E0), yielding approximately 30 yolk sacs per prep. We then gently homogenized with 5 slow triturations each across successively smaller outlets (p1000 tip p200 tip 18g needle 27g), and passed the homogenate over a 70 μm cell strainer. We next centrifuged for 5 min at 175g, resuspended in 40 μl cold PBS, and injected 1 μL per pup (~0.75 yolk sacs per mice) into 30-40 pups across 5-7 litters.
Fetal brain
We manually dissected fetal brain tissue from E12-13 embryos into cold PBS, dissociated by 20 gentle triturations using a p1000 tip, and passed over a 40 μm cell strainer. To enrich for macrophages, we either used CD11B+ MACS positive selection as described for microglia or FACS, using the sort strategy shown in figure S2B. We injected 2.5-8.5 x 103 cells per host as limited by cell yield.
Fetal liver
We manually dissected fetal liver from E13-14 embryos, homogenized by gentle trituration using a p1000, passed over a 40 μm strainer, and stained cells for FACS, using the sort strategy shown in figure S2C. We injected 5-20 x 103 cells per host as limited by cell yield.
Bone marrow
We dissected femurs and tibia from 4-8 week old FVB WT, Osb-GFP and Ccr2 KO (Rfp/Rfp) animals of mixed sex, isolated whole bone marrow by flushing bones with PBS, and lysed RBCs using ACK as previously described (Bennett et al., 2016). For ICTs, we injected 100-200 x 103 cells per host. For IP BMTs we injected 2-5x106 cells per animal.
Bone marrow monocytes
We isolated bone marrow as described above, then enriched for bone marrow monocytes using the bone marrow monocyte isolation kit (Miltenyi), which enriches for monocytes by depletion of other cell types. We then stained this enriched population for CD45, c-kit, Ly6c, NK1.1, CD3, B220, Il7ra, Siglecf, Ly6g using antibodies and dilutions shown in the key resources table, and sorted for monocytes using the sort strategy in figure S3F. We injected 0.5-2 x 106 cells per mouse by the intraperitoneal route.
Blood
We collected whole blood from 6-8 week old FVB WT animals of mixed sex in 0.5M EDTA coated syringes by cardiac puncture, pooled blood from all animals in 0.05M final concentration of EDTA, and centrifuged at 1000 RPM for 12 minutes at RT. We then collected the buffy coat layer, lysed RBCs in 10-20 volumes ACK buffer (Gibco) for 10 min at RT, centrifuged for 5 min at 200g, washed x 1 and then resuspended in PBS. We injected 100-200 x 103 cells per host.
Flow cytometry
MG/MLC isolation
We generated single cell suspensions of MG/MLCs using dounce homogenization and MACS myelin depletion as described above, then followed methods described in (Bennett et al., 2016). Briefly, we stained with a vital dye to exclude dead cells (Thermo Fisher), then for CD45, CD11b, and Tmem119 at dilutions shown in key resources table, adding RNAsin (Promega) and DNAse (Worthington) to our sort tubes, following the sort strategy shown in Figure S1C.
Donor tissue isolation and analysis by FACS
For FACS sorting of donor tissues described above, we used identical staining protocols except performed centrifugation steps at 300g for 5 minutes, and stained for different markers as described above. We performed all flow cytometry experiments using a large diameter (100 μm) nozzle at rates of 1-2.5 on BD FACSAria instruments in the Stanford Shared FACS Facility core. We sorted into FACS buffer for transplantation experiments. We used Flowjo software (Treestar) to analyze and visualize data. Gating strategies are shown wherever applicable in Figures S1, S2 and S7.
RNA and DNA extraction
We sorted engrafted cells directly into RNA extraction buffer. For BMT experiments we used Trizol LS (Sigma) according to manufacturer’s protocols, collecting RNA and sometimes genomic DNA for genotyping of sorted cells. For one MG ICT experiment we sorted into RLT Plus (Qiagen) and isolated both RNA and genomic DNA using the Allprep Micro kit. For remaining experiments we sorted into RLT buffer (Qiagen) and isolated RNA using the RNAeasy micro kit with on column DNAse digestion. We measured RNA quality by Agilent Bioanalyzer, and only processed samples with RIN >7. We found that in order to reliably obtain adequate quantity and quality of RNA, we required 30 x 103 sorted cells.
RNAseq library construction and sequencing
We constructed and quality controlled libraries as described previously (Bennett et al., 2016), using the Nugen Ovation RNA-seq system V2, and the NEB Next Ultra RNAseq kit for Illumina, with 9-10 cycles of PCR enrightment. High quality libraries were sequenced by Miseq (Illumina), using 75 bp paired end reads. Aside from one WT control sample with 0.9x106, we obtained at least 1x106 paired reads per sample, with a range of 1-6x106. At least 70% of reads were mapped in all samples.
Anti-human Tmem119 antibody generation
To generate mouse anti-human TMEM119 FACS antibodies, we cloned the extracellular domain (ECD) after the signal peptide for human Tmem119 (corresponding residues: 26-95) into a custom pMAL vector for periplasmic MBP-ECD-8xHis fusion expression (gift from A. Ring, Yale University, New Haven, CT). We purified recombinant fusion proteins by Ni-NTA columns. BioLegend (San Diego, CA) immunized mice with the recombinant ECD proteins and screened positive multi-clone supernatants by ELISA. We tested multi- and then single-clone supernatants first by staining HEK cells transfected with His-tagged human Tmem119 expressed in pCMV-SPORT6 mammalian expression vector. For promising clones we verified that staining was blocked by pre-incubation with immunizing peptide, and tested them on primary human brain and blood tissue samples. We found that clone A16075D stained 70-98% of CD45+/CD11B+ human brain cells with the highest signal to noise ratio; public release of clone A16075D by Biolegend is forthcoming.
Tissue Immunostaining
We performed immunostaining on 4% PFA perfusion- or immersion-fixed samples depending on whether whole animals or brain pieces were processed. Samples were cryoprotected in 30% sucrose-PBS, embedded in OCT (Fisher), cryosectioned (12-16 μm), mounted on Superfrost Plus slides (Fisher) and stored at -80 until use. For mouse TMEM119 and IBA1 staining, we dried slides at 60 °C, rehydrated in PBS, blocked f or 1 hour at room temperature (RT) in PBSTx (PBS with 0.3-0.5% Triton X-100) with 10% serum. We then incubated with primary antibodies in PBSTx/1% serum (staining buffer) overnight at 4 °C. After washing, we incubated slides in staining buffer with Alexa-conjugated secondary antibodies (Life Technologies) for 2 hr at RT, washed, Antibodies for MS4A7 (Atlas) and human TMEM119 (Abcam) required antigen retrieval prior to blocking. After rehydration, we boiled samples for 4 minutes in 10mM sodium citrate/0.05% Tween 20, pH 6, then incubated for 15 more minutes at RT in hot buffer. For MS4A7 peptide blocking studies, we obtained immunizing peptide from the antibody manufacturer (Atlas). We preincubated primary antibody with 40 molar excess of blocking peptide in 31.5 μl PBS overnight at 4 °C prior to staining. To measu re percent of parenchymal IBA1+ cells that were also TMEM119+, we provided numerically coded images obtained by FCB to FY, who also was not told the purpose of the experiment, but was instructed to mark all green cells (IBA1 channel), and then the number of these that had any co-localized red (TMEM119 channel) staining. To measure cell density, we counted the number of nucleated TMEM119 positive cells in the same dataset, expressed as cells per unit area. To measure albumin and IgG accumulation in the brain parenchyma, we stained 30 micron sections from animals perfused at approximately 70% of cardiac output (0.07 x mass in grams) for albumin (Abcam) or IgG (Life Technologies), and compared to unstained tissue (IgG) or secondary only (albumin) controls processed in parallel for each sample. We examined all samples for qualitative evidence of local staining, and quantified mean florescence intensity in identical ROIs from the cortex. We expressed positive staining as a fold change over the MFI from negative control samples, propagating error accordingly.
RNA in situ hybridization
We performed in situ hybridizations on fixed frozen samples using the RNAscope system (ACDbio) with RNAscope 2.5 HD Duplex Reagent kit for colorimetric and Fluorescent Multiplex Reagent kit V2 for fluorescent development, according to manufacturer’s protocols using TSA reagents (Perkin Elmer). When staining concurrently for IBA1 protein, we used our standard immunostaining protocol, omitting Triton-x100, with fluorescent secondary antibodies or HRP-conjugated secondary antibodies (Jackson Immuno-research) and DAB development kit (Thermo Scientific). Sections were counterstained with hematoxylin (colorimetric) or DAPI (fluorescent) for nuclei.
Image acquisition and processing
We acquired epifluorescence images using an Axio Imager M1 (Zeiss), except for stitched images in Figures 2, 3, and S2 for which we used a BZ-X700 Fluorescent Microscope (Keyence), confocal images for which we used an LSM710 (Zeiss) and DNA gels for which we used an Alpha Imager (Innotech). We acquired color images using an Axio Imager A2 (Zeiss). We analyzed images in Fiji (https://imagej.net/Fiji) or ICY (icy.bioimageanalysis.org), adjusting for brightness and black values (notes and raw images available upon request). We performed no other image math or processing.
Analysis of RNAseq data
We mapped, assembled transcripts, estimated FPKM, and analyzed differential gene expression as described previously (Bennett et al., 2016), using the tuxedo pipeline and edgeR, and identical reference genome. To avoid misinterpretation of expression differences in lowly expressed genes, we focused most analyses on genes with moderate or high expression (FPKM >20), and only interpreted gene expression differences with log2(FC)>1 and FDR <0.05. To generate a correlation heatmap, we selected the top 1000 most variant genes across the datasets studied and used the R package gplots cor function to generate a map of Spearman coefficients for each comparison. When other published datasets were used, we normalized their reads to the average by a simple scalar. To make heatmaps, we used gplots heatmap.2 function. For PCAs, we used Clustvis. We ran analyses on the top 2500 most variant genes based on log2(FPKM+1 values), applying unit variance scaling to rows, and using SVD with imputation to calculate principal components. In PCA plots, ellipses predict cluster boundaries with probability 0.95. We used JVenn to create Venn diagrams showing shared and distinct differential gene expression. For hierarchical clustering and p-value estimation, we used the pvclust package (Suzuki and Shimodaira, 2014). We clustered data based on Spearman correlation of the top 1000 most variant genes (using the unofficial version available at www.sigmath.es.osaka-u.ac.jp/shimo-lab/prog/pvclust/), bootstrapping 10000x to arrive at the final p value. GSEA was performed using GSEA software V3, running 1000 permutations by phenotype, calculating weight enrichment and ranking genes by Signal2Noise. ALS UP, AD UP, LPS UP 1/2, DEV UP 1/2, CULTURE UP, and CULTURE DOWN were obtained from (Bohlen et al., 2017). The Sall1 UP gene list represents the top 49 most upregulated genes in Sall1 KO microglia by fold change with FDR < 0.05 from (Buttgereit et al., 2016). The NRROS UP gene list contains genes with log2 (NRROSKO/WT)>1.5 in sorted microglia/macrophages and adjusted p-value <0.05 from (Wong et al., 2017). For the correlation plot in panel 5B, we merged gene lists from Sall−/− microglia (log2(FC/WT)>1 or <-1) with those from HSC-MLCs, omitting genes for which edgeR was unable to obtain Log2FC values or did not appear in 1 of the 2 datasets.
Replicates
Number of biological replicates used in immunostaining and RNAseq experiments are specified below:
Adult microglia; Immunostaining: 6, RNAseq: 2
P5 microglia; Immunostaining: 5, RNAseq: 2
Cultured microglia; Immunostaining: 7, RNAseq: 4
Yolk sac; Immunostaining: 6, RNAseq: 3
Fetal Brain; Immunostaining: 3, RNAseq: 5
Bone marrow ICT; Immunostaining: 7, RNAseq: 7
Blood; Immunostaining; 9, RNAseq: 3
Bone marrow IP; Immunostaining; 9 RNAseq: 5
WT control; Immunostaining; 7, RNAseq: 7
QUANTIFICATION AND STATISTICAL ANALYSIS
To quantify gene expression from RNAseq data for comparison across samples (as shown in Figures 4G, 5C, 6A, S4A, S5A-C, Tables S1-5), we used FPKM values as generated by the Tuxedo pipeline. To measure relative gene expression between samples, we used Log2(fold change) as measured in edgeR (Figures 1C, 1D, 4D-F, 5B, S1G, Tables S1-5, and supplemental data sheets). We defined the statistical significance of differential gene expression between samples by false discovery rate (FDR) as calculated by edgeR, at a significance cutoff of 0.05 (Figures 1C, 1D, 4A, 4D-G, 5C, 6A, S1G, S5A-C, Tables S1-5, Supplemental Data Sheets). In some cases we also provided p-values as reported by edgeR (Figures S2, S3, Supplemental Data Sheets). We sometimes used FPKM cutoffs to focus analysis on genes of moderate or higher expression, as indicated in figure legends and results. To measure statistical significance of differences in macrophage density, we used Graphpad to perform either a student’s t test (unpaired, two tails, Figure S1B), applying Dunnett’s test for multiple comparisons, or ANOVA (Figure S2E) with post-hoc analysis of pairwise differences, adjusting for multiple comparisons. To quantify the correlation between Sall1−/− microglial and HSC-MLC gene expression, we performed linear regression in Graphpad and reported r and p values. To measure statistical significance of differences in average florescence intensity of IgG and Albumin stains (Figure S3C), we used Graphpad to perform student t tests (unpaired, two tails). All error bars (Figures 4G, 5C, 6A, S1B, S2E. S3C, S5A-C) represent standard error of the mean, calculated either in Excel or Graphpad. To estimate statistical significance of hierarchical clustering branches shown in Figure 4C, we calculated p-values using PVclust as described.
DATA AND SOFTWARE AVAILABILITY
All raw sequencing data are publicly available at National Center for Biotechnology Information (NCBI) BioProject, https://www.ncbi.nlm.nih.gov/bioproject (accession no. PRJNA453419). Differential gene expression by EdgeR is available in Data Sheet 1. Mapped data (expressed as FPKM), and gene lists used in figure 5 are available in Data Sheet 2.
Supplemental Data Spreadsheets
Data sheet 1: Differential gene expression comparisons by edgeR.
Data sheet 2: FPKM values for individual replicates, group averages with SEM, and GSEA lists.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Donkey anti-rabbit BV421 IgG (1:300) | Biolegend | Cat# 406410; RRID:AB_10897810 |
| Rat monoclonal anti-Mouse CD45 (1:200) | ebiosciences | Cat# 25-0451-82; RRID:AB_469625 |
| Anti-CD11b microbeads | Miltinenyi | Cat# 130-049-601 |
| Rat anti-mouse Ly6g PercpCy5.5 (1:100) | Biolegend | Cat# 127615; RRID:AB_1877272 |
| Rat anti-Mouse f4/80 APC (1:20) | Biorad | MCA497APC; RRID:AB_324435 |
| PerCP/Cy5.5 anti-mouse/human CD45R/B220 (1:100) | Biolegend | Cat#103235; RRID:AB_893356 |
| PE-CyTM 7 mouse anti-Human CD45 (10ul/test) | BD Biosciences | Cat#557748; RRID:AB_396854 |
| Anti-Mouse Tmem119 (0.25-0.5 ug/mL) Clone [28-3] | Abcam | Cat# 209064 RRID:AB_2728083 |
| Rabbit anti-human Tmem119 (1:200) | Abcam | Cat# 185333; RRID:AB_2687894 |
| PerCP/Cy5.5 anti-mouse/human CD11b (1:100-200) | Biolegend | Cat#101228 RRID:AB_893232 |
| PerCP/Cy5.5 anti-mouse CD3 (1:100) | Biolegend | Cat#100217; RRID:AB_1595597 |
| Human BD Fc Block (20 ul/300ul cells) | BD Biosciences | Cat#564220 RRID:AB_2728082 |
| Brilliant Violet 421 goat anti-mouse IgG (1:250) | Biolegend | Cat#405317; RRID:AB_10900419 |
| PerCP/Cy5.5 anti-mouse CD127(IL-7Ra) (1:100) | Biolegend | Cat#135021; RRID:AB_1937274 |
| BV421 Goat anti-rabbit IgG polyclonal (1:300) | BD Biosciences | Cat#565014; RRID:AB_2716308) |
| PE anti-mouse CD117(c-kit) (1:200) | Biolegend | Cat#105807; RRID:AB_313216 |
| Purified rat anti-mouse CD16/CD32 (10 ul/300ul stain) | BD Biosciences | Cat#553142 RRID:AB_394657 |
| PerCP/Cy5.5 anti-mouse Ly-6G (1:100) | Biolegend | Cat#127615; RRID:AB_1877272 |
| PerCP/Cy5.5 anti-mouse NK-1.1 (1:100) | Biolegend | Cat#108727; RRID:AB_2132706 |
| Purified mouse IgG2b, k Isotype Ctrl (concentration matched to stain) | Biolegend | Cat#401201 RRID:AB_326631 |
| Purified anti-human TMEM119 clone A16075D, (0.3 ug/ml) | Biolegend | Forthcoming |
| Goat anti-mouse/human IBA1 (1:500) | Abcam | Cat# 5076; RRID:AB_2224402 |
| Anti-Human cytoplasm (2ug/mL) | Takara | Cat# Y40410 RRID:AB_2632385 |
| Brilliant Violet 421 anti-mouse Ly-6C (1:100) | Biolegend | Cat#128031; RRID:AB_2562177 |
| Rabbit polyclonal anti-Human Ms4a7 (1:30) | Atlas | Cat# HPA017418; RRID:AB_1854137 |
| PE anti-mouse/human CD11b (1:100) | Biolegend | Cat#101207; RRID:AB_312790 |
| PerCP/Cy5.5 anti-mouse/human CD11b (1:100) | Biolegend | Cat#101228; RRID:AB_893232 |
| Anti-mouse CD45 PE-Cyanine7 (1:200) | eBioscience | Cat#25-0451-82 RRID: AB_469625 |
| Goat anti-mouse IgG Alexa 647 (1:500) | Life Technologies | Cat#A21236 RRID:AB_2535805 |
| Rat anti-mouse albumin (1:750) | Abcam | Cat#Ab24325 RRID:AB_448009 |
| Critical Commercial Assays | ||
| AllPrep Micro Kit | Qiagen | Cat#80284 |
| RNAeasy Micro Kit | Qiagen | Cat# 74004 |
| NEB Next Ultra RNAseq Kit | New England Biolabs | Cat# E7530S |
| RNAscope® Fluorescent Multiplex Detection Reagents | ACDbio | Cat#320851 |
| RNAscope Multiplex Fluorescent Detection Kit v2 | ACDbio | Cat# 323110 |
| RNAscope 2.5 HD Duplex Detection Reagents Kit | ACDbio | Cat# 322430 |
| RNAscope® H202 & Protease Plus Reagents | ACDbio | Cat#322330 |
| RNAscope® 2.5 Duplex Positive Control probe | ACDbio | Cat# 321641 |
| TSA detection reagents; TSA plus Fluorescein | PerkinElmer | Cat# NEL741001KT |
| TSA detection reagents; TSA plus Cyanine 3 | PerkinElmer | Cat# NEL744E001KT |
| TSA detection reagents; TSA plus Cyanine 5 | PerkinElmer | Cat# NEL513001KT |
| Ovation-RNA-seq system V2 | NuGEN | Cat# 7102 |
| Probe:Hs-Tmem119 | ACDbio | Cat# 478911 |
| Probe: Mm-Ms4a7-C2 | ACDbio | Cat#314601-C2 |
| Probe: Mm-Clec12a-C2 | ACDbio | Cat# 514351-C2 |
| Probe: Mm-Sall1 | ACDbio | Cat# 469661 |
| Probe: Mm-Tmem119-C2 | ACDbio | Cat# 472901-C2 |
| Probe: Hs-Sall1 | ACDbio | Cat# 514331 |
| Bone marrow monocytes isolation kit | Miltenyi Biotec | Cat# 130-100-629 |
| Biological Samples | ||
| FVB Embryonic tissues | Charles River | Strain code: 207 |
| Fetal brain tissue (human) | Stemexpress | N/A |
| Adult peripheral blood samples | Stanford Blood Center | N/A |
| Intraoperative brain tissue | Stanford Brain Bank | N/A |
| Post-mortem human brain samples | Stanford Brain Bank | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ms4a7 Peptide (40 molar excess) | Atlas | Cat# APrEST72174 |
| TIC medium | Bohlen et al., 2017 | N/A |
| Heat inactivated FCS | Gibco | Cat#10437-028 |
| ACK buffer | Gibco | Cat#A10492-01 |
| RNAsin | Promega | Cat# N2615 |
| DNAseI | Worthington | Cat# DPRFS |
| Trizol LS | Sigma | Cat# T3934 |
| RLT buffer | Qiagen | Cat# 79216 |
| RLT Plus | Qiagen | Cat# 1053393 |
| DAB development Kit | Thermo Scientific | Cat# 34002 |
| Z-4-Hydroxytamoxifen | Sigma-Aldrich | Cat# H7904 |
| Human TGFβ2 | Peprotech | Cat#100-35B |
| Human Mcsf | Peprotech | Cat#300-25 |
| Mouse Mcsf | Peprotech | Cat#315-02 |
| Ovine wool cholesterol | Avanti Polar Lipids | Cat# 700000P |
| LIVE/DEAD Green (1 ul/1ml cells) | Thermo Fisher | Cat# L34969 |
| Deposited Data | ||
| Microglia and MLC RNA-seq | This Paper | http://www.ncbi.nlm.nih.gov/bioproject/453419 |
| Gene lists ALS UP, AD UP, LPS UP 1/2, DEV UP 1/2, CULTURE UP, and CULTURE DOWN | (Bohlen et al., 2017) | https://www.cell.com/action/showImagesData?pii=S0896-6273%2817%2930400-2 |
| Gene list SALL1 UP | (Buttgereit et al., 2016) | https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5077/ |
| Gene list NRROS UP | (Wong et al., 2017) | https://www.nature.com/articles/ni.3743#supplementary-information |
| Mouse reference genome UCSC annotation mm9 | UCSC Genome Browser | http://hgdownload.soe.ucsc.edu/downloads.html#mouse |
| Immune cell RNA-seq data | (Lavin et al., 2014); (Zhang et al., 2014) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63340; https://web.stanford.edu/group/barres_lab/brain_rnaseq.html |
| Experimental Models: Organisms/Strains | ||
| Mouse: FVB.129X1-Csf1rtmErs (also back crossed to C57BL6 in this paper) | (Dai et al., 2002)/Dr. Richard Stanley | RRID:MGI:3810944 |
| Mouse: FVB/NCr | Charles River | RRID:MGI:2160605 |
| Mouse: C57BL/6-Tg (CAG-EGFP)131Osb/LeySopJ- | The Jackson Laboratory | RRID:IMSR_JAX:006567 |
| Mouse: Rag2tm1.1Flv CSF1tm1 (CSF1)Flv IL2rgtm1.1FLv/J | The Jackson Laboratory | RRID:IMSR_JAX:017708 |
| Mouse: Rag2tm1.1Flv CSF1tm1 (CSF1)Flv IL2rgtm1.1FLv/J X Csf1rtmErs | This paper | N/A |
| Mouse: Cx3cr1CreER; B6 | The Jackson Laboratory | RRID:IMSR_JAX:021160 |
| Mouse: Csf1rflox; B6 | The Jackson Laboratory | RRID:IMSR_JAX: 021212 |
| Mouse: Cx3cr1CreER;Csf1rflox ; B6, CX3Cr1CreER mice were bred with Csf1rflox mice | This paper | N/A |
| Mouse: Ccr2RFP; B6 | The Jackson Laboratory | RRID:IMSR_JAX: 017586 |
| Mouse: C57BL6 WT | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Oligonucleotides | ||
| Primers for Csf1r mice genotyping: | ||
| F1; 5′-AGACTCATTCCAGAACCAGAGC-3′ | This Paper | N/A |
| F2; 5′-CCGGTAGAATTCCTCGAGTCTA-3′ | This Paper | N/A |
| R1; 5′-GAATTTGGAGTCCTCACCTTTG-3′ | This Paper | N/A |
| Human Tmem119-6xhis Forward primer 5′- AAAAGTCGACGCCGCCACCAtggtttcggcggcag 3′ | This Paper | N/A |
| Human Tmem119-6xhis Reverse primer 5′ -TTTTGCGGCCGCTTAGTGGTGATGGTGATGATGACCAGAACCACCGACACTGGGGTGGACACTGC - 3′ | This Paper | N/A |
| Human Tmem119-ECD Forward primer 5′- AaaaaaggatccCgctctgtgcccctg -3′ | This Paper | N/A |
| Human Tmem119-ECD Reverse primer 5′ - TTT TTT TTG CGG CCG CCA TCA CGT ACT GGC -3′ | This Paper | N/A |
| Software and Algorithms | ||
| edgeR | (Robinson et al., 2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Tuxedo Pipeline | Galaxyproject.org | |
| ICY | http://icy.bioimageanalysis.org/ | |
| Graphpad Prism (version 7.0) | GraphPad | https://www.graphpad.com |
| Clustvis | http://biit.cs.ut.ee/clustvis_large/ | |
| Flowjo (version 10.4) | Treestar | https://www.flowjo.com |
| R (version 3.3.1) | The R foundation | https://www.r-project.org |
| Miseq | Illumina | N/A |
| JVenn | (Bardou et al.,2014) | http://jvenn.toulouse.inra.fr/app/example.html |
| Fiji | https://imagej.net/Fiji | |
| PVclust | (Suzuki et al., 2014) | www.sigmath.es.osaka-u.ac.jp/shimo-lab/prog/pvclust/ |
| GSEA V3 | http://software.broadinstitute.org/gsea/index.jsp | |
| Other | ||
| Dounce homogenizer, 7 ml | Wheaton | 357542 |
| Microcapillary tube | WPI | Cat#1B100f-4 |
| DietGel Boost | ClearH20 | Cat#72-04-5022 |
TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and Virus Strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al., 2011 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al., 2016) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological Samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children's Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical Commercial Assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | Perkin-Elmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; and Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental Models: Cell Lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al., 2013 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal. | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
Supplementary Material
Highlights.
Brain signals induce and sustain homeostatic gene expression in microglia
Hematopoietic stem cell (HSC) derived macrophages attain a microglia-like identity
Stable markers and gene signatures betray parenchymal macrophage origin
Macrophages with HSC origin markers are found in human neurodegeneration
Bennett et al create a macrophage transplantation system to measure how origin and brain environment contribute to microglial identity. Although diverse macrophage types survive in the brain, only those sharing developmental origins with microglia express microglial genes normally.
Acknowledgments
We thank the B.A.B. laboratory, particularly C. Bohlen, S. Sloan, L. Clarke, S. Liddelow, L. Sun, T. Li for feedback and consultation; G. Smerin, K. Guttenplan for tissue samples; L.B. Torres for Ccr2 Rfp and R. Stanley for Csf1r −/− animals; T. Wyss-Coray and H. Nakauchi for feedback and mentoring; the Stanford Shared FACS Facility, supported by NIH S10RR025518-01; A. Olson and the Stanford Neuroscience Microscopy Service, supported by NIH NS069375; the Stanford Alzheimer Disease Research Center for providing human samples, supported by NIH P50AG047366; P. Taylor, C. Wang, and K. Cohane for TMEM119 antibody collaboration. This work was funded by NIH R37DA01504317 (to B.A.B.); NRSA predoctoral Fellowship F31 NS078813 (to M.L.B.); training Grants T32MH019938-22 and K08MH112120 (to F.C.B.), K08NS901527 and R01CA216054-01 (to M.H.G.) and K08 NS075144 (to G.A.G.); JPB Foundation, AMRF, and Vincent and Stella Coates. To B.A.B, who forever changed our lives, science, and society: thank you, and we miss you.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions
Conceptualization, FCB, BAB, MLB; Methodology, FCB, MLB; Formal Analysis, FCB, MLB; Investigation FCB, MLB, SBM, FY; Resources GAG, MHG, EDP; Data Curation FCB, Writing – Original Draft, FCB; Writing – Review and Editing, FCB, MLB; Visualization, FCB, MLB; Supervision BAB, FCB; Project Administration FCB; Funding Acquisition BAB, FCB, MLB.
Declaration of interests
The authors declare no competing interests
References
- Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron. 2017;94:278–293.e279. doi: 10.1016/j.neuron.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FMV. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Bain CC, Bravo-Blas A, Scott CL, Gomez Perdiguero E, Geissmann F, Henri S, Malissen B, Osborne LC, Artis D, Mowat AM. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nature Immunology. 2014;15:929–937. doi: 10.1038/ni.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113:E1738–E1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158–1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres Ben A. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined- Medium Cultures. Neuron. 2017;94:759–773.e8. doi: 10.1016/j.neuron.2017.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruttger J, Karram K, Wörtge S, Regen T, Marini F, Hoppmann N, Klein M, Blank T, Yona S, Wolf Y, et al. Genetic Cell Ablation Reveals Clusters of Local Self- Renewing Microglia in the Mammalian Central Nervous System. Immunity. 2015;43:92–106. doi: 10.1016/j.immuni.2015.06.012. [DOI] [PubMed] [Google Scholar]
- Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, Nishinakamura R, Becher B, Greter M. Sall1 is a transcriptional regulator defining microglia identity and function. Nature Immunology. 2016;17:1397–1406. doi: 10.1038/ni.3585. [DOI] [PubMed] [Google Scholar]
- Chiu IM, Morimoto ETA, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, et al. A Neurodegeneration-Specific Gene-Expression Signature of Acutely Isolated Microglia from an Amyotrophic Lateral Sclerosis Mouse Model. Cell Rep. 2013;4:385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. 2018;115:E1896–E1905. doi: 10.1073/pnas.1800165115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, Ji E, Goldman DH, Smirnov I, Geraci N, Acton S, et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med. 2018;47 doi: 10.1084/jem.20180247. jem.20180247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, Stanley ER. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99:111–120. doi: 10.1182/blood.v99.1.111. [DOI] [PubMed] [Google Scholar]
- Dzenko KA, Andjelkovic AV, Kuziel WA, Pachter JS. The chemokine receptor CCR2 mediates the binding and internalization of monocyte chemoattractant protein-1 along brain microvessels. Journal of Neuroscience. 2001;21:9214–9223. doi: 10.1523/JNEUROSCI.21-23-09214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, et al. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017;356 doi: 10.1126/science.aal3222. eaal3222-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Heng TSP, Painter MW, Immunological Genome Project Consortium The Immunological Genome Project: networks of gene expression in immune cells. Nature Immunology. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, Khoury JE. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013:1–12. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC, et al. C-Myb+ Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity. 2015;42:665–678. doi: 10.1016/j.immuni.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nature Genetics. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, Wang J, Zhao L, Liang YX, Wu T, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. 2018;21:530–540. doi: 10.1038/s41593-018-0090-8. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, Fatimy El R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017;47:566–581.e569. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen K, Zhu L, Pollard JW. Conditional deletion of the colony stimulating factor-1 receptor (c-fms proto-oncogene) in mice. Genesis. 2006;44:328–335. doi: 10.1002/dvg.20219. [DOI] [PubMed] [Google Scholar]
- Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nature Reviews Immunology. 2017;123:229. doi: 10.1038/nri.2017.125. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, Jacome-Galarza CE, Händler K, Klughammer J, Kobayashi Y, et al. Specification of tissue-resident macrophages during organogenesis. Science. 2016;353:aaf4238–aaf4238. doi: 10.1126/science.aaf4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada González F, Perrin P, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353:aad8670–aad8670. doi: 10.1126/science.aad8670. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgidani M, Kato TA, Setoyama D, Sagata N, Hashimoto R, Shigenobu K, Yoshida T, Hayakawa K, Shimokawa N, Miura D, et al. Direct induction of ramified microglia-like cells from human monocytes: Dynamic microglial dysfunction in Nasu-Hakola disease. Sci Rep. 2014;4 doi: 10.1038/srep04957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiguero EG, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2014:1–17. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priller J, Flugel A, Wehner T, Boentert M, Haas CA, Prinz M, Fernandez-Klett F, Prass K, Bechmann I, de Boer BA, et al. Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat Med. 2001;7:1356–1361. doi: 10.1038/nm1201-1356. [DOI] [PubMed] [Google Scholar]
- Rathinam C, Poueymirou WT, Rojas J, Murphy AJ, Valenzuela DM, Yancopoulos GD, Rongvaux A, Eynon EE, Manz MG, Flavell RA. Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood. 2011;118:3119–3128. doi: 10.1182/blood-2010-12-326926. [DOI] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010;5:e13693. doi: 10.1371/journal.pone.0013693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar D, Singh NP, Ginwala R, Huang X, Philip R, Nagarkatti M, Nagarkatti P, Neumann K, Ruland J, Andrews AM, et al. Antibody blockade of CLEC12A delays EAE onset and attenuates disease severity by impairing myeloid cell CNS infiltration and restoring positive immunity. Sci Rep. 2017;7:409–416. doi: 10.1038/s41598-017-03027-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23:1018–1027. doi: 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, Lippens S, Abels C, Schoonooghe S, Raes G, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nature Communications. 2016;7:10321. doi: 10.1038/ncomms10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellgren CM, Sheridan SD, Gracias J, Xuan D, Fu T, Perlis RH. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. 2016:1–8. doi: 10.1038/mp.2016.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523–527. doi: 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Shimodaira H. pvclust: Hierarchical Clustering with p-Values via Multiscale Bootstrap Resampling. R package version 1.2-2. 2014 URL http://CRAN.R-project.org/package=pvclust.
- van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, Hoffmann E, Beyaert R, Saeys Y, Lambrecht BN, et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity. 2016;44:755–768. doi: 10.1016/j.immuni.2016.02.017. [DOI] [PubMed] [Google Scholar]
- Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, Brawek B, Thal DR, Charo IF, Heppner FL, Aguzzi A, et al. Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci U S A. 2012;109:18150–18155. doi: 10.1073/pnas.1210150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Noubade R, Manzanillo P, Ota N, Foreman O, Hackney JA, Friedman BA, Pappu R, Scearce-Levie K, Ouyang W. Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nature Immunology. 2017;18:633–641. doi: 10.1038/ni.3743. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. Journal of Neuroscience. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.