

Supplemental Digital Content is available in the text.
Keywords: atherosclerosis, β-catenin, inflammation, low-density lipoprotein receptor, macrophage
Abstract
Objective—
The Wnt/β-catenin signaling is an ancient and evolutionarily conserved pathway that regulates essential aspects of cell differentiation, proliferation, migration and polarity. Canonical Wnt/β-catenin signaling has also been implicated in the pathogenesis of atherosclerosis. Macrophage is one of the major cell types involved in the initiation and progression of atherosclerosis, but the role of macrophage β-catenin in atherosclerosis remains elusive. This study aims to investigate the impact of β-catenin expression on macrophage functions and atherosclerosis development.
Approach and Results—
To investigate the role of macrophage canonical Wnt/β-catenin signaling in atherogenesis, we generated β-cateninΔmyeLDLR−/− mice (low-density lipoprotein receptor–deficient mice with myeloid-specific β-catenin deficiency). As expected, deletion of β-catenin decreased macrophage adhesion and migration properties in vitro. However, deficiency of β-catenin significantly increased atherosclerotic lesion areas in the aortic root of LDLR−/− (low-density lipoprotein receptor–deficient) mice without affecting the plasma lipid levels and atherosclerotic plaque composition. Mechanistic studies revealed that β-catenin can regulate activation of STAT (signal transducer and activator of transcription) pathway in macrophages, and ablation of β-catenin resulted in STAT3 downregulation and STAT1 activation, leading to elevated macrophage inflammatory responses and increased atherosclerosis.
Conclusions—
This study demonstrates a critical role of myeloid β-catenin expression in atherosclerosis by modulating macrophage inflammatory responses.
The Wnt signaling pathway plays essential roles in the regulation of cell proliferation, migration, and differentiation.1,2 Wnt signaling is traditionally classified into the canonical β-catenin-dependent pathway (Wnt/β-catenin pathway) and noncanonical β-catenin-independent pathway.2 For the canonical Wnt/β-catenin pathway, Wnt ligands bind to the membrane-spanning receptor proteins Frizzled and LRP (low-density lipoprotein receptor–related protein) family and subsequently trigger the phosphorylation of the Dishevelled protein, which further activates a signaling cascade that results in the stabilization and nuclear localization of β-catenin.2 Nuclear-localized β-catenin interacts with the T cell–specific factor/lymphoid enhancer–binding factor to induce the transcription of canonical Wnt signaling target genes. In the absence of Wnt ligands, several kinases including GSK3β (glycogen synthase kinase-3β) and priming kinase CKIα (casein kinase Iα) can phosphorylate the degron motif of β-catenin to prime it for β-TrCP-mediated ubiquitination and degradation.2,3
Canonical Wnt/β-catenin signaling has been implicated in the pathogenesis of human atherosclerosis.4 For example, the levels of active β-catenin were increased in disrupted atherosclerotic plaques as compared with stable plaques.5 The expression levels of LRP6 were also reduced in carotid atherosclerotic lesions of human LRP6 1062V variant carriers, and 1062V variant of LRP6 and carotid atherosclerosis were strongly associated in hypertensive patients, indicating that reduced activation of canonical Wnt signaling may contribute to the increased atherogenesis.6 Animal studies have also identified the activation of β-catenin in vascular endothelium before and during early atherogenesis.7 Activation of β-catenin can increase vascular smooth muscle cell proliferation and migration and has been suggested as a key component of atherosclerotic physiology.8–11 By contrast, Wnt signaling inhibitor Sclerostin inhibited angiotensin II–induced aortic aneurysm and atherosclerosis formation in ApoE−/− (apolipoprotein E–deficient) mice.12 However, other studies found that excess plasma lipid can alter LRP5 expression in vessel wall,13 and LRP5 deficiency can downregulate Wnt signaling and promote aortic lipid infiltration in mice,14 suggesting that canonical Wnt signaling may exert a protective defense mechanism against atherosclerosis development.
Macrophage accumulation within the vascular wall is a hallmark of atherosclerosis, yet the role of macrophage Wnt/β-catenin signaling in atherosclerosis remains elusive. Wnt/β-catenin pathway has been known to regulate macrophage adhesion and migration properties,15 which play an important role in the initiation and progression of atherosclerosis.16 Activation of monocyte Wnt/β-catenin signaling increased its adhesion to endothelial cells.17 By contrast, myeloid-derived β-catenin deletion decreased the migration and adhesion ability of macrophages and impaired the wound-healing process.15 Thus, macrophage Wnt/β-catenin signaling may contribute to atherogenesis by promoting the cell migration and adhesion. However, inflammatory responses are also the driving force of atherosclerosis development, and macrophages are the major inflammatory cells involved in the progression of atherosclerosis.16,18 Wnt/β-catenin signaling has been demonstrated to regulate both proinflammatory19–21 and anti-inflammatory22–24 responses in various cell types depending on the context or environment. For example, activation of canonical Wnt/β-catenin by Wnt5a and Wnt3a treatment significantly induced proinflammatory cytokine production secretion in murine macrophages.25 However, knockdown of β-catenin also increased the proinflammatory cytokine IL (interleukin)-6 expression in RAW264.7 macrophages, indicating the anti-inflammatory effects of β-catenin in those cells.26 Therefore, the functions of β-catenin in the regulation of macrophage functions related to atherosclerosis are complex. To our knowledge, the impact of macrophage β-catenin expression on atherosclerosis development in appropriate animal models has not been reported.
To explore the role of macrophage-derived β-catenin in atherogenesis, we developed a β-cateninΔmyeLDLR−/− mouse model (low-density lipoprotein receptor–deficient mouse model with myeloid-specific β-catenin deficiency). Our results demonstrate that β-catenin deficiency exacerbate atherosclerosis in LDLR−/− (low-density lipoprotein receptor-deficient) mice by elevating macrophage inflammatory responses.
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Animals
Myeloid-specific β-catenin knockout (β-cateninΔMye) mice were generated by crossing mice carrying loxP-flanked β-catenin alleles (β-cateninF/F)27 with LysM-Cre transgenic mice.28 To increase susceptibility to atherosclerotic lesion development, the β-cateninΔMye mice were crossed to LDLR−/− mice (The Jackson Laboratory) to generate β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− mice. All mice used in this study had β-cateninF/FLDLR−/− double-mutant background, and β-cateninΔMyeLDLR−/− mice carried heterozygous knock-in for LysM-Cre. For atherosclerosis study, 4-week-old male β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− littermates were fed a high-fat Western diet (21.2% fat, 0.2% cholesterol; TD88137, Harlan Teklad) for 12 weeks until euthanization at 16 weeks old. Body weight was measured weekly, and body composition was measured by EchoMRI (Echo Medical System). Intraperitoneal glucose tolerance test was performed as described previously.29 All experimental mice used in this study were male, partially because of the crosstalk between Wnt/β-catenin and estrogen signaling.30,31 However, studying a single sex has limitations because sex differences have been widely reported in mouse atherosclerosis studies.32 All the procedures were approved by the University of Kentucky Institutional Animal Care and Use Committee.
Blood Analysis
Plasma total cholesterol and triglyceride concentrations were determined enzymatically by a colorimetric method.18 Plasma from multiple mice (n=6) was pooled, and plasma lipoprotein cholesterol distributions were determined by fast-performance liquid chromatography.33 Plasma cytokine levels were determined by a mouse cytokine multiplex assay kit and a BioPlex 200 system (Bio-Rad Laboratories).34
Atherosclerosis Lesion Analyses
The atherosclerosis study and analysis were performed following the American Heart Association statement on atherosclerosis.35 Optimum cutting temperature compound–embedded hearts were sectioned and stained with Oil red O (Sigma, O0625-100G), and atherosclerotic lesion sizes at aortic roots were quantified as previously described.18,29,36 The aortic root sections were also stained with hematoxylin & eosin, and necrotic core size were quantified.37 Lesional collagen content and smooth muscle cell composition were determined with Masson’s trichrome staining and quantified by using image pro plus software as previously described.38 Immunohistochemical staining of atherosclerotic lesions was performed on 12-μm sections of aortic roots embedded in optimum cutting temperature.18,39 Sections were first fixed in 100% ice-cold acetone for 15 minutes and then washed with PBS for 20 minutes. Sections were permeabilized with PBS+0.1% Triton X-100 for 10 minutes. Nonspecific binding was reduced by incubating slides in 10% rabbit sera diluted in PBS+0.1% Triton X-100 for 20 minutes at room temperature. Sections were then incubated with antibodies against MCP-1 (monocyte chemoattractant protein 1; Abcam, ab7202), TNFα (tumor necrosis factor-α; Abcam, ab6671), STAT (signal transducer and activator of transcription)-3, Ki67 (eBioscience, 465698), Ly6G(1A8) (BD Bioscience, 551459) at 4°C for 12 to 15 h. Sections were rinsed with PBS and incubated with fluorescein-labeled secondary antibodies (Invitrogen, A11072). The nuclei were stained by mounting the slides with DAPI (4’,6-diamidino-2-phenylindole) medium (Vector Laboratories, H-1200). Normal IgG antibodies were used as negative controls. Dual staining of MCP-1, TNFα, STAT3, and MOMA2 (monocyte/macrophage marker antibody; Bio-Rad, MCA519a) were performed to demonstrate the colocalization of these factors and macrophages. For p65 nuclear translocation analysis, bone marrow–derived macrophages (BMM) were seeded on to 4-well chamber; after being treated with lipopolysaccharides (LPS), cells were fixed with 4% paraformaldehyde at room temperature for 15 minutes, then washed with PBS for 15 minutes. Nonspecific binding was reduced by incubating cells with 10% rabbit sera diluted in PBS+0.1% Triton X-100 for 20 minutes at room temperature. Cells were then incubated with antibody against p65 at 4°C for 12 to 15 h. Sections were rinsed with PBS and incubated with fluorescein-labeled secondary antibodies (Invitrogen, A11072). The nuclei were stained by mounting the slides with DAPI medium. Images were acquired with a Nikon fluorescence microscopy (Nikon).
Macrophage Isolation and Function Assays
BMM and peritoneal macrophages (PM) were isolated as previously described.18 Scratch wound healing assays were performed using 200 μL pipette tip to make a straight scratch on the 100% confluent BMM cell plate. Cells were starved for 24 hours and washed with PBS twice before scratching assay. During the assay, culture medium was replaced with minimal essential medium containing 0.5% FBS to prevent proliferation.40,41 Images were taken at day 0 and day 1 after scratching. For adhesion assay, calcein acetoxymethyl–labeled PMs were incubated with primary porcine endothelial cells, and attached cells were fixed and counted.42 Migration assays were performed using transwells with 8.0-μm pore polycarbonate membrane inserts (Fisher Scientific, 07-200-165).43 Two hundred microliter matrigel (Corning, 356327, final concentration: 300 μg/mL) was added to a 24-well transwell insert and solidified in a 37°C incubator for 2 hours to form a thin gel layer. Macrophages were seeded on top of the matrigel-coated transwell filters, and the lower chambers were filled with non-FBS minimal essential media supplemented with or without 500 ng/mL LPS (Sigma, L4391). After 24 hours, cells were removed from the upper surface of the insert by scraping using Q-Tips. The membranes were fixed with 100% cold methanol (Fisher Scientific, A412-4), stained with hematoxylin (Leica, 3801575), and mounted on the slides using glycerol gelatin. Hematoxylin-stained cells were counted under the microscope.
In Vitro Cell Culture and Treatment
The murine monocyte/macrophage cell line RAW264.7 was obtained from American Type Culture Collection and maintained in 10% FBS containing minimal essential medium supplemented with penicillin and streptomycin (Invitrogen, 15140122). The CRISPR/Cas9 (clustered regularly interspaced short palindromic repeat/CRISPR-associated protein 9) method was used to delete β-catenin in RAW264.7 cells as we previously described.44 Briefly, the lentiCRISPR plasmid (Addgene plasmid No. 52961), containing hCas9 and single-guide RNA, was digested with BbsI, and a pair of annealed oligonucleotides for β-catenin (5′-CACCGAGCCAAGCGCTGGACATTAG-3′) for targeting site was cloned into plasmid. The Transgenomic Surveyor Mutation Detection Kit (Thermo Fisher Scientific No. 706020, Waltham, MA, USA) and genomic DNA sequencing were used to verify genomic mutations within the locus of β-catenin. To study the effects of Wnt3a treatment on STAT3 activation and target gene expression, 100 ng/mL Wnt3a recombinant protein (R&D system, 5036-WN-010) was added to serum-free cultured RAW264.7 cells and BMM for 6 hours before analysis. To study the impact of β-catenin ablation on the nuclear translocation of p65, 500 ng/mL LPS was added to the serum-free cultured BMM cells for 30 minutes before analysis. To knock down STAT3 in macrophages, 100 pmol siRNA targeting STAT3 (Sigma-Aldrich, SASI_Mm01_00041176) was transfected into BMM using Fugene 6 transfection reagent (VWR, E2692) according to manufacturer’s protocol. After transfection, cells were cultured for 48 hours before analysis.
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction Analysis
Total RNA was isolated from mouse tissues or cells using TRIzol Reagent (Life Technologies, 15596018), and quantitative real-time polymerase chain reaction was performed using gene-specific primers and SYBR Green PCR kit (BioRad, 1725125) as previously described.44 The sequences of primer sets used in this study are listed in the Table in the online-only Data Supplement.
Immunoprecipitation and Immunoblotting Analysis
Proteins were isolated from cells or mouse tissues by homogenization in RIPA (radioimmunoprecipitation assay) buffer with complete mini protease inhibitor cocktail (Roche, 11836170001). Protein concentrations were determined by the Pierce BCA protein assay kit (Thermo Fisher Scientific, 23225). Three hundred microgram PM proteins from 3 identical genotype mice pooled per group per immunoprecipitation, proteinase inhibitor cocktail, Protein A agarose beads (Roche, 11134515001), anti-STAT1 antibody (Cell Signaling Technology, 14994T), and Rabbit normal IgG (Cell Signaling Technology, 2729P) were used. For immunoblotting, equal amount of proteins (20 or 30 μg) from PM per mouse were resolved by SDS-PAGE and transferred onto Immunobilon-P membranes (Millipore, IPVH00010). Precision Plus Protein Standards (Bio-Rad Laboratories, 161–0394) were loaded into one lane of the gel. Membranes were incubated in 5% nonfat milk for 45 minutes and then were incubated for 18 h at 4°C with the following primary antibodies in 5% nonfat milk: anti-IL-1β (Abcam, ab9722), anti-IL-6 (Biorad, MCA1490), anti-MCP1 (Abcam, ab7202), anti-phospho-STAT1 (9167S), anti-phospho-STAT3 (9145s), anti-STAT3 (12640S), anti-STAT1 (Cell Signaling Technology), anti-phospho-p65 (Cell Signaling Technology, 3033P), anti-p65 (Santa Cruz, SC-372), anti-β-catenin (Sigma, C2206), and anti-actin (Sigma, A2066). For detailed information of antibodies, please see the Major Resources Table in the online-only Data Supplement. Signals were detected using the Pierce ECL Western Blotting Substrate (32106).
Statistical Analysis
All data are presented as the mean±SEM. Statistically significant differences between 2 groups were analyzed by 2-tailed Student’s t test for data normally distributed and by the Mann–Whitney test for data not normally distributed using Prism software. A P value <0.05 was considered significant.
Results
Generation of LDLR−/− Mice With Myeloid-Specific β-Catenin Deficiency
To investigate the role of β-catenin in macrophage functions, we generated myeloid-specific β-catenin knockout mice (β-cateninΔMye) by breeding β-catenin flox mice (β-cateninF/F)27 with LysMCre transgenic mice.18,28 To increase susceptibility to atherosclerotic lesion development, β-cateninΔMye mice were further crossed with LDLR−/− mice to generate β-cateninΔMyeLDLR−/− mice. Polymerase chain reaction analysis of genomic DNA indicates that the recombination was specific to the PM and BMM of β-cateninΔMye mice (Figure I in the online-only Data Supplement). As expected, the mRNA levels of β-catenin were significantly decreased in both PM and BMM but not in other major tissues of β-cateninΔMyeLDLR−/− mice as compared with β-cateninF/FLDLR−/− mice (Figure 1A). Consistently, β-catenin protein levels were also substantially reduced in both PM and BMM of β-cateninΔMyeLDLR−/− mice (Figure 1B). These results demonstrated the specific and efficient β-catenin deletion in macrophage of β-cateninΔMyeLDLR−/− mice.
Figure 1.

Generation of LDLR−/− mice with myeloid-specific β-catenin deficiency. A, mRNA levels of β-catenin in major tissues and macrophages of β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice (**P<0.01, n=5). B, Protein levels of β-catenin in heart, liver, spleen, kidney, and macrophages of β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice. C, Macrophage migration assay. Peritoneal macrophages (PM) isolated from β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice were seeded on the matrigel-coated transwell filters for 24 hours. Cells that infiltrated and migrated to the underside of transwell were stained with hematoxylin and counted under the microscope (**P<0.01, n=5; bar=200 μm). D, Macrophage wound healing assay. Bone marrow–derived macrophages (BMM) collected from β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice underwent in vitro scratch wound healing assay. Representative images of each group were captured at day 0 and 1 after scratching. Number of cells migrated to the scratch gap was calculated and quantified (**P<0.01, n=5; bar=500 μm). LDLR indicates low-density lipoprotein receptor.
Canonical Wnt/β-catenin signaling has been well established to play an important role in the regulation of cell migration.15,45,46 To determine the impact of deficiency of β-catenin on macrophage migration properties, we performed both transwell migration and scratch wound healing assays. As expected, β-catenin deficiency significantly reduced migration ability of PM as determined by transwell assays (Figure 1C). Scratch wound healing assays also demonstrated that deletion of β-catenin significantly decreases the number of migrated BMM into the scratching gap (Figure 1D). Next, we also incubated freshly isolated PM with primary endothelia cells and found that macrophages from β-cateninΔMyeLDLR−/− mice had reduced adhesion ability to endothelia cells (Figure IIA in the online-only Data Supplement). β-Catenin has been shown to positively regulate the expression of genes that mediate macrophage migration and adhesion.15 Consistently, we also found that deficiency of β-catenin significantly reduced the expression of those genes, including Itga4, Adam9, and Cdh1 (Figure IIB in the online-only Data Supplement). Taken together, these results confirmed that ablation of β-catenin can indeed reduce macrophages adhesion and migration abilities.
Deficiency of β-Catenin in Macrophages Does Not Affect Plasma Lipid Levels but Increases Atherosclerosis in LDLR−/− Mice
To determine the role of macrophage β-catenin in atherosclerosis development, 4-week-old male β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− littermates were fed with Western diet for 12 weeks. At 16 weeks of age, β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− mice had similar body weight, fat mass, and lean mass (Figure IIIA and IIIB in the online-only Data Supplement). Deficiency of myeloid β-catenin did not affect the fasting glucose levels and glucose tolerance of β-cateninΔMyeLDLR−/− mice (Figure IIIC and IIID in the online-only Data Supplement). Both β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− mice had diet-induced hyperlipidemia, and myeloid β-catenin deletion did not alter plasma total cholesterol and triglyceride levels (Figure 2A). Further, fast-performance liquid chromatography analysis also showed similar cholesterol distribution pattern between β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− mice (Figure 2B).
Figure 2.
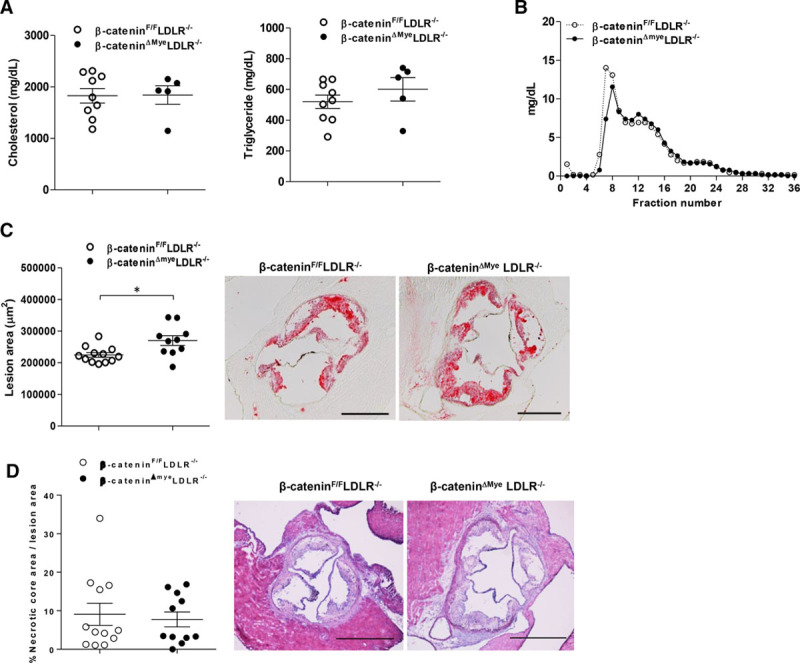
Deficiency of myeloid β-catenin increases atherosclerosis of LDLR−/− mice without affecting lipid profile. Four-week-old male β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− littermates were fed a Western diet for 12 weeks. A and B, The plasma levels of total cholesterol and triglyceride (A) were measured by standard method, and plasma cholesterol distribution was analyzed by fast-performance liquid chromatography (B). C, Quantitative analysis of atherosclerotic lesion areas in the aortic root of β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice (n=10–12 per group; *P<0.05). Representative Oil-red-O–stained sections from each genotypes are displayed next to the quantification data (bar=500 μm). D, Quantitative analysis of necrotic core areas in the aortic root of β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice (n=11–12 per group). Representative images of hematoxylin & eosin staining at aortic root sections from β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice are displayed next to the quantification data (bar=500 μm). LDLR indicates low-density lipoprotein receptor.
Although deficiency of myeloid β-catenin did not affect metabolic phenotypes and plasma lipid levels, quantification of cross-sectional lesion areas at the aortic root revealed that β-cateninΔMyeLDLR−/− mice had 26% increased lesion sizes (286 590.4±43 656.63 μm2) as compared with β-cateninF/FLDLR−/− littermates (227 247.1±26 061.89 μm2; P=0.013; Figure 2C). Immunostaining results revealed that deficiency of β-catenin did not significantly affect the proportion of macrophage content in atherosclerotic lesions of β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− mice (Figure IVA in the online-only Data Supplement). Further, Masson’s trichrome staining also demonstrated that the plaque composition was similar between β-cateninΔmyeLDLR−/− and β-cateninF/FLDLR−/− mice (Figure IVB in the online-only Data Supplement). Although β-catenin signaling can regulate cell proliferation, β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− mice had similar proliferating Ki67-positive cells in the lesions (Figure V in the online-only Data Supplement). In addition, deficiency of β-catenin did not affect the proportion of necrotic core areas in atherosclerotic lesions of β-cateninΔMyeLDLR−/− mice (Figure 2D). Because LysM is also expressed by neutrophils47,48 that contribute to the atherosclerotic lesion formation,49 we also performed immunofluorescence staining with antibodies against neutrophil marker Ly6G(1A8) and found that LysM-Cre-mediated β-catenin deletion did not affect lesional neutrophil numbers (Figure VI in the online-only Data Supplement). Taken together, deletion of β-catenin in macrophages exacerbates diet-induced atherosclerosis in LDLR−/− mice without altering plasma lipid level and plaque composition.
Ablation of β-Catenin Elevates Macrophage Inflammatory Responses Without Affecting NF-κB Activities
Atherosclerosis is an inflammatory disease, and macrophages are the major inflammatory cells contributing to atherosclerotic lesion formation and progression.16,50 We next investigated whether deficiency of β-catenin affected macrophage inflammatory responses in β-cateninΔMyeLDLR−/− mice. Quantitative real-time polymerase chain reaction analyses demonstrated that the mRNA levels of proinflammatory cytokines, including IL-6 and TNFα, were significantly increased in freshly isolated PMs of β-cateninΔMyeLDLR−/− mice (Figure 3A). However, the expression levels of anti-inflammatory genes were not affected by β-catenin deficiency (Figure VII in the online-only Data Supplement). In addition, protein levels of IL-1β, MCP1, and IL-6 were also elevated in PM of β-cateninΔMyeLDLR−/− mice as compared with littermate controls (Figure 3B). Consistent with gene and protein expression analyses, immunofluorescence staining showed that the expression of key inflammatory cytokines, TNFα and MCP1, was increased in the atherosclerotic lesional macrophages of β-cateninΔMyeLDLR−/− mice (Figure 3C; Figure VIII in the online-only Data Supplement). We next measured plasma cytokine levels to determine whether β-cateninΔMyeLDLR−/− mice have increased systemic inflammation. Indeed, the plasma levels of proinflammatory cytokines including TNFα, IL-6, and IFNγ were significantly increased in β-cateninΔMyeLDLR−/− mice (Figure 3D). Taken together, deficiency of β-catenin elevated macrophage inflammatory responses and increased lesional and systemic inflammation in β-cateninΔMyeLDLR−/− mice.
Figure 3.

Ablation of myeloid β-catenin increases macrophage and systemic inflammation in β-cateninΔMyeLDLR−/− mice. A, Quantitative real-time polymerase chain reaction (QPCR) analysis of proinflammatory cytokine expression in peritoneal macrophages (PM) of β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice (**P<0.01, n=8). B, Immunoblotting images and quantification data of proinflammatory cytokines in PM of β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice (*P<0.05, **P<0.01, n=3). C, Representative images and quantification data of TNFα (tumor necrosis factor-α) and MCP1 (monocyte chemoattractant protein 1) immunofluorescent staining in the aortic root of β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice (**P<0.01, n=6; bar=100 μm). D, Plasma protein levels of indicated cytokines from β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice (**P<0.01, n=11–14). CXCL1 indicates C-X-C motif chemokine ligand 1; GPR18, G protein–coupled receptor 18; IL, interleukin; LDLR, low-density lipoprotein receptor; MIP1β, macrophage inflammatory protein-1 beta; and RANTES, regulated on activation, normal T-cell–expressed and secreted.
Wnt/β-catenin signaling has been previously shown to repress NF-κB (nuclear factor-κB) activity in several cell types.51,52 Since NF-κB plays an important role in mediating macrophage inflammatory responses during atherosclerosis development,18 we then investigated whether β-catenin deletion can affect NF-κB activities in macrophages. The total active form of NF-κB subunit p65 (phosphorylated p65) was not affected by β-catenin deletion in PM of β-cateninΔMyeLDLR−/− mice (Figure IXA in the online-only Data Supplement), indicating the unchanged NF-κB activity in those cells. We also isolated BMM from β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− mice and treated them with LPS to stimulate the nuclear translocation of p65. Consistently, deficiency of β-catenin did not affect LPS-elicited p65 translocation as determined by both immunoblotting and immunofluorescence staining (Figure IXB and IXC in the online-only Data Supplement). Therefore, the increased inflammatory response of macrophage from β-cateninΔMyeLDLR−/− mice was unlikely through NF-κB signaling.
β-Catenin Regulates JAK-STAT Signaling and Inflammatory Responses in Macrophages
One of the downstream target genes of canonical Wnt/β-catenin signaling is STAT3,53 which is a part of the JAK (Janus kinase)-STAT signaling, another central pathway controlling the expression of proinflammatory and anti-inflammatory cytokines in macrophages.54 Canonical Wnt/β-catenin signaling has been reported to positively regulate STAT3 signaling and to repress inflammatory responses in several different murine and human cell types.23,43,53,55,56 To investigate whether the STAT signaling contributes to the increased inflammatory responses in β-catenin-depleted macrophages, PM were isolated from β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− mice, and the expression levels of STAT1, STAT3, and the downstream target genes57 were analyzed. Consistent with previous studies,43,53,58 deficiency of β-catenin significantly reduced the mRNA levels of STAT3 and its targets such as Cited2, Sbno2, BCL3, and Nfil3 but did not affect the expression of STAT1 (Figure 4A). The protein levels of STAT3 were also substantially decreased in β-catenin-deficient macrophages (Figure 4B). Immunofluorescence staining confirmed the decreased STAT3 expression in the lesions of β-cateninΔMyeLDLR−/− mice (Figure 4C).
Figure 4.

β-Catenin deficiency affects STAT3 expression and STAT1 activity in macrophages. A, Quantitative real-time polymerase chain reaction (QPCR) analysis of STAT3, STAT1, and STAT3 target genes in peritoneal macrophages (PM) of β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice (**P<0.01, n=8). B, Immunoblotting of STAT3, STAT1, phosphorylated STAT1, and phosphorylated STAT3 in PM. C, Representative images and quantification data of STAT3 immunofluorescent staining in the aortic root of β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice (*P<0.05, n=5; bar=100 μm). D, PM isolated from β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice were lysed and immunoprecipitated with antibodies against STAT1 and IgG. Total STAT1, phosphorylated STAT1 and STAT3 proteins were further immunoblotted with specific antibodies. Ten percent cell lysate were used as input for immunoblotting. LDLR indicates low-density lipoprotein receptor; STAT, signal transducer and activator of transcription.
STAT3 has been demonstrated to suppress the activation of STAT1 by forming STAT3/STAT1 heterodimer and inhibiting its tyrosine phosphorylation in a cellular context- and signal-dependent manner.59–61 Indeed, β-catenin-deficient macrophages had unchanged total STAT1 protein levels but increased STAT1 phosphorylation (Figure 4B). Co-immunoprecipitation assays also confirmed a direct interaction between STAT1 and STAT3 in control macrophages, but the interaction was substantially reduced in β-catenin-deficient macrophages (Figure 4D). Further, phosphorylated STAT1 proteins were increased in the protein complex pulled down by anti-STAT1 antibodies in β-catenin-deficient macrophages (Figure 4D). In addition, siRNA-mediated STAT3 knockdown also led to increased STAT1 phosphorylation without affecting the total STAT1 protein levels (Figure X in the online-only Data Supplement). Collectively, these results suggest that decreased STAT3 levels may contribute to the activation of STAT1 and increased inflammatory responses in β-catenin-deficient macrophages.
To further explore the understudied functions of canonical Wnt/β-catenin signaling in the regulation of macrophage inflammatory responses, BMM isolated from β-cateninΔMyeLDLR−/− and β-cateninF/FLDLR−/− mice were treated with Wnt3a that activates canonical Wnt/β-catenin signaling.44 Wnt3a treatment significantly upregulated STAT3 and its downstream genes in control macrophages but not in β-catenin-deficient macrophages (Figure 5A). As expected, increased STAT3 expression was associated with decreased proinflammatory cytokine expression in control macrophages but deficiency of β-catenin abolished Wnt3a-elicited downregulation of inflammatory genes (Figure 5B). In addition to primary macrophages, we also used CRISPR/Cas9 approach to generate β-catenin-deficient murine RAW264.7 macrophage cell line (Figure XI in the in the online-only Data Supplement). As shown in Figure 5C and 5D, CRISPR/Cas9-mediated β-catenin deletion also inhibited Wnt3a-stimulated upregulation of STAT3 and inflammatory responses in RAW264.7 cells. Taken together, these results demonstrated an important role of β-catenin-STAT3/STAT1 signaling cascade in the regulation of macrophage inflammatory responses, which likely contributes to the increased atherosclerosis in β-cateninΔMyeLDLR−/− mice.
Figure 5.

Deletion of β-catenin abolishes the impact of Wnt3a on STAT3 and proinflammatory cytokine expression in macrophages. A and B, Quantitative real-time polymerase chain reaction (QPCR) analysis of STAT3, STAT1, and STAT3 target genes (A) and proinflammatory genes (B) in bone marrow–derived macrophages (BMM) isolated from β-cateninF/FLDLR−/− and β-cateninΔMyeLDLR−/− mice treated with vehicle control or Wnt 3a (*P<0.05, **P<0.01, n=8). C and D, QPCR analysis of STAT3, STAT1, and STAT3 target genes (C) and proinflammatory genes (D) in control or β-catenin-deficient RAW264.7 macrophages treated with vehicle control or Wnt3a (*P<0.05, **P<0.01, n=8). BCL3 indicates B-cell lymphoma 3; GPR18, G protein–coupled receptor 18; IL, interleukin; LDLR, low-density lipoprotein receptor; STAT, signal transducer and activator of transcription; and TNFα, tumor necrosis factor-α.
Discussion
Emerging evidence has demonstrated the potential role of Wnt/β-catenin signaling in atherogenesis by regulating endothelial functions7,62 and smooth muscle cell proliferation and migration.9–11 Myeloid-derived β-catenin has also been shown to promote macrophage migration and adhesion.15 Therefore, it has been suggested that attenuation of Wnt/β-catenin signaling may have beneficial effects for the cardiovascular system.12,63,64 However, there have been no studies on the impact of macrophage β-catenin expression on atherosclerosis development in hyperlipidemic mouse models. In the current study, we generated myeloid-specific β-catenin-deficient LDLR−/− mice to investigate the role of macrophage β-catenin in atherogenesis. Consistent with previous studies,15 we found that deficiency of β-catenin decreased macrophage migration and adhesion abilities. Therefore, one would expect that deficiency of myeloid β-catenin should decrease atherosclerosis development in vivo. Unexpectedly, atherosclerosis analyses revealed that β-cateninΔMyeLDLR−/− mice had increased rather than decreased aortic root atherosclerotic lesion sizes as compared with control β-cateninF/FLDLR−/− mice. Interestingly, deletion of β-catenin increased macrophage inflammatory responses and elevated plasma proinflammatory cytokine levels, which may lead to the increased atherosclerosis. To our knowledge, this study is the first to demonstrate the impact of myeloid β-catenin expression on the atherosclerosis development in an appropriate small animal model.
Atherosclerosis is a chronic inflammatory disease, and the key regulator of the innate and adaptive immune responses, NF-κB signaling has been implicated in the atherosclerosis development.16,18,65,66 We previously demonstrated that inhibition of myeloid NF-κB activation by deletion of IKKβ (IκB kinase β) attenuated macrophage inflammatory responses and decreased atherosclerosis development in LDLR−/− mice.18 In the current study, deficiency of β-catenin did not affect NF-κB translocation and activation in macrophages, which cannot explain the elevated inflammatory responses in those cells. In addition to NF-κB signaling, the JAK-STAT signaling is another key pathway regulating cellular inflammatory responses,67 and canonical Wnt/β-catenin signaling can transcriptionally regulate STAT3 expression.53,55 Indeed, we found that the expression of STAT3 was decreased in both macrophages and atherosclerotic lesions of β-cateninΔMyeLDLR−/− mice. The balance between activation of STAT1 and STAT3 coordinately regulates macrophage polarization and inflammation in responding to different stimuli.68,69 While activation of STAT1 promotes macrophage proinflammatory responses, activation of STAT3 results in suppressed immune responses.56,68–71 Further, studies have also demonstrated that STAT3 can attenuate STAT1-mediated proinflammatory cytokine expression through direct interaction with STAT1 to suppress its phosphorylation or the formation of DNA-binding STAT1 homodimers.59,60,68 Consistent with those studies, we also detected a direct interaction between STAT3 and STAT1 in macrophages, and decreased STAT3 protein levels led to increased STAT1 phosphorylation without affecting total STAT1 protein levels in β-catenin-deficient macrophages. Further, the impact of Wnt3a treatment on STAT3 and inflammatory gene expression was abolished by β-catenin deficiency. Therefore, it is likely that deficiency of β-catenin increases macrophage inflammatory responses through modulation of STAT3/STAT1 signaling cascade.
The role of Wnt/β-catenin signaling in the regulation of inflammatory responses in different cell types has not been completely understood. For example, some studies demonstrated that β-catenin is a negative regulator of inflammation in certain immune cells, such as dendritic cells.23,24 By contrast, other studies suggested that β-catenin can increase inflammatory cytokine production in different cell types.19,20 Thus, the functions of β-catenin in immunity are complex, and β-catenin may have both proinflammatory and anti-inflammatory effects depending on the cell type, stimulus, and cellular environment. Nevertheless, our studies demonstrated that canonical Wnt/β-catenin signaling has anti-inflammatory effects in macrophages of Western diet–fed LDLR−/− mice, and deficiency of β-catenin led to increased macrophage inflammatory responses and systemic inflammation. Future studies will be required to determine the cell type–specific functions of Wnt signaling in inflammation and the detailed underlying mechanisms.
In summary, we demonstrate that β-catenin can regulate multiple macrophage functions related to atherosclerosis, and myeloid-specific β-catenin deficiency increased diet-induced atherosclerosis at aortic root of LDLR−/− mice. Although deletion of β-catenin decreased macrophage adhesion and migration properties, β-catenin deficiency increased macrophage inflammatory responses, leading to exacerbated atherosclerosis in LDLR−/− mice. Wnt/β-catenin signaling is an evolutionarily conserved pathway, and findings from this study will hopefully stimulate further investigations of the function of Wnt/β-catenin signaling in atherogenesis.
Acknowledgments
We thank Dr Alan Daugherty and Deborah Howatt for fast-performance liquid chromatography analysis.
Sources of Funding
This work was supported in part by National Institutes of Health (NIH) grants (R01HL123358, R01HL131925, R01ES023470, R21ES022745, and P20GM103527).
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- ApoE−/−
- apolipoprotein E–deficient
- BMM
- bone marrow–derived macrophage
- CKIα
- casein kinase Iα
- CRISPR/Cas9
- clustered regularly interspaced short palindromic repeat/CRISPR-associated protein 9
- GSK3β
- glycogen synthase kinase-3β
- IKKβ
- IκB kinase β
- IL
- interleukin
- JAK
- Janus kinase
- LDLR
- low-density lipoprotein receptor
- LRP
- low-density lipoprotein receptor-related protein
- MCP-1
- monocyte chemoattractant protein 1
- NF-κB
- nuclear factor-κB
- PM
- peritoneal macrophage
- STAT
- signal transducer and activator of transcription
- TNFα
- tumor necrosis factor-α
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.118.311059/-/DC1.
Highlights
Canonical Wnt/β-catenin signaling has both proatherogenic and antiatherogenic effects in macrophages.
Deficiency of myeloid β-catenin decreases macrophage adhesion and migration properties but increases atherosclerosis in LDLR−/− (low-density lipoprotein receptor-deficient) mice.
Ablation of β-catenin elevates macrophage inflammatory responses and systemic inflammation in LDLR−/− mice.
β-Catenin regulates STAT3 (signal transducer and activator of transcription 3)/STAT1 signaling cascade in macrophages, and deficiency of β-catenin results in elevated STAT1 activation and increased macrophage inflammation.
References
- 1.Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4:68–75. doi: 10.4161/org.4.2.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 4.Badimon L, Borrell-Pages M. Wnt signaling in the vessel wall. Curr Opin Hematol. 2017;24:230–239. doi: 10.1097/MOH.0000000000000336. doi: 10.1097/MOH.0000000000000336. [DOI] [PubMed] [Google Scholar]
- 5.Bedel A, Nègre-Salvayre A, Heeneman S, Grazide MH, Thiers JC, Salvayre R, Maupas-Schwalm F. E-cadherin/beta-catenin/T-cell factor pathway is involved in smooth muscle cell proliferation elicited by oxidized low-density lipoprotein. Circ Res. 2008;103:694–701. doi: 10.1161/CIRCRESAHA.107.166405. doi: 10.1161/CIRCRESAHA.107.166405. [DOI] [PubMed] [Google Scholar]
- 6.Sarzani R, Salvi F, Bordicchia M, Guerra F, Battistoni I, Pagliariccio G, Carbonari L, Dessì-Fulgheri P, Rappelli A. Carotid artery atherosclerosis in hypertensive patients with a functional LDL receptor-related protein 6 gene variant. Nutr Metab Cardiovasc Dis. 2011;21:150–156. doi: 10.1016/j.numecd.2009.08.004. doi: 10.1016/j.numecd.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Gelfand BD, Meller J, Pryor AW, Kahn M, Bortz PD, Wamhoff BR, Blackman BR. Hemodynamic activation of beta-catenin and T-cell-specific transcription factor signaling in vascular endothelium regulates fibronectin expression. Arterioscler Thromb Vasc Biol. 2011;31:1625–1633. doi: 10.1161/ATVBAHA.111.227827. doi: 10.1161/ATVBAHA.111.227827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng SL, Shao JS, Halstead LR, Distelhorst K, Sierra O, Towler DA. Activation of vascular smooth muscle parathyroid hormone receptor inhibits Wnt/beta-catenin signaling and aortic fibrosis in diabetic arteriosclerosis. Circ Res. 2010;107:271–282. doi: 10.1161/CIRCRESAHA.110.219899. doi: 10.1161/CIRCRESAHA.110.219899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Xiao Y, Mou Y, Zhao Y, Blankesteijn WM, Hall JL. A role for the beta-catenin/T-cell factor signaling cascade in vascular remodeling. Circ Res. 2002;90:340–347. doi: 10.1161/hh0302.104466. [DOI] [PubMed] [Google Scholar]
- 10.Slater SC, Koutsouki E, Jackson CL, Bush RC, Angelini GD, Newby AC, George SJ. R-cadherin:beta-catenin complex and its association with vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol. 2004;24:1204–1210. doi: 10.1161/01.ATV.0000130464.24599.e0. doi: 10.1161/01.ATV.0000130464.24599.e0. [DOI] [PubMed] [Google Scholar]
- 11.Mill C, George SJ. Wnt signalling in smooth muscle cells and its role in cardiovascular disorders. Cardiovasc Res. 2012;95:233–240. doi: 10.1093/cvr/cvs141. doi: 10.1093/cvr/cvs141. [DOI] [PubMed] [Google Scholar]
- 12.Krishna SM, Seto SW, Jose RJ, Li J, Morton SK, Biros E, Wang Y, Nsengiyumva V, Lindeman JH, Loots GG, Rush CM, Craig JM, Golledge J. Wnt signaling pathway inhibitor sclerostin inhibits angiotensin II-induced aortic aneurysm and atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37:553–566. doi: 10.1161/ATVBAHA.116.308723. doi: 10.1161/ATVBAHA.116.308723. [DOI] [PubMed] [Google Scholar]
- 13.Borrell-Pages M, Romero JC, Badimon L. Cholesterol modulates LRP5 expression in the vessel wall. Atherosclerosis. 2014;235:363–370. doi: 10.1016/j.atherosclerosis.2014.05.922. doi: 10.1016/j.atherosclerosis.2014.05.922. [DOI] [PubMed] [Google Scholar]
- 14.Borrell-Pagès M, Romero JC, Badimon L. LRP5 deficiency down-regulates Wnt signalling and promotes aortic lipid infiltration in hypercholesterolaemic mice. J Cell Mol Med. 2015;19:770–777. doi: 10.1111/jcmm.12396. doi: 10.1111/jcmm.12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amini-Nik S, Cambridge E, Yu W, Guo A, Whetstone H, Nadesan P, Poon R, Hinz B, Alman BA. β-Catenin-regulated myeloid cell adhesion and migration determine wound healing. J Clin Invest. 2014;124:2599–2610. doi: 10.1172/JCI62059. doi: 10.1172/JCI62059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee DK, Nathan Grantham R, Trachte AL, Mannion JD, Wilson CL. Activation of the canonical Wnt/beta-catenin pathway enhances monocyte adhesion to endothelial cells. Biochem Biophys Res Commun. 2006;347:109–116. doi: 10.1016/j.bbrc.2006.06.082. doi: 10.1016/j.bbrc.2006.06.082. [DOI] [PubMed] [Google Scholar]
- 18.Park SH, Sui Y, Gizard F, Xu J, Rios-Pilier J, Helsley RN, Han SS, Zhou C. Myeloid-specific IκB kinase β deficiency decreases atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32:2869–2876. doi: 10.1161/ATVBAHA.112.254573. doi: 10.1161/ATVBAHA.112.254573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blumenthal A, Ehlers S, Lauber J, Buer J, Lange C, Goldmann T, Heine H, Brandt E, Reiling N. The wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood. 2006;108:965–973. doi: 10.1182/blood-2005-12-5046. doi: 10.1182/blood-2005-12-5046. [DOI] [PubMed] [Google Scholar]
- 20.Halleskog C, Mulder J, Dahlström J, Mackie K, Hortobágyi T, Tanila H, Kumar Puli L, Färber K, Harkany T, Schulte G. Wnt signaling in activated microglia is proinflammatory. Glia. 2011;59:119–131. doi: 10.1002/glia.21081. doi: 10.1002/glia.21081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halleskog C, Schulte G. WNT-3A and WNT-5A counteract lipopolysaccharide-induced pro-inflammatory changes in mouse primary microglia. J Neurochem. 2013;125:803–808. doi: 10.1111/jnc.12250. doi: 10.1111/jnc.12250. [DOI] [PubMed] [Google Scholar]
- 22.Suryawanshi A, Tadagavadi RK, Swafford D, Manicassamy S. Modulation of inflammatory responses by Wnt/β-catenin signaling in dendritic cells: a novel immunotherapy target for autoimmunity and cancer. Front Immunol. 2016;7:460. doi: 10.3389/fimmu.2016.00460. doi: 10.3389/fimmu.2016.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duan Y, Liao AP, Kuppireddi S, Ye Z, Ciancio MJ, Sun J. Beta-Catenin activity negatively regulates bacteria-induced inflammation. Lab Invest. 2007;87:613–624. doi: 10.1038/labinvest.3700545. doi: 10.1038/labinvest.3700545. [DOI] [PubMed] [Google Scholar]
- 24.Swafford D, Manicassamy S. Wnt signaling in dendritic cells: its role in regulation of immunity and tolerance. Discov Med. 2015;19:303–310. [PMC free article] [PubMed] [Google Scholar]
- 25.Yu CH, Nguyen TT, Irvine KM, Sweet MJ, Frazer IH, Blumenthal A. Recombinant Wnt3a and Wnt5a elicit macrophage cytokine production and tolerization to microbial stimulation via toll-like receptor 4. Eur J Immunol. 2014;44:1480–1490. doi: 10.1002/eji.201343959. doi: 10.1002/eji.201343959. [DOI] [PubMed] [Google Scholar]
- 26.Lee H, Bae S, Choi BW, Yoon Y. Wnt/β-catenin pathway is modulated in asthma patients and LPS-stimulated RAW264.7 macrophage cell line. Immunopharmacol Immunotoxicol. 2012;34:56–65. doi: 10.3109/08923973.2011.574704. doi: 10.3109/08923973.2011.574704. [DOI] [PubMed] [Google Scholar]
- 27.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 28.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 29.Sui Y, Park SH, Xu J, Monette S, Helsley RN, Han SS, Zhou C. IKKβ links vascular inflammation to obesity and atherosclerosis. J Exp Med. 2014;211:869–886. doi: 10.1084/jem.20131281. doi: 10.1084/jem.20131281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kouzmenko AP, Takeyama K, Ito S, Furutani T, Sawatsubashi S, Maki A, Suzuki E, Kawasaki Y, Akiyama T, Tabata T, Kato S. Wnt/beta-catenin and estrogen signaling converge in vivo. J Biol Chem. 2004;279:40255–40258. doi: 10.1074/jbc.C400331200. doi: 10.1074/jbc.C400331200. [DOI] [PubMed] [Google Scholar]
- 31.Xiong W, Zhang L, Yu L, Xie W, Man Y, Xiong Y, Liu H, Liu Y. Estradiol promotes cells invasion by activating β-catenin signaling pathway in endometriosis. Reproduction. 2015;150:507–516. doi: 10.1530/REP-15-0371. doi: 10.1530/REP-15-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from ATVB council. Arterioscler Thromb Vasc Biol. 2018;38:292–303. doi: 10.1161/ATVBAHA.117.309524. doi: 10.1161/ATVBAHA.117.309524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sui Y, Xu J, Rios-Pilier J, Zhou C. Deficiency of PXR decreases atherosclerosis in apoE-deficient mice. J Lipid Res. 2011;52:1652–1659. doi: 10.1194/jlr.M017376. doi: 10.1194/jlr.M017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park SH, Liu Z, Sui Y, Helsley RN, Zhu B, Powell DK, Kern PA, Zhou C. IKKβ is essential for adipocyte survival and adaptive adipose remodeling in obesity. Diabetes. 2016;65:1616–1629. doi: 10.2337/db15-1156. doi: 10.2337/db15-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, García-Cardeña G, Lusis AJ, Owens AP, III, Rosenfeld ME, Virmani R American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; and Council on Basic Cardiovascular Sciences. Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. doi: 10.1161/ATV.0000000000000062. doi: 10.1161/ATV.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 36.Sui Y, Park SH, Wang F, Zhou C. Perinatal bisphenol a exposure increases atherosclerosis in adult male PXR-humanized mice. Endocrinology. 2018;159:1595–1608. doi: 10.1210/en.2017-03250. doi: 10.1210/en.2017-03250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu G, Hu W, Shahsafaei A, Song W, Dobarro M, Sukhova GK, Bronson RR, Shi GP, Rother RP, Halperin JA, Qin X. Complement regulator CD59 protects against atherosclerosis by restricting the formation of complement membrane attack complex. Circ Res. 2009;104:550–558. doi: 10.1161/CIRCRESAHA.108.191361. doi: 10.1161/CIRCRESAHA.108.191361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du M, Wang X, Tan X, Li X, Huang D, Huang K, Yang L, Zhang F, Wang Y, Huang D, Huang K. Nkx2-5 is expressed in atherosclerotic plaques and attenuates development of atherosclerosis in apolipoprotein e-deficient mice. J Am Heart Assoc. 2016;5:e004440. doi: 10.1161/JAHA.116.004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou C, Pridgen B, King N, Xu J, Breslow JL. Hyperglycemic Ins2AkitaLdlr-/- mice show severely elevated lipid levels and increased atherosclerosis: a model of type 1 diabetic macrovascular disease. J Lipid Res. 2011;52:1483–1493. doi: 10.1194/jlr.M014092. doi: 10.1194/jlr.M014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xuan YH, Huang BB, Tian HS, Chi LS, Duan YM, Wang X, Zhu ZX, Cai WH, Zhu YT, Wei TM, Ye HB, Cong WT, Jin LT. High-glucose inhibits human fibroblast cell migration in wound healing via repression of bFGF-regulating JNK phosphorylation. PLoS One. 2014;9:e108182. doi: 10.1371/journal.pone.0108182. doi: 10.1371/journal.pone.0108182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nyegaard S, Christensen B, Rasmussen JT. An optimized method for accurate quantification of cell migration using human small intestine cells. Metab Eng Commun. 2016;3:76–83. doi: 10.1016/j.meteno.2016.03.002. doi: 10.1016/j.meteno.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han SG, Eum SY, Toborek M, Smart E, Hennig B. Polychlorinated biphenyl-induced VCAM-1 expression is attenuated in aortic endothelial cells isolated from caveolin-1 deficient mice. Toxicol Appl Pharmacol. 2010;246:74–82. doi: 10.1016/j.taap.2010.04.009. doi: 10.1016/j.taap.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anand M, Lai R, Gelebart P. β-catenin is constitutively active and increases STAT3 expression/activation in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica. 2011;96:253–261. doi: 10.3324/haematol.2010.027086. doi: 10.3324/haematol.2010.027086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sui Y, Liu Z, Park SH, Thatcher SE, Zhu B, Fernandez JP, Molina H, Kern PA, Zhou C. Ikkbeta is a beta-catenin kinase that regulates mesenchymal stem cell differentiation. JCI Insight. 2018;3:e96660. doi: 10.1172/jci.insight.96660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwai S, Yonekawa A, Harada C, Hamada M, Katagiri W, Nakazawa M, Yura Y. Involvement of the Wnt-β-catenin pathway in invasion and migration of oral squamous carcinoma cells. Int J Oncol. 2010;37:1095–1103. doi: 10.3892/ijo_00000761. [DOI] [PubMed] [Google Scholar]
- 46.Yang CM, Ji S, Li Y, Fu LY, Jiang T, Meng FD. β-catenin promotes cell proliferation, migration, and invasion but induces apoptosis in renal cell carcinoma. Onco Targets Ther. 2017;10:711–724. doi: 10.2147/OTT.S117933. doi: 10.2147/OTT.S117933. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods. 2014;408:89–100. doi: 10.1016/j.jim.2014.05.009. doi: 10.1016/j.jim.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albarrán-Juárez J, Kaur H, Grimm M, Offermanns S, Wettschureck N. Lineage tracing of cells involved in atherosclerosis. Atherosclerosis. 2016;251:445–453. doi: 10.1016/j.atherosclerosis.2016.06.012. doi: 10.1016/j.atherosclerosis.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 49.Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012;110:875–888. doi: 10.1161/CIRCRESAHA.111.257535. doi: 10.1161/CIRCRESAHA.111.257535. [DOI] [PubMed] [Google Scholar]
- 50.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, Li Y, Lin SY, Hung MC. beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell. 2002;2:323–334. doi: 10.1016/s1535-6108(02)00154-x. [DOI] [PubMed] [Google Scholar]
- 52.Nejak-Bowen K, Kikuchi A, Monga SP. Beta-catenin-NF-κB interactions in murine hepatocytes: a complex to die for. Hepatology. 2013;57:763–774. doi: 10.1002/hep.26042. doi: 10.1002/hep.26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan S, Zhou C, Zhang W, Zhang G, Zhao X, Yang S, Wang Y, Lu N, Zhu H, Xu N. Beta-catenin/TCF pathway upregulates STAT3 expression in human esophageal squamous cell carcinoma. Cancer Lett. 2008;271:85–97. doi: 10.1016/j.canlet.2008.05.035. doi: 10.1016/j.canlet.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 54.Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614. doi: 10.3389/fimmu.2014.00614. doi: 10.3389/fimmu.2014.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hao J, Li TG, Qi X, Zhao DF, Zhao GQ. Wnt/beta-catenin pathway up-regulates Stat3 and converges on LIF to prevent differentiation of mouse embryonic stem cells. Dev Biol. 2006;290:81–91. doi: 10.1016/j.ydbio.2005.11.011. doi: 10.1016/j.ydbio.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 56.Matsukawa A, Kudo S, Maeda T, Numata K, Watanabe H, Takeda K, Akira S, Ito T. Stat3 in resident macrophages as a repressor protein of inflammatory response. J Immunol. 2005;175:3354–3359. doi: 10.4049/jimmunol.175.5.3354. [DOI] [PubMed] [Google Scholar]
- 57.Hutchins AP, Takahashi Y, Miranda-Saavedra D. Genomic analysis of LPS-stimulated myeloid cells identifies a common pro-inflammatory response but divergent IL-10 anti-inflammatory responses. Sci Rep. 2015;5:9100. doi: 10.1038/srep09100. doi: 10.1038/srep09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao C, Zhang M, Liu W, Wang C, Zhang Q, Li W. β-catenin knockdown inhibits pituitary adenoma cell proliferation and invasion via interfering with AKT and gelatinases expression. Int J Oncol. 2015;46:1643–1650. doi: 10.3892/ijo.2015.2862. doi: 10.3892/ijo.2015.2862. [DOI] [PubMed] [Google Scholar]
- 59.Ho HH, Ivashkiv LB. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J Biol Chem. 2006;281:14111–14118. doi: 10.1074/jbc.M511797200. doi: 10.1074/jbc.M511797200. [DOI] [PubMed] [Google Scholar]
- 60.Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, Murray-Tait V, Chiarle R, Poli V. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol. 2004;5:401–409. doi: 10.1038/ni1052. doi: 10.1038/ni1052. [DOI] [PubMed] [Google Scholar]
- 61.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Förster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 62.Ueland T, Otterdal K, Lekva T, Halvorsen B, Gabrielsen A, Sandberg WJ, Paulsson-Berne G, Pedersen TM, Folkersen L, Gullestad L, Oie E, Hansson GK, Aukrust P. Dickkopf-1 enhances inflammatory interaction between platelets and endothelial cells and shows increased expression in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1228–1234. doi: 10.1161/ATVBAHA.109.189761. doi: 10.1161/ATVBAHA.109.189761. [DOI] [PubMed] [Google Scholar]
- 63.Matthijs Blankesteijn W, Hermans KC. Wnt signaling in atherosclerosis. Eur J Pharmacol. 2015;763(pt A):122–130. doi: 10.1016/j.ejphar.2015.05.023. doi: 10.1016/j.ejphar.2015.05.023. [DOI] [PubMed] [Google Scholar]
- 64.Marinou K, Christodoulides C, Antoniades C, Koutsilieris M. Wnt signaling in cardiovascular physiology. Trends Endocrinol Metab. 2012;23:628–636. doi: 10.1016/j.tem.2012.06.001. doi: 10.1016/j.tem.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 65.Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8:372–383. doi: 10.1016/j.cmet.2008.08.016. doi: 10.1016/j.cmet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 67.Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77:521–546. doi: 10.1007/s40265-017-0701-9. doi: 10.1007/s40265-017-0701-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Regis G, Pensa S, Boselli D, Novelli F, Poli V. Ups and downs: the STAT1:STAT3 seesaw of interferon and gp130 receptor signalling. Semin Cell Dev Biol. 2008;19:351–359. doi: 10.1016/j.semcdb.2008.06.004. doi: 10.1016/j.semcdb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 69.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pattison MJ, Mackenzie KF, Arthur JS. Inhibition of JAKs in macrophages increases lipopolysaccharide-induced cytokine production by blocking IL-10-mediated feedback. J Immunol. 2012;189:2784–2792. doi: 10.4049/jimmunol.1200310. doi: 10.4049/jimmunol.1200310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McAlpine CS, Huang A, Emdin A, Banko NS, Beriault DR, Shi Y, Werstuck GH. Deletion of myeloid GSK3α attenuates atherosclerosis and promotes an M2 macrophage phenotype. Arterioscler Thromb Vasc Biol. 2015;35:1113–1122. doi: 10.1161/ATVBAHA.115.305438. doi: 10.1161/ATVBAHA.115.305438. [DOI] [PubMed] [Google Scholar]