

Supplemental Digital Content is available in the text.
Keywords: carotid arteries; mass spectrometry; muscle, smooth; proteome; vascular remodeling
Abstract
Objective—
Vascular remodeling is associated with complex molecular changes, including increased Notch2, which promotes quiescence in human smooth muscle cells. We used unbiased protein profiling to understand molecular signatures related to neointimal lesion formation in the presence or absence of Notch2 and to test the hypothesis that loss of Notch2 would increase neointimal lesion formation because of a hyperproliferative injury response.
Approach and Results—
Murine carotid arteries isolated at 6 or 14 days after ligation injury were analyzed by mass spectrometry using a data-independent acquisition strategy in comparison to uninjured or sham injured arteries. We used a tamoxifen-inducible, cell-specific Cre recombinase strain to delete the Notch2 gene in smooth muscle cells. Vessel morphometric analysis and immunohistochemical staining were used to characterize lesion formation, assess vascular smooth muscle cell proliferation, and validate proteomic findings. Loss of Notch2 in smooth muscle cells leads to protein profile changes in the vessel wall during remodeling but does not alter overall lesion morphology or cell proliferation. Loss of smooth muscle Notch2 also decreases the expression of enhancer of rudimentary homolog, plectin, and annexin A2 in vascular remodeling.
Conclusions—
We identified unique protein signatures that represent temporal changes in the vessel wall during neointimal lesion formation in the presence and absence of Notch2. Overall lesion formation was not affected with loss of smooth muscle Notch2, suggesting compensatory pathways. We also validated the regulation of known injury- or Notch-related targets identified in other vascular contexts, providing additional insight into conserved pathways involved in vascular remodeling.
Notch ligands and receptors participate in vascular remodeling in response to injury, and specific roles have been identified using targeted mouse models. Mice heterozygous for a mutant Jagged1 allele in endothelial cells respond with enhanced neointimal lesion formation after carotid artery injury.1 Soluble Jagged1, an inhibitor of Jagged1-mediated Notch activation, inhibits neointima formation after endothelial denudation by balloon injury in rat carotid arteries.2 Notch1 heterozygous deficient mice display a 70% reduction in neointimal lesion formation after carotid artery ligation, but no change was observed in homozygous Notch3 knockout mice in the same study.3 To date, no in vivo studies have been published studying the isolated loss of Notch2 in neointimal lesion formation. Given that global deletion of Notch2 results in embryonic lethality by E11.5 because of cardiovascular defects,4,5 one goal of this study was to characterize the impact of conditional and inducible loss of Notch2 signaling in vascular smooth muscle cells (VSMC) in neointimal lesion formation.
We previously showed that Notch2 activation by Jagged1 mediates a unique function in human VSMC to suppress proliferation.6 Subsequent studies have confirmed the antiproliferative effects of Notch2 by demonstrating Notch2-specific inhibition of PDGF-B (platelet-derived growth factor B)–dependent proliferation in human aortic VSMC and increased ex vivo proliferation of VSMC isolated from mice with targeted smooth muscle deletion of Notch2 compared with wild-type cells.7 We hypothesized that loss of function of smooth muscle Notch2 signaling would result in a hyperproliferative response in vivo with increased neointimal lesion formation after vascular injury.
Vascular occlusive disorders are characterized by extensive biological changes in the vessel wall in addition to the proliferation and abluminal migration of VSMC. Many genes and pathways have been targeted, particularly in mouse models of vascular injury, demonstrating that complex gene network activation is required for vascular remodeling. However, there is limited information on how global protein levels change during specific stages of neointimal lesion formation in comparison to uninjured vessels. Thus, a second goal of this study was to define unique vascular proteomic signatures that distinguish different stages of neointimal lesion formation in response to altered blood flow. A recent innovation in protein analysis workflow is sequential window acquisition of all theoretical spectra (SWATH8–11) ion scanning technique for isotope-free protein analysis. SWATH is innovative because it matches experimental mass spectra with spectral libraries instead of theoretical spectra derived from protein sequence information. It is powerful because it meets the challenge of simultaneous protein identification and quantification.10,12 Recently, the utility of SWATH was demonstrated in the analysis of proteomic changes in the vascular endothelium after irradiation of brain arteriovenous malformations.13 In the context of carotid vasculature, non-SWATH–based proteomic analysis has been used to determine the molecular signature of symptomatic carotid plaques.14 These studies demonstrate the value of proteomics-based approaches in elucidating the elaborate molecular-level changes occurring in vascular injury and ultimately in spurring the development of novel approaches for treating vascular disease. To our knowledge, our study is the first use of SWATH proteomics analysis to identify injury signatures in the carotid artery remodeling process, as well as Notch-related protein responses.
Materials and Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Mouse Models
Experimental protocols using mice were approved by the Maine Medical Center Institutional Animal Care and Use Committee. Mice were housed in our clean barrier facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. In the Notch2 conditional null line, loxP sites flank exon 3 of the Notch2 allele.4 Cre recombination results in excision of exon 3 and a frameshift-mediated premature truncation of Notch2 protein, generating a null allele. Global modification of this Notch2 allele is embryonic lethal by E11.5.4,5 The SM-MHC (smooth muscle myosin heavy chain)-CreERT2 driver strain15 was obtained by request from Dr Stefan Offermanns (University of Heidelberg) via Dr Joseph M. Miano (University of Rochester) and was induced with tamoxifen to express Cre recombinase in smooth muscle cells. The transgene is located on the Y chromosome, and thus a single copy of the transgene is maintained for passage through male breeders only. The crossed floxed Notch2 and SM-MHC-CreERT2 lines were maintained on the C57BL/6J background. The Rosa26 Cre reporter16 line was obtained from The Jackson Laboratory (Bar Harbor, ME). Male mice were started in all in vivo experiments at ≈10 weeks of age.
Human Atherosclerotic Lesion Collection
All research involving human subjects was approved by the Maine Medical Center Institutional Review Board, which is accredited by the Association for the Accreditation of Human Research Protection Programs, Inc. Atherosclerotic plaque was obtained from human subjects undergoing endarterectomy surgery under our institutional review board–approved protocol 4530NR with written informed consent from study participants. Specimens included in this article were obtained from an 80-year-old male donor (Figure 6C and 6D; enhancer of rudimentary homolog [ERH] and serpin H1) and a 72-year-old male donor (Figure 6C and 6D; vitronectin). Collected plaque was processed for paraffin embedding and sectioned at 5 μm intervals.
Genomic DNA Isolation
Genomic DNA was isolated from murine toe or tail samples or carotid arteries. Lysis buffer was 0.1 mol/L Tris (pH=8.8), 0.2 mol/L NaCl, 5 mmol/L EDTA, and 0.5% SDS. Immediately before use, proteinase K was added to a final concentration of 0.4 mg/mL, and samples incubated at 55°C overnight. DNA was precipitated with an equal volume of isopropanol and pelleted at 13 400 rpm. The supernatant was discarded, and the pellet was washed with 70% ethanol and centrifuged at 13 400 rpm. The supernatant was discarded and the pellet air dried. The DNA was resuspended in 50 μL distilled water.
Genotyping and Cre Excision Analysis
Polymerase chain reaction (PCR) primers used for this study are listed below and in the Major Resources Table in the online-only Data Supplement. Standard protocols used 2 pmol/μL primers in MasterMix (4PRIME, Inc). For genotyping the Notch2 allele, the cycling conditions were 95°C for 3 minutes, followed by 40 cycles of 95°C for 45 seconds, 60°C for 45 seconds, and 72°C for 75 seconds, followed by a final 72°C for 2 minutes. This yielded a 161 bp band from the wild-type allele and a 201 bp product from the floxed allele. For genotyping the SM-MHC-CreERT2 transgene, cycling conditions were 93°C for 3 minutes, and 35 cycles of 93°C for 30 seconds, 58°C for 30 seconds, and 72°C 1 minute, followed by a final 72°C for 7 minutes. This yielded a 455 bp product corresponding to the transgene. Negative controls of the reaction without template were used for each reaction.
Primers used were as follows:
SM-MHC-CreERT2: TCCAACCTGCTGACTGTG, TCAGAGTTCTCCAGGG
Notch2 floxed: TAGGAAGCAGCTCAGCTCACAG, ATAACGCTAAACGTGCACTGGAG
N2-L3: GCTCAGCTAGAGTGTTGTTCTTG
N2-L5; AGAACCATTGGTTTAGTGTCTCC
Cre recombination of the floxed Notch2 allele was detected by PCR using genomic DNA isolated from carotid arteries with the N2-L3 and N2-L5 primers. These primers flank the 2 loxP sites, yielding a nonrecombined 1.9 kb product from the floxed allele and an 887 bp product after Cre recombinase-mediated excision.
Tamoxifen Induction
Mice received 5 consecutive days of intraperitoneal injections of 100 μL of a 10 mg/mL solution of tamoxifen (Sigma) dissolved in corn oil (Sigma), a daily dose of 1 mg/mouse. The stock solution was prepared by warming the solution to 55°C to dissolve. Administration of 100 μL corn oil served as the vehicle control. The tamoxifen induction period was followed by a 2 week Cre-mediated recombination period before arterial ligation or sham surgery. Mice receiving tamoxifen were housed separately from mice receiving corn oil.
Ligation of the Mouse Common Carotid Artery and Tissue Collection and Processing
This procedure was performed as described.17,18 In brief, each mouse was anesthetized, and the left carotid artery was exposed. The left common carotid artery was completely tied off using suture just proximal to the carotid bifurcation. The skin incision was closed, and the mouse was allowed to recover. Mice were maintained for 6 or 14 days after arterial ligation. For sham surgeries, mice were anesthetized, and the left carotid artery was exposed before closing the wound with sutures. To collect vessels for paraffin embedding, arteries were perfusion fixed with 4% paraformaldehyde, dissected, and maintained in fixative overnight at 4°C. Tissues were then processed for paraffin embedding. Each ligated carotid artery was sectioned using the ligation as a reference point, and sections at specific distances (200 μm, 350 μm, 500 μm, 1 mm, 1.5 mm, and 2 mm) from the ligature were collected for quantification and additional immunostaining. To collect vessels for SWATH proteomic analysis, vessels were perfused with PBS, dissected, and immediately frozen and stored at −80°C until protein extraction.
Detection of β-Galactosidase Activity
For detection of β-galactosidase activity, carotid vessels were perfusion fixed with 4% paraformaldehyde, harvested, and kept in 4% paraformaldehyde for 2 hours at 4°C. Vessels were washed in PBS and were given two 30-minute incubations with detergent rinse (1 mol/L magnesium chloride, 1% sodium deoxycholate, and Nonidet P-40 in PBS). The specimens were then wrapped in foil overnight in a 37°C incubator in staining solution, which contained detergent rinse ingredients plus 1 mol/L potassium ferricyanide, 0.5 mol/L potassium ferrocyanide, 1 mol/L Tris, and 1 mg/mL of x-gal staining solution (Bioline). The next morning, specimens were washed 3× in PBS before fixation in 4% paraformaldehyde overnight. Specimens were then processed, sectioned at 5 μm, and counter stained with nuclear fast red.
Morphometric Analysis
The importance of location in morphometric analysis of vascular lesion formation is established.17,19,20 Strain differences in neointimal lesion formation should also be taken into consideration.21 Multiple valid models of quantification have been used, from intermittent sections along the length of the injured artery,19 to sections across a 500 μm to 3 mm segment proximal to the ligation,22 to sections from a single point 3 mm from the ligation.23 Some groups have chosen to obtain measurements from the apex of the lesion as determined by serial sections at 150 μm intervals across the entire length of the artery.24 Alternatively, some groups have performed longitudinal sectioning,17 with quantification by lesion length or area,25 or as a percentage of total vessel area.26,27 We chose to analyze morphometric data in aggregate for 6 distances along the vessel (200 μm, 350 μm, 500 μm, 1 mm, 1.5 mm, and 2 mm from the ligature) to capture changes observed across the region of highest remodeling.20 The widest variation in neointimal area is typically observed at the closest distance of 200 µm because of the increased likelihood of clotting near the ligature site.20 Morphometric analysis was performed by tracing of anatomic features of cross-sections of the carotid artery at known distances from the ligature. Measurements were made for the lumen circumference (defining lumen area), the internal elastic lamina circumference, the circumference of the external elastic lamina, and the outermost adventitial circumference using a pen tablet (Intuos 4 Professional, Wacom). Measurements were quantified using National Institutes of Health ImageJ software.28 A stage micrometer was used to determine the linear conversion rate of number of pixels per mm. The neointimal area was determined by subtracting the luminal area from the area bound by the internal elastic lamina. The medial area was determined by subtracting the area bound by the internal elastic lamina from the area bound by the external elastic lamina. The adventitial area was determined by subtracting the area bound by the external elastic lamina from the outermost adventitial area. Measurements were performed with the observer blinded to experimental group. For the morphometric analysis, 13 Notch2VEH-CTL mice (Notch2fl/fl;SM-MHC-CreERT2 mice treated with corn oil) were compared with 13 Notch2SMC-null mice (Notch2fl/fl;SM-MHC-CreERT2 mice treated with tamoxifen) and 13 Notch2SMC-WT mice (Notch2wt/wt;SM-MHC-CreERT2 mice treated with tamoxifen) at the 14-day time point after carotid artery ligation.
Histology and Immunostaining of Paraffin-Embedded Tissue Sections
Formalin-fixed paraffin-embedded slides containing 5 cross-sections of 5 μm each were baked for 45 minutes at 60°C. The sections were then rehydrated in a rundown of AmeriClear (Cardinal Health) followed by decreasing percentages of ethanol (100%, 95%, and 70%). Once rehydrated, some sections were stained with hematoxylin/eosin or Verhoeff stain. Some sections underwent antigen retrieval by steaming in 0.01 mol/L sodium citrate buffer pH=6.0 for 30 minutes. After cooling, the slides were washed in water and then transferred to Tris-buffered saline with Tween (TBS-T) before quenching endogenous peroxidases in a 3% solution of hydrogen peroxide in PBS for 30 minutes. Slides were washed twice in TBS-T before the sections were permeabilized in a solution of 0.5% Triton X-100 in PBS for 45 minutes on an orbital shaker. The slides were then washed once in distilled water and once in TBS-T before blocking in 2% BSA with 2% goat serum in PBS overnight at 4°C. The next day, primary antibodies or primary antibody IgG controls (please see the Major Resources Table in the online-only Data Supplement) were diluted in blocking solution and incubated on the sections overnight at 4°C. Secondary only controls were incubated with blocking solution for the duration of the primary antibody step. After 3 washes in TBS-T, the sections were incubated with SignalStain Boost IHC Detection Reagent (Cell Signaling) for 30 minutes at room temperature. Slides were then washed 3 more times in TBS-T before reacting with SignalStain diaminobenzidine substrate kit (Cell Signaling). The reaction was quenched with tap water before counterstaining with hematoxylin. Finally, the sections were dehydrated through increasing ethanol (95% and 100%) and 3 AmeriClear baths before coverslipping. Primary antibody IgG controls and secondary only control sections were examined for all antibodies (except for the Ki-67 antibody) to confirm lack of staining in the absence of primary antibody incubation. The Ki-67 staining was performed using Avidin/Biotin Blocking Kit (Vector Laboratories, Cat #SP-2001), Biotin-SP AffiniPure goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Cat #111-065-144), and the VECTASTAIN Elite ABC-HRP Kit (Vector Laboratories, Cat #PK-6100). Ki-67 quantification was performed with National Institutes of Health ImageJ software,28 and a control slide containing a section of mouse spleen was analyzed to confirm staining specificity. For staining of mouse tissue with mouse monoclonal antibodies, the Vector M.O.M. Immunodetection Kit (Vector Laboratories Cat #PK-2200) and protocol were used with the following modifications. Antigen retrieval was performed using 0.01 mol/L sodium citrate buffer (pH=6.0) with 0.05% tween for 27 minutes, and endogenous peroxidases were blocked with a solution of 3% hydrogen peroxide in PBS for 15 minutes. M.O.M Block (5 drops in 2.5 mLs of PBS) was incubated overnight at 4°C with an additional 30 minutes the next morning at room temperature. The biotinylated anti-mouse IgG reagent was used at one quarter of the recommended dilution (1 µL per 1 mL).
Proteomic Analysis of Carotid Arteries Using SWATH
Carotid arteries were weighed and proteins extracted using the Qproteome kit (Qiagen). On ice, 10 μL protease inhibitor plus 0.04 μL benzonase nuclease was added to 1 mL of lysis buffer. A total of 180μL of lysis solution was then added to each tube containing 1 artery, along with 2 small magnetic beads per sample. Samples were then homogenized at maximum speed for 60 seconds using an Autodisruptor. The tubes were then rechilled on ice, and the process was repeated until the samples were sufficiently homogenized. The tubes were then centrifuged at 14 000 rpm for 10 minutes in a precooled microcentrifuge at 4°C. The supernatant was transferred into a new tube, and 4 volumes (720 μL) of ice cold acetone were added. The tubes were then frozen at −70°C before mass spectrometry.
For this study, SWATH was used as a comprehensive strategy for analysis of all detectable analytes in samples via a data-independent acquisition method29 using nanospray infusion of tryptic peptides, after separation on a ThermoFisher/Dionex U3000 nanoscale liquid chromatograph as described,30,31 on a Sciex 5600 TripleTOF mass spectrometer. A mouse blood vessel ion library comprised 4091 proteins was constructed using ProteinPilot software. For identification of peptides generated by SWATH, multiple fragment ion chromatograms were retrieved from the spectral library for each peptide of interest. These spectra were compared with the extracted fragment ion traces for the corresponding isolation window to identify the transitions that best identify and quantify the target peptide. SWATH protein responses were determined using PeakView software, and this information was extracted for principle component analysis and t test comparisons using MarkerView software.32
For the Protein Analysis Through Evolutionary Relationships (PANTHER) gene ontology classification analyses, lists of significantly regulated proteins generated by SWATH proteomics were analyzed using PANTHER version 13.0 (released November 12, 2017).33 For the molecular function categorization (Figures 2B and 5D), differentially expressed proteins after injury were classified based on molecular function and displayed by the percent of proteins in each category. The PANTHER Overrepresentation Tests (released December 5, 2017; Table 2) were generated with an input text file with all Notch2-regulated genes as determined by the 6- and 14-day Notch2SMC-NULL versus Notch2SMC-WT SWATH proteomic comparisons with their respective fold changes and the Mus musculus Reference Gene List. Annotation Data Sets used included PANTHER Pathways, PANTHER GO-Slim Molecular Function, and REACTOME pathways. Unclassified data were not included in Table 2, and only a selected portion (14 of 55, or ≈25%) of significantly overrepresented REACTOME pathways was included because of space constraints.
Statistical Analysis
Morphometric data were graphed and analyzed with GraphPad Prism 7.0 (GraphPad Software Inc, San Diego, CA), and comparisons of normally distributed variables between multiple experimental groups were made using 2-way ANOVA. Ki-67 staining quantification was also analyzed in GraphPad Prism 7.0 using multiple t tests with the 2-stage step-up method of Benjamini, Krieger, and Yekutieli for a false discovery rate approach. SWATH data were analyzed in MarkerView using t test comparisons with P<0.05 considered significant. Statistical analysis in PANTHER was performed by Fisher exact test with false discovery rate multiple test correction in each case.
Results
Characterization of the SM-MHC-CreERT2 Strain in Vascular Injury and VSMC Loss of Notch2
In the original SM-MHC-CreERT2 mouse model characterization,15 no significant Cre recombinase activity was noted in the absence of tamoxifen when crossed with the Rosa26LacZ Cre reporter strain (B6.129S4-Gt(ROSA)26Sortm1Sor/J).16 We repeated this cross to assess Cre-mediated recombination in carotid artery VSMC and to determine the contribution of SM-MHC–expressing medial VSMC to the neointimal layer after injury. In this cross, Cre-mediated recombination to induce β-galactosidase activity is a permanent mark of differentiated VSMC within the vessel. First, Cre expression was validated in mature carotid VSMC in uninjured carotid arteries from double transgenic mice. β-Galactosidase activity was detected in medial VSMC after induction treatment with tamoxifen but not with corn oil vehicle (Figure 1A). After tamoxifen induction and carotid artery ligation, both neointimal and medial cells were highly stained (Figure 1B). In this model, the strong presence of positively stained cells in the neointimal and medial layers of tamoxifen-induced mice indicates that the neointima is predominantly derived from differentiated medial VSMC. Of note, occasional blue cells (0–2 per high power field) were observed in ligated vessel cross-sections from double transgenic mice in the absence of tamoxifen (Figure 1C). Although this recombination in the corn oil vehicle group is negligible in comparison to the widespread positive staining observed in VSMC after tamoxifen treatment, this did suggest a low level of baseline Cre recombinase activity in some cells in the vessel wall.
Figure 1.
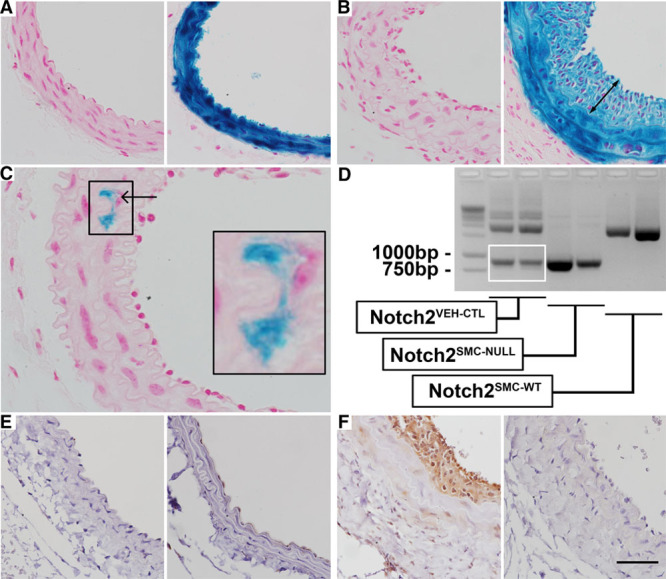
Validation of inducible Notch2 targeting in vascular smooth muscle cell (VSMC). A–C, The SM-MHC (smooth muscle myosin heavy chain)-CreERT2 Cre strain was crossed to the ROSA26LacZ Cre reporter strain, and uninjured double transgenic mice (A) were treated with corn oil vehicle (left) or tamoxifen (right). Whole-mount tissue detection of β-galactosidase activity was performed and sections prepared to examine cellular distribution of Cre recombination. After tamoxifen induction, VSMC were highly stained. B, Ligated carotid arteries from double transgenic mice treated with corn oil vehicle (left) or tamoxifen (right) were collected 14 d after ligation, and staining of both neointimal (double arrow) and medial VSMC was observed in vessels from tamoxifen-induced mice. C, Occasional low levels of recombination were noted in ligated vessel cross-sections from double transgenic mice treated with corn oil vehicle (arrow in C, higher magnification in inset). D, Analysis of the efficiency of Cre recombination was performed by polymerase chain reaction (PCR) of genomic DNA isolated from corn oil vehicle–treated Notch2fl/fl; SM-MHC-CreERT2 (Notch2VEH-CTL), tamoxifen-treated Notch2fl/fl; SM-MHC-CreERT2 (Notch2SMC-NULL), and tamoxifen-treated Notch2wt/wt; SM-MHC-CreERT2 (Notch2SMC-WT) carotid arteries 14 d after ligation. PCR was performed with the N2-L3: N2-L5 primer pair, which amplifies a 1.9 kb fragment from the nonrecombined floxed allele, an 887 bp product from the Cre-recombined locus, and an ≈1450 bp fragment from the wild-type allele. Some 887 bp product was detected in the Notch2VEH-CTL ligated carotid genomic DNA samples (white box). Immunostaining to detect Notch2 protein in uninjured carotid arteries (E) from Notch2VEH-CTL (left) and Notch2SMC-NULL (right) mice and from injured carotid arteries (F) collected 14 d after ligation in Notch2VEH-CTL (left) and Notch2SMC-NULL (right) mice. Scale bar, 50 μm in A and B and E and F, and 25 μm in C.
For our loss of Notch2 studies, we used a previously characterized floxed Notch2 allele4 in combination with the SM-MHC-CreERT2 strain.15 A PCR-based assay to measure Cre recombination efficiency was performed in all 3 experimental conditions used for this study. Corn oil–treated Notch2fl/fl; SM-MHC-CreERT2 mice served as a vehicle control and are subsequently designated as Notch2VEH-CTL. Tamoxifen-treated Notch2fl/fl; SM-MHC-CreERT2 mice, which undergo loss of Notch2 in smooth muscle cells, are designated as Notch2SMC-NULL. Tamoxifen-treated Notch2wt/wt; SM-MHC-CreERT2 mice do not have the potential for Cre-mediated loss of the wild-type Notch2 allele and are subsequently designated as Notch2SMC-WT. To analyze Cre recombination in these experimental groups, genomic DNA was isolated from carotid arteries 14 days after carotid artery ligation, and PCR was performed using the N2-L3:N2-L5 primer pair. This primer pair amplifies a 1.9 kb product in the nonrecombined floxed allele, an 887 bp product in the recombined allele, and an ≈1450 bp product in a wild-type Notch2 allele (Figure 1D). In Notch2SMC-WT mice, only the ≈1450 bp product corresponding to the wild-type Notch2 allele was detected. In Notch2SMC-NULL mice, loss of the 1.9 kb nonrecombined floxed allele product and predominant presence of the 887 bp product resulting from Cre recombination was noted. In Notch2VEH-CTL mice, evidence of the 887 bp Cre-recombined locus was detected (Figure 1D, white box) in addition to the 1.9 kb nonrecombined floxed allele product. This detection of recombination in the absence of tamoxifen was consistent with the observation of occasional recombination in vehicle-treated double transgenic Rosa26LacZ Cre reporter mice. Due to the sensitivity of PCR amplification, even a minimal amount of recombination could be expected to result in a detection of a band corresponding to the Cre-recombined locus. To evaluate the effects of Cre recombination on Notch2 protein, immunostaining was also performed. No Notch2 staining was detected in uninjured Notch2VEH-CTL or Notch2SMC-NULL mice (Figure 1E), consistent with our previous reports of low Notch2 expression in quiescent carotid arteries and increased expression of Notch2 after vascular injury.6,34 Notch2 immunostaining was also used to verify loss of VSMC Notch2 protein expression in Notch2SMC-null carotid arteries after injury (Figure 1F).
Protein Profiling of Remodeling Vessels in Notch2SMC-WT Mice
To characterize the overall proteomic signature of the vessel wall to understand molecular changes during neointimal lesion formation, we performed protein profiling to compare uninjured carotid arteries with injured carotid arteries at 6 and 14 days after ligation using a SWATH proteomics approach in Notch2SMC-WT mice. SWATH analysis of injured Notch2SMC-WT vessels showed a protein profile response that included 2 distinct waves of protein regulation, as well as sustained changes characteristic of injury (Figure 2A). In comparison to uninjured carotid arteries, Notch2SMC-WT remodeling arteries had 341 distinct proteins upregulated at day 6, with 35 downregulated (Figure 2A, first wave; Table I in the online-only Data Supplement) and 116 proteins uniquely upregulated at day 14 with 13 downregulated (second wave; Table II in the online-only Data Supplement). In addition, we found 157 proteins that remained elevated and sustained throughout the 2 weeks of remodeling, and 9 that remained at lower levels (Figure 2A, sustained; Table III in the online-only Data Supplement). The numbers displayed for the first and second waves include 5 proteins that were downregulated at 6 days and upregulated at 14 days, and 3 proteins that were upregulated at 6 days and downregulated at 14 days (Table IV in the online-only Data Supplement). We additionally used the PANTHER ontology classification system35 to provide a more detailed molecular analysis of these protein profiles. The PANTHER GeneOntology tool showed that in Notch2SMC-WT carotid arteries, 45% of the regulated proteins at 6 and 14 days after injury fall into the catalytic activity category and 33% correspond to binding activity (Figure 2B). These data are consistent with a model where active changes in proteins involved in enzymatic biochemical reactions and binding (including transcription factors and receptor binding) are regulated strongly during the remodeling response. From the Notch2SMC-WT injury-related proteomics analysis, several of the proteins identified already have established functions in vascular injury and remodeling, and these are summarized in Table 1.
Figure 2.
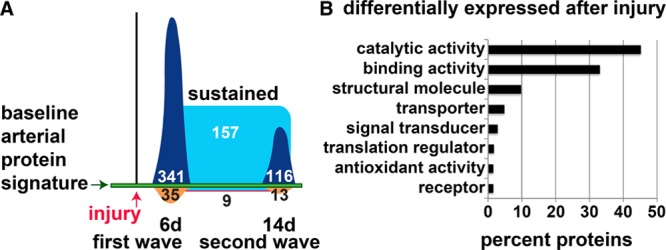
Global proteomic analysis of the vascular injury response. A, Schematic overview of changes in the baseline arterial protein signature in Notch2SMC-WT mice (Notch2wt/wt;SM-MHC (smooth muscle myosin heavy chain)-CreERT2 mice treated with tamoxifen) at 6 d (n=3) and 14 d (n=3) after carotid artery ligation injury compared with uninjured Notch2SMC-WT mice (no surgery, n=6) detected by sequential window acquisition of all theoretical spectra analysis performed on single mouse carotid arteries. Proteins upregulated at 6 d (first wave, dark blue, 341) and 14 d (second wave, dark blue, 116); proteins downregulated at 6 d (orange, 35) and 14 d (orange, 13); proteins upregulated at both 6 and 14 d (sustained, light blue, 157); and proteins downregulated at both 6 and 14 d (light blue with red border, 9), not drawn to scale. The displayed numbers include 5 proteins that were downregulated at 6 d and upregulated at 14 d, and 3 proteins that were upregulated at 6 d and downregulated at 14 d. B, Proteins differentially expressed after injury were analyzed in the Protein Analysis Through Evolutionary Relationships database, categorized by molecular function, and displayed by the percent of proteins in each category.
Table 1.
Related In Vivo Vascular Functions of Selected Proteomic Hits
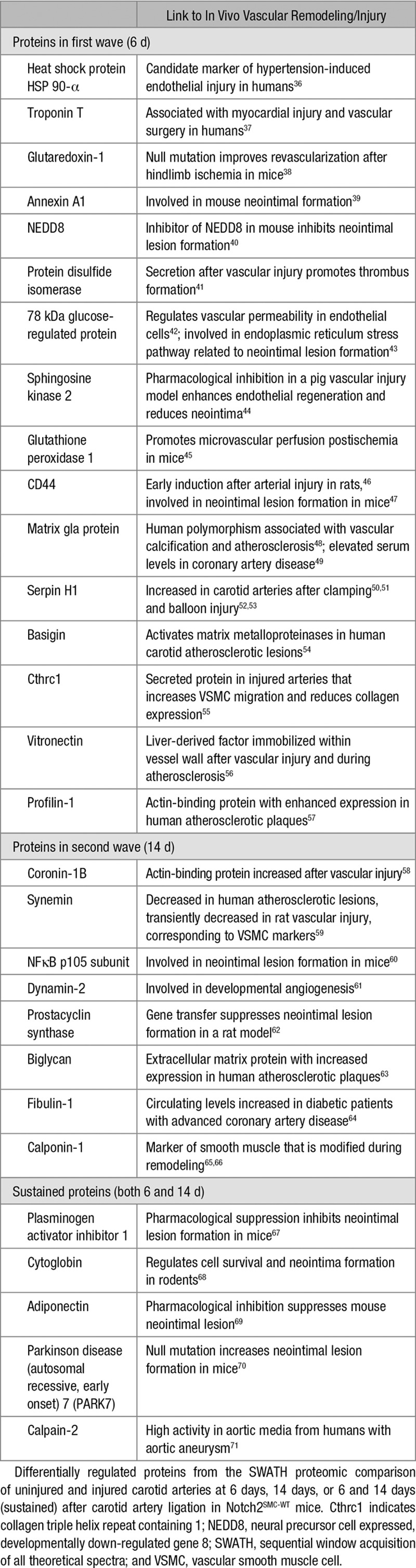
The identification of several proteins from the Notch2SMC-WT injury-related proteomics analysis as established contributors to vascular remodeling indicates the power of the SWATH proteomic approach for detecting known injury response proteins and identifying new candidates for further analysis. To identify changes in vascular remodeling in Notch2VEH-CTL, Notch2SMC-NULL, and Notch2SMC-WT vessels, morphometric analysis was performed to characterize lesion formation, and SWATH proteomic analysis addressed changes in protein profiles.
Analysis of the Remodeling Response in the Presence or Absence of Notch2
To quantify effects of the loss of VSMC Notch2 on neointimal lesion formation, we analyzed morphometric data in aggregate for 6 distances along the vessel (200 μm, 350 μm, 500 μm, 1 mm, 1.5 mm, and 2 mm from the ligature) at 14 days after ligation. Representative sections used for quantification at the 2 mm distance are shown in Figure 3A. A sham-operated Notch2SMC-null vessel section is included for comparison. There was no significant difference between Notch2VEH-CTL, Notch2SMC-NULL, or Notch2SMC-WT vessels in neointimal area, medial area, or neointimal/medial ratio (Figure 3B). Likewise, no significant difference in proliferation was noted in Notch2SMC-NULL vessels (Figure 3C and 3D). Although this finding was surprising because of prior work by our group and others in demonstrating the role of Notch2 in mediating VSMC growth arrest,6,7 the overarching complexity of regulators of the neointimal lesion formation response17,18 suggests that several different pathways could be compensating for the loss of VSMC Notch2 in this model. For example, Notch3 is the predominant Notch receptor in carotid VSMC6 and is highly expressed in the vessel wall throughout the remodeling process both in the presence and absence of Notch2 (data not shown).
Figure 3.
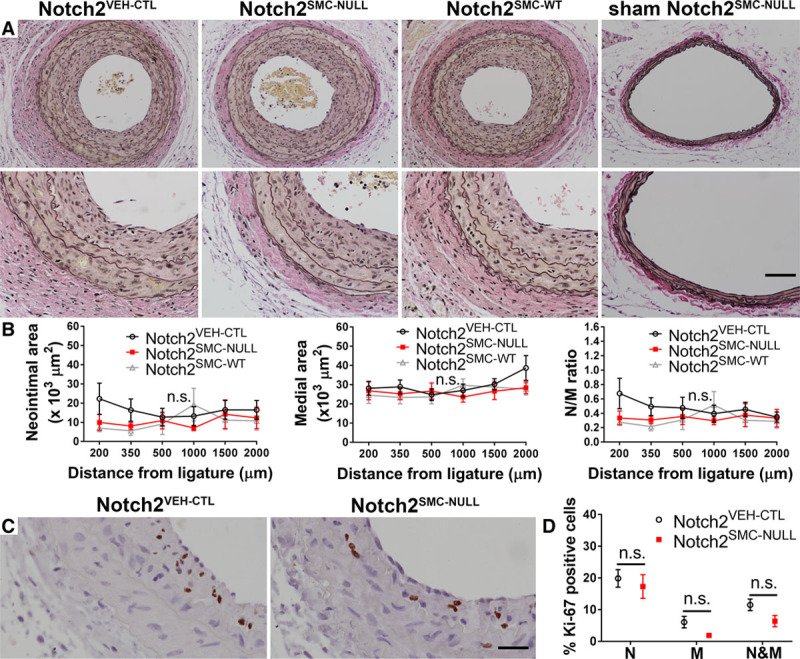
Lesion formation and cell proliferation in the absence of smooth muscle Notch2. A, Representative lesion formation in Notch2VEH-CTL (Notch2fl/fl;SM-MHC (smooth muscle myosin heavy chain)-CreERT2 mice treated with corn oil), Notch2SMC-NULL (Notch2fl/fl;SM-MHC-CreERT2 mice treated with tamoxifen), and Notch2SMC-WT (Notch2wt/wt;SM-MHC-CreERT2 mice treated with tamoxifen) 14 d after carotid artery ligation or sham (far right) surgery at 2000 µm from the ligature or estimated sham ligature site. n=13 for each experimental group, sham n=1. Scale bar, 100 µm in top row, 50 µm in bottom row. B, Quantification of neointimal area, medial area, and neointimal/medial (N/M) ratio for 6 set distances ranging from 200 to 2000 µm from the ligature, n.s. indicates not significant by 2-way ANOVA. C, Representative Ki-67 staining from Notch2VEH-CTL and Notch2SMC-NULL lesions, scale bar, 25 µm. D, Percent Ki-67–positive cells in the neointimal layer (N), medial layer (M), or combined neointimal and medial layers (N&M), n=7 per experimental group for Ki-67 staining, n.s. indicates not significant by multiple t tests with the 2-stage step-up method of Benjamini, Krieger, and Yekutieli for a false discovery rate approach.
Further characterization of lesion formation in Notch2SMC-NULL vessels was performed by immunohistochemical staining. Consistent with previous observations,17 the carotid artery ligation model demonstrates low levels of infiltrating immune cells in the vessel wall, as determined by CD45 staining (Figure 4A). No qualitative difference was observed in level of macrophage infiltration in Notch2SMC-NULL vessels compared with Notch2VEH-CTL and Notch2SMC-WT vessels as measured by CD68 staining (Figure 4B). Likewise, staining patterns for VSMC markers SMA (smooth muscle actin; Figure 4C) and SM-MHC (Figure 4D) were similar across experimental groups. No change in apoptosis, as measured by caspase-3 staining, was noted across experimental groups after injury (data not shown). No qualitative difference in collagen deposition was observed by Masson trichrome (Figure 4E) nor was there any significant difference in quantification of the collagen-rich adventitial area surrounding the vessel (data not shown).
Figure 4.
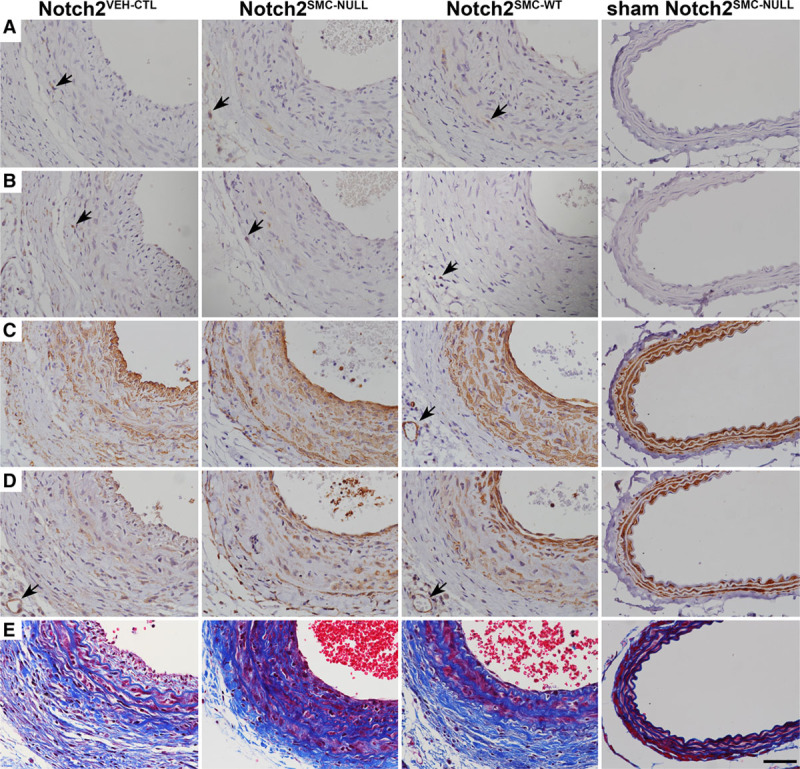
Cellular composition of vascular lesions after injury. Representative immunohistochemical staining of Notch2VEH-CTL, Notch2SMC-NULL, Notch2SMC-WT, and sham Notch2SMC-NULL carotid arteries 14 d after ligation injury or sham surgery. CD45 staining for immune cells (A, black arrows), CD68 staining for macrophages (B, black arrows), staining for vascular smooth muscle cell (VSMC) markers SMA (smooth muscle actin; C) and SM-MHC (smooth muscle myosin heavy chain; D), and Masson trichrome staining (E). Black arrows in C and D indicate cross-sections of small adventitial vessels to show staining specificity for VSMC, scale bar, 50 µm.
To learn more about global proteomic changes during the loss of Notch2 in neointimal lesion formation, we performed SWATH analysis of single vessels at the 6- and 14-day time points after carotid artery ligation.
Protein Profiling of Remodeling Vessels in Notch2SMC-NULL Mice
For primary identification of proteins differentially regulated after carotid ligation injury in the absence of VSMC Notch2, we highlight the SWATH comparison of Notch2SMC-NULL vessels with Notch2SMC-WT vessels because both of these experimental groups are SM-MHC-CreERT2 positive and have undergone tamoxifen induction, which specifically controls for any changes directly resulting from tamoxifen administration or the presence of Cre recombinase. Modified Venn diagrams summarizing this comparison at day 6 (Figure 5A) correspond to 66 proteins that are higher in Notch2SMC-WT vessels (Table V in the online-only Data Supplement), 1493 proteins that are not significantly changed, and 53 proteins that are higher in Notch2SMC-NULL vessels (Table VI in the online-only Data Supplement). For a secondary goal of identifying proteins directly related to vascular ligation injury in the context of loss of VSMC Notch2, we compared Notch2SMC-NULL vessels with sham Notch2SMC-NULL vessels (Figure 5A). Both groups are SM-MHC-CreERT2 positive, tamoxifen induced, and have undergone surgical isolation of the carotid artery. The difference in this comparison is that sham Notch2SMC-NULL carotid vessels have not undergone ligation before wound closure, which provides a highly controlled view of differences resulting specifically from carotid vessel injury. This comparison identified 561 proteins higher in Notch2SMC-NULL vessels (Table VII in the online-only Data Supplement), 1030 proteins not significantly changed, and 21 proteins higher in sham Notch2SMC-NULL vessels (Table VIII in the online-only Data Supplement). The summary of the corresponding day 14 analysis is presented in Figure 5B (Tables IX–XII in the online-only Data Supplement). An alternative schematic overview of changes specific to loss of Notch2 is provided in Figure 5C. Of additional interest, glutaredoxin-1 was the only protein identified as upregulated at both the 6- and 14-day time points in Notch2SMC-NULL vessels compared with Notch2SMC-WT vessels. Glutaredoxin-1 is a regulator of redox signaling that has previously been linked to ischemic limb revascularization72 but not to Notch signaling. Analysis of the molecular function of all proteins differentially expressed in Notch2SMC-NULL vessels compared with Notch2SMC-WT vessels reveals a similar profile to that generated from the Notch2SMC-WT analysis with the majority of proteins in the catalytic or binding activity categories (Figure 5D).
Figure 5.
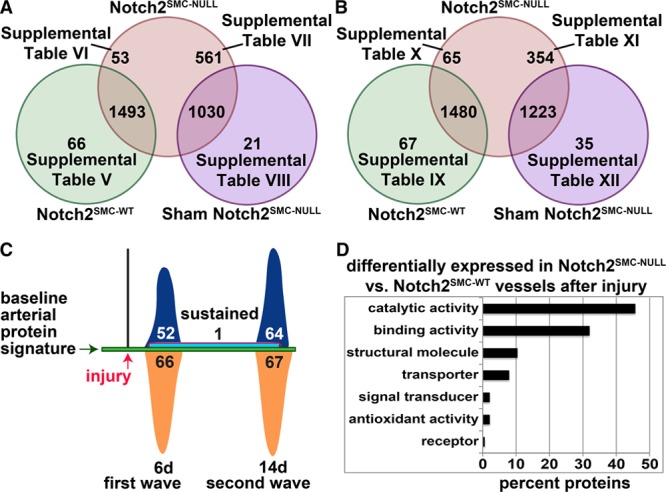
Proteomic analysis of loss of vascular smooth muscle cells (VSMC) Notch2 in the carotid artery injury response. A and B, Modified Venn diagrams representing sequential window acquisition of all theoretical spectra (SWATH) proteomic analysis performed on single mouse carotid arteries (n=3 per group) at 6 d (A) or 14 d (B) after ligation or sham ligation surgery. A, Sixty-six proteins have significantly higher levels in Notch2SMC-WT vessels (green circle, Notch2wt/wt;SM-MHC-CreERT2 mice treated with tamoxifen and ligated) compared with Notch2SMC-NULL vessels (red circle, Notch2fl/fl;SM-MHC (smooth muscle myosin heavy chain)-CreERT2 mice treated with tamoxifen and ligated). A total of 1493 proteins are not significantly changed between these 2 groups, and 53 proteins have significantly higher levels in Notch2SMC-NULL vessels. Five hundred sixty-one proteins are significantly higher in Notch2SMC-NULL vessels compared with sham Notch2SMC-NULL vessels (purple circle, Notch2fl/fl;SM-MHC-CreERT2 mice treated with tamoxifen and sham ligated). Of note, 6 of these 561 proteins are redundant to the 53 identified from the Notch2SMC-WT comparison. One thousand thirty proteins are not significantly changed between the Notch2SMC-NULL and the sham Notch2SMC-NULL groups, and 21 proteins are significantly higher in sham Notch2SMC-NULL vessels. Significance defined by P<0.05, with P values generated by SWATH proteomic analysis. References to tables with corresponding protein data are provided. B, Corresponding analysis at 14 d after ligation or sham ligation surgery. Of the 354 significantly higher Notch2SMC-NULL proteins identified in comparison to sham Notch2SMC-NULL vessels, 41 are redundant to the 65 identified from the Notch2SMC-WT comparison. C, Alternative schematic overview of changes in the baseline arterial protein signature in Notch2SMC-NULL mice at 6 and 14 d after carotid artery ligation injury compared with injured Notch2SMC-WT mice detected by the SWATH vessel analysis described in A and B. Proteins upregulated at 6 d (first wave, dark blue, 52) and 14 d (second wave, dark blue, 64); proteins upregulated at both 6 and 14 d (sustained, light blue with red border, 1); and proteins downregulated at 6 d (orange, 66) and 14 d (orange, 67). No proteins were downregulated at both 6 and 14 d. The displayed numbers include 5 proteins that were downregulated at 6 d and upregulated at 14 d, and 1 protein that was upregulated at 6 d and downregulated at 14 d (Tables V and X in the online-only Data Supplement, biphasically regulated). D, Proteins differentially expressed in Notch2SMC-NULL vessels compared with Notch2SMC-WT vessels after injury were analyzed in the PANTHER database, categorized by molecular function, and displayed by the percent of proteins in each category.
Protein Analysis of Remodeling Vessels in Notch2SMC-NULL Mice
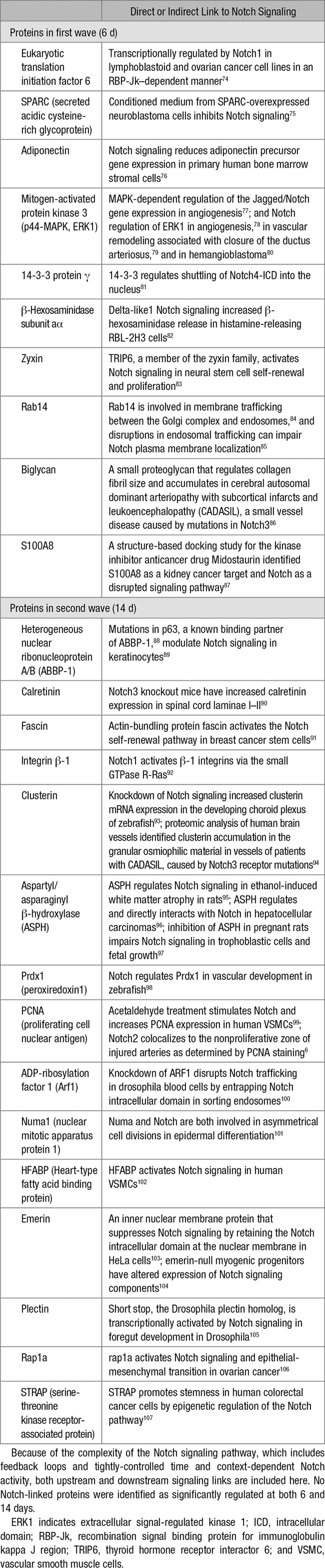
To further characterize the proteomic profiling changes observed in Notch2SMC-NULL vessels as compared with Notch2SMC-WT vessels, additional PANTHER overrepresentation and enrichment analyses were performed to determine significantly altered pathways and molecular functions as determined by fold changes of proteins identified by SWATH proteomic analysis (Table 2). PANTHER pathway analysis identified 2 significantly overrepresented pathways, glycolysis and the cholecystokinin receptor signaling pathway, which include several matrix metalloproteinases, phosphoinositide 3-kinase signaling mediators, and other regulators of migration, adhesion, cell growth and proliferation, and inflammation.35 Of the significantly overrepresented molecular functions, protein disulfide isomerase activity had the highest fold enrichment in Notch2SMC-NULL vessels as compared with Notch2SMC-WT vessels. Protein disulfide isomerase, a known endoplasmic reticulum protein that assists with protein folding, has already been identified as a downstream target of the Notch signaling pathway in Notch intracellular domain-transfected K562 cells.73 PANTHER supports enrichment analysis using pathway classifications from the REACTOME resource, and thus significantly enriched metabolic, smooth muscle contraction, transport and mitosis-related pathways identified by the REACTOME resource are also included in Table 2. The PANTHER and REACTOME analyses for significant differences in Notch2SMC-NULL vessels as compared with Notch2SMC-WT vessels are largely consistent with the types of changes expected during vascular remodeling and suggest that metabolic changes are an important feature resulting from loss of Notch2 in VSMC. Differentially regulated proteins in Notch2SMC-NULL vessels compared with Notch2SMC-WT vessels were then searched in PubMed to identify known links to the Notch signaling pathway, and selected links to Notch signaling are summarized in Table 3.
Table 2.
PANTHER Overrepresentation and Enrichment Analyses of Notch2-Regulated Proteins After Injury
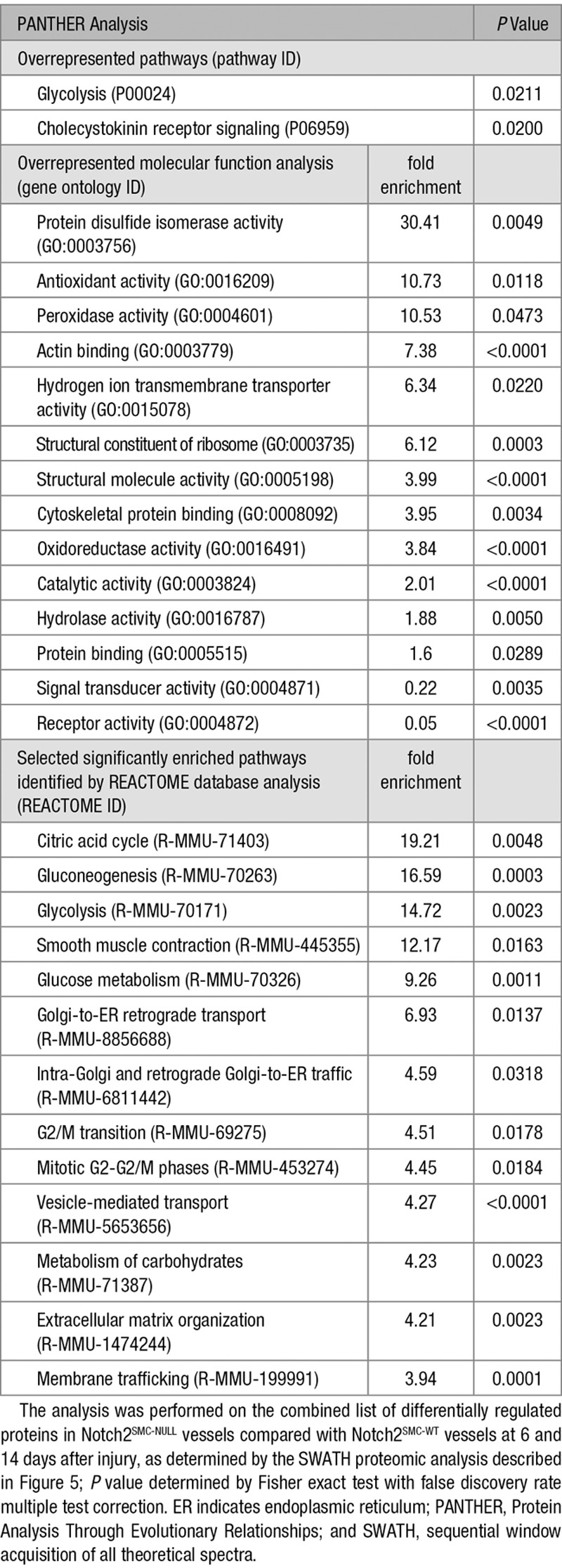
Table 3.
Differentially Regulated Proteins in Notch2SMC-NULL Vessels Compared With Notch2SMC-WT Vessels With Links to Notch Signaling
Protein Expression Changes With Loss of VSMC Notch2 During Early Remodeling and Correlation With Human Atherosclerotic Lesions
Validation of selected early protein-level changes identified by the SWATH comparison of Notch2SMC-NULL (Figure 6A) and Notch2SMC-WT (Figure 6B) vessels at 6 days after injury was performed by immunohistochemical staining. Notch signaling culminates in activation of gene expression. Thus, we postulate that proteins decreased in the absence of Notch2 may be normally activated by Notch signaling, and we chose 3 proteins for validation that fit this pattern. From the SWATH analysis, protein expression of ERH is higher in Notch2SMC-WT vessels compared with Notch2SMC-NULL vessels (Table V in the online-only Data Supplement; fold change, 5.71; P<0.01). Corresponding with this analysis, more staining is observed in Notch2SMC-WT vessel sections compared with Notch2SMC-NULL vessels sections. ERH is highly conserved in eukaryotes and contributes to cell cycle regulation through its mRNA splicing activity.108 At 6 days after injury, plectin protein expression was also detected at higher levels in Notch2SMC-WT vessel sections compared with Notch2SMC-NULL vessels sections (Table V in the online-only Data Supplement; fold change, 2.87; P<0.05). Similarly, plectin staining was higher in Notch2SMC-WT vessel sections than in Notch2SMC-NULL vessels sections. Plectin is a scaffolding protein that interacts with vimentin and actin filaments to promote vascular integrity.109 Of note, there is suggestive evidence that short stop, the Drosophila homolog of plectin, may be transcriptionally regulated by Notch signaling (also referenced in Table 3).105 The third protein selected for immunohistochemical staining validation was annexin A2. The SWATH analysis demonstrated higher expression in Notch2SMC-WT vessels compared with Notch2SMC-NULL vessels (Table V in the online-only Data Supplement; fold change, 2.58; P<0.05). Immunohistochemical staining also revealed higher protein expression in Notch2SMC-WT vessel sections compared with Notch2SMC-NULL vessels. Like plectin, annexin A2 is linked to vascular homeostasis.110
Figure 6.
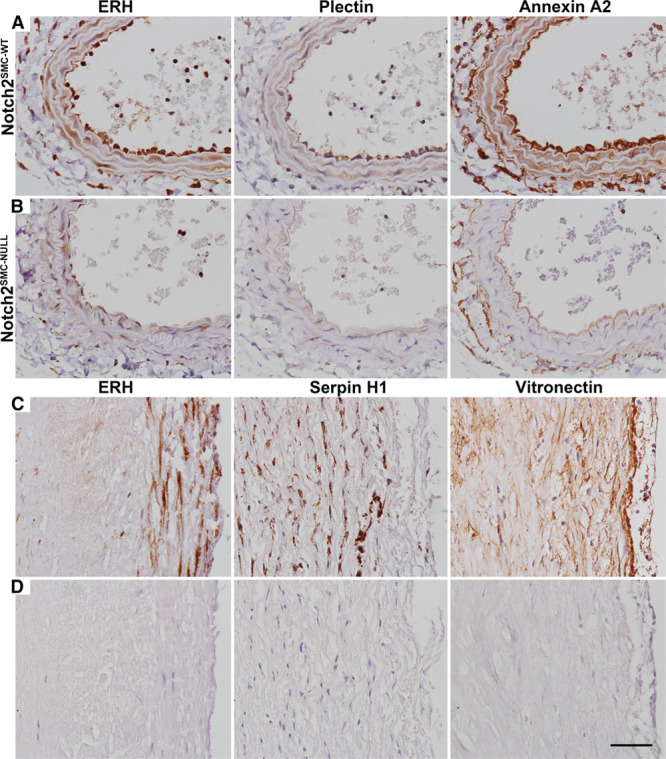
Protein expression during the early phase of vascular remodeling in murine vessels and in atherosclerotic plaque isolated from human vessels. Evaluation of changes in protein expression of enhancer of rudimentary homolog (ERH), plectin, and annexin A2 at 6 d after carotid ligation in Notch2SMC-WT (A) and Notch2SMC-NULL (B) vessel sections. C, Protein expression of ERH, serpin H1, and vitronectin in atherosclerotic plaque collected from human vessels during carotid artery endarterectomy. D, Nonspecific IgG-treated adjacent sections of human atherosclerotic plaque. Scale bar, 50 µm.
We also selected abundant injury-related proteins from our mouse proteomic screen to evaluate correspondence to human diseased vessels. To confirm that some of the targets identified by SWATH proteomics were present in detectable levels in human atherosclerotic lesions, plaque was collected from consented human donors at the time of carotid endarterectomy surgery. Representative staining for ERH, serpin H1, and vitronectin validates that these proteins are expressed in human atherosclerotic lesions (Figure 6C). Because of lack of an appropriate control specimen for atherosclerotic plaque, nonspecific IgG-treated control sections are included (Figure 6D). From the SWATH analysis, Serpin H1 was upregulated in both Notch2SMC-WT vessel sections after injury (Table I in the online-only Data Supplement; fold change, 5.36; P<0.01) and in injured Notch2SMC-NULL vessels compared with sham Notch2SMC-NULL vessels (Table VII in the online-only Data Supplement; fold change, 18.80; P<0.01×10−12). Vitronectin was upregulated after injury in Notch2SMC-WT vessels (Table I in the online-only Data Supplement; fold change, 3.42; P<0.01).
Discussion
Our study provides a detailed and comprehensive comparison of protein signatures associated with vascular injury in the mouse. In addition, we evaluated a specific hypothesis that loss of VSMC Notch2 would result in increased neointimal lesion formation after vascular injury. We previously found that Notch2 signaling in human VSMC activates a quiescence pathway, suggesting that loss of VSMC Notch2 signaling would yield a hyperproliferative response in vivo. Contrary to our hypothesis, loss of VSMC Notch2 does not alter overall lesion size nor does it alter proliferation rates in the carotid ligation injury model, either at 6 or 14 days after injury. Although strain differences contribute to some variation in overall lesion size,21 14 days are sufficient for neointimal lesion formation as previously published17 and as demonstrated by our morphometric analysis. Because VSMC proliferation is a relatively early feature of this model,17 we would have expected to capture any significant proliferation changes at the 6- or 14-day time points and thus did not design our study to examine lesion formation beyond this time. The SM-MHC-CreERT2 driver strain is commonly used to study inducible gene expression in VSMC because of its high lineage specificity relative to other smooth muscle Cre drivers. A known limitation of this model is that insertion of the Cre transgene into the Y chromosome excludes the incorporation of female mice into the study design. Fortunately, translocation of the Cre transgene to the X chromosome has recently been published,111 opening up this field for future studies in female mice to address the issue of sex as a biological variable in vascular disease pathogenesis. An additional consideration in carotid ligation model study design is the wide variety of approaches for measuring and reporting lesion size in this model. Details of different common approaches are outlined in the methods section along with our rationale for performing morphometric assessments at 6 set distances across the largest area of the lesion. One distinct advantage of the carotid ligation model is that it triggers less of an inflammatory response than other arterial injury models, such as endothelial denudation,112 which simplifies the analysis and characterization of VSMC phenotypic changes. As expected, loss of Notch2 did not alter VSMC apoptosis as determined by caspase-3 staining. This is supported by in vitro observations that there are no significant differences in ultraviolet-mediated apoptosis with overexpression or knockdown of Notch2 in VSMC as determined by caspase-3 activity or by expression of prosurvival genes.7
Our results suggest that other pathways and mediators are able to compensate for the loss of Notch2 during vascular remodeling. One possibility is that VSMC Notch3 is partially compensating for the loss of Notch2 in this context. Indeed, functional redundancy between Notch2 and Notch3 has been demonstrated during embryonic development of the vasculature.113,114 Studies in mouse models of patent ductus arteriosus, which results from failure of the ductus arteriosus to close in the transition between embryonic and postnatal circulation, demonstrate that VSMC Notch signaling via lateral induction through the vessel wall is required for closure.115 Patent ductus arteriosus can result from VSMC deletion of Jagged1115 and by disruption of all canonical Notch signal reception in smooth muscle via deletion of the Rbpj gene in VSMC.116 Patent ductus arteriosus occurs in 40% of mice with VSMC deletion of Notch2 and in 100% of mice with VSMC deletion of Notch2 combined with global heterozygous deletion of Notch3.114 Taken together, these results indicate a compelling role for Notch2 in the contractile VSMC differentiation required for ductus arteriosus closure and provide a developmental model for functional overlap between Notch2 and Notch3. The extent of the functional overlap in neointimal lesion formation could be elucidated using a similar approach to Baeten et al114 by performing carotid ligation studies in mice with different combinations of conditional mutant and wild-type Notch2 and Notch3 alleles in VSMC. Even so, neointimal lesion formation after arterial injury is a complex process governed by many different regulators that control the extent of the neointimal lesion response. We have previously reviewed genes that contribute to neointimal lesion formation after complete ligation of the common carotid artery.18 These include adhesion molecules, growth factors, cytokines, hormones, cytoskeletal components, blood components, reactive oxygen pathways, secreted proteins, transcriptional regulators, extracellular matrix proteins, transmembrane signaling molecules, intracellular enzymes, and microRNAs.18 Within each of these categories, multiple mouse models have identified numerous specific regulators of the neointimal lesion response. These regulators significantly alter lesion size or morphology when targeted by genetic ablation, transgenic expression, or by administration of inhibitors, antibodies, mimics, adenoviral vectors, or other recombinant proteins. Although single-gene studies have made important contributions to the canon of neointimal lesion response literature, many studies have focused on quantification of lesion size as the primary or exclusive outcome measure. Because of the number of genes regulating neointimal lesion size, more complex outcome analyses are needed to grasp resultant protein-level changes in an unbiased and comprehensive manner. In this study, we have described methodology for proteomics analysis by SWATH to characterize neointimal lesion changes both in the presence and absence of Notch2. This comprehensive approach was validated by the identification of several known regulators of neointima formation and opens up new opportunities for further exploration with the identification of proteins previously not known to be involved in vascular remodeling or responses to Notch signaling.
Proteomic comparison of remodeling Notch2SMC-WT vessels to uninjured Notch2SMC-WT vessels identifies proteins responsive to vascular injury. We observed 2 distinct waves of protein regulation, 376 in the first wave and 129 in the second wave, that occur in the setting of 166 additional sustained changes in protein expression. In total, this injury-related analysis yielded 671 protein-level changes. In contrast, comparison of remodeling Notch2SMC-NULL vessels to remodeling Notch2SMC-WT vessels identifies changes specific to the loss of Notch2 and includes 118 protein changes in the first wave, 131 in the second wave, and only 1 sustained change for a total of 250 protein-level changes. This indicates that more protein-level changes can be attributed to vascular injury than to the loss of VSMC Notch2. This conclusion holds true for the comparison of remodeling Notch2SMC-NULL vessels with sham Notch2SMC-NULL vessels which highlights injury-related changes, with the loss of VSMC Notch2 and surgical isolation of the carotid artery held as constants. This analysis identified 582 protein-level changes at 6 days and 389 changes at 14 days for a total of 971 protein-level changes. Of additional interest, the injury response proteomes of both Notch2SMC-WT and Notch2SMC-NULL vessels demonstrate higher numbers of proteins that are upregulated in injury compared with numbers of proteins that are downregulated in injury. The comparison of remodeling Notch2SMC-NULL vessels to remodeling Notch2SMC-WT vessels, however, demonstrates waves of protein regulation that are roughly equivalent in amplitude for both upregulated and downregulated proteins at both 6 and 14 days after injury. These observations are consistent with one role of Notch as a component of a transcriptional activation complex.118 PANTHER and REACTOME pathway analysis of differentially regulated genes in the absence of Notch2 highlighted several significantly enriched pathways connected to cell metabolism. This suggests that Notch regulation of cell metabolism may prove to be an interesting area for future exploration. Notch signaling has already been linked to regulation of glucose metabolism in hepatocytes119 and in activated hepatic macrophages,120 and associated with suppression of the browning of adipocytes, a transition involving significant cellular metabolic changes.121,122
Novel candidates uncovered in our discovery approach will be useful for future continued analysis and comparison with human vascular disease. In moving toward this goal, we validated protein expression in human atherosclerotic plaque of ERH, serpin H1, and vitronectin, 3 injury-associated proteins identified in this analysis. Recently, a 4-biomarker signature of carotid atherosclerotic plaque correlating with predictive risk for rupture was identified by proteomic comparison of symptomatic and asymptomatic human carotid plaques.14 This predictive panel includes matrix metalloproteinase 9, S100A8/S100A9, cathepsin D, and galectin-3-binding protein.14 The study is a prominent example of the trend toward using unbiased and comprehensive proteomic approaches to characterize vascular changes in human atherosclerotic lesion formation. In relating these recent findings to our study, we noted that cathepsin D was identified as differentially regulated in Notch2SMC-WT vessels after injury (Table I in the online-only Data Supplement), and S100A8/S100A9 were differentially regulated in Notch2SMC-NULL vessels (Tables V and VII in the online-only Data Supplement). This overlap highlights the timeliness of this study in identifying mediators of neointimal lesion formation with established therapeutic relevance both in the presence and absence of Notch2.
The concept that Notch signaling may be of therapeutic benefit for patients with cardiovascular disease has been supported by studies addressing inflammation, particularly monocyte/macrophage activation. Both in vivo and in vitro studies (reviewed in123) suggest that the Notch ligand Dll4, potentially via Notch1, establishes a proinflammatory M1 phenotype relevant to disease conditions such as atherosclerosis. The same Dll4/Notch1 pathway in human endothelial cells promotes quiescence,124 which is required under homeostatic conditions. Thus, unique signaling based on the target cell type is important to consider. Studies focused on VSMC have shown that Jagged1/Notch3 signaling is required for development and maintenance of the mature, contractile phenotype,125,126 and we showed that Jagged1/Notch2 signaling in human smooth muscle cells supports cellular quiescence.6 We also found that Jagged1 stimulation of primary human smooth muscle cells derived from diseased atherosclerotic arteries decreased cell proliferation.127 Based on these collective observations, signaling via Notch2 and Notch3 in smooth muscle cells is predicted to be beneficial in pathologies that initiate hyperproliferation of smooth muscle cells. Thus, therapeutic approaches involving targeting Notch pathways in cardiovascular disease are likely to be most successful under conditions where specific Notch ligands or receptors can be selectively targeted in discrete cell populations.128–130
Acknowledgments
We are grateful for the expert technical assistance of Grazina Mangoba and Mayasah Al Hashimi for tissue processing and histopathology, and Barbara Conley and Eric Tweedie for preparation of samples for mass spectrometry.
Sources of Funding
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Numbers 1R01H109652 and R01HL070865 (L. Liaw, Principal Investigator [PI]) and F32HL136076 (S. Peterson, PI). Early research support was also provided by American Heart Association predoctoral fellowship 14PRE17820000 (S. Peterson, PI). Core facilities that assisted with mouse strains (Mouse Transgenic Core Facility), mass spectrometry (Proteomics and Lipidomics Core Facility), and tissue processing, and sectioning (Histopathology and Histomorphometry Core Facility) were supported by National Institutes of Health (NIH) P30GM103392 (R. Friesel, D. St. Germain PIs) and NIH U54GM115516, (C. Rosen and G. Stein, PIs). The Proteomics and Lipidomics and Histopathology and Histomorphometry Core Facility are funded by NIH P20GM121301 (L. Liaw, PI). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the American Heart Association.
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- ERH
- enhancer of rudimentary homolog
- PANTHER
- Protein Analysis Through Evolutionary Relationships
- PCR
- polymerase chain reaction
- SWATH
- sequential window acquisition of all theoretical spectra
- VSMC
- vascular smooth muscle cell
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.118.311092/-/DC1.
Highlights
Neointimal lesion formation corresponds to unique proteomic signatures that define distinct waves of remodeling; our study identifies these unique signatures.
Loss of Notch2 in vascular smooth muscle cells alters the proteomic signatures defining the vascular remodeling response.
Despite these changes in protein profiles with loss of Notch2, the overall morphological remodeling response was not altered in the vascular smooth muscle–specific Notch2 knockout.
Our proteomics analysis uncovered many proteins not previously identified in vascular remodeling and identified potential novel targets of Notch signaling.
References
- 1.Wu X, Zou Y, Zhou Q, Huang L, Gong H, Sun A, Tateno K, Katsube K, Radtke F, Ge J, Minamino T, Komuro I. Role of Jagged1 in arterial lesions after vascular injury. Arterioscler Thromb Vasc Biol. 2011;31:2000–2006. doi: 10.1161/ATVBAHA.111.225144. doi: 10.1161/ATVBAHA.111.225144. [DOI] [PubMed] [Google Scholar]
- 2.Caolo V, Schulten HM, Zhuang ZW, Murakami M, Wagenaar A, Verbruggen S, Molin DG, Post MJ. Soluble Jagged-1 inhibits neointima formation by attenuating Notch-Herp2 signaling. Arterioscler Thromb Vasc Biol. 2011;31:1059–1065. doi: 10.1161/ATVBAHA.110.217935. doi: 10.1161/ATVBAHA.110.217935. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Takeshita K, Liu PY, Satoh M, Oyama N, Mukai Y, Chin MT, Krebs L, Kotlikoff MI, Radtke F, Gridley T, Liao JK. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation. 2009;119:2686–2692. doi: 10.1161/CIRCULATIONAHA.108.790485. doi: 10.1161/CIRCULATIONAHA.108.790485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCright B, Lozier J, Gridley T. Generation of new Notch2 mutant alleles. Genesis. 2006;44:29–33. doi: 10.1002/gene.20181. doi: 10.1002/gene.20181. [DOI] [PubMed] [Google Scholar]
- 5.Hamada Y, Kadokawa Y, Okabe M, Ikawa M, Coleman JR, Tsujimoto Y. Mutation in ankyrin repeats of the mouse Notch2 gene induces early embryonic lethality. Development. 1999;126:3415–3424. doi: 10.1242/dev.126.15.3415. [DOI] [PubMed] [Google Scholar]
- 6.Boucher JM, Harrington A, Rostama B, Lindner V, Liaw L. A receptor-specific function for Notch2 in mediating vascular smooth muscle cell growth arrest through cyclin-dependent kinase inhibitor 1B. Circ Res. 2013;113:975–985. doi: 10.1161/CIRCRESAHA.113.301272. doi: 10.1161/CIRCRESAHA.113.301272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baeten JT, Lilly B. Differential regulation of NOTCH2 and NOTCH3 contribute to their unique functions in vascular smooth muscle cells. J Biol Chem. 2015;290:16226–16237. doi: 10.1074/jbc.M115.655548. doi: 10.1074/jbc.M115.655548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schubert OT, Gillet LC, Collins BC, Navarro P, Rosenberger G, Wolski WE, Lam H, Amodei D, Mallick P, MacLean B, Aebersold R. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc. 2015;10:426–441. doi: 10.1038/nprot.2015.015. doi: 10.1038/nprot.2015.015. [DOI] [PubMed] [Google Scholar]
- 9.Korwar AM, Vannuruswamy G, Jagadeeshaprasad MG, Jayaramaiah RH, Bhat S, Regin BS, Ramaswamy S, Giri AP, Mohan V, Balasubramanyam M, Kulkarni MJ. Development of diagnostic fragment ion library for glycated peptides of human serum albumin: targeted quantification in prediabetic, diabetic, and microalbuminuria plasma by parallel reaction monitoring, SWATH, and MSE. Mol Cell Proteomics. 2015;14:2150–2159. doi: 10.1074/mcp.M115.050518. doi: 10.1074/mcp.M115.050518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Q, Yang L, Luo J, Guo L, Wang Z, Yang X, Jin W, Fang Y, Ye J, Shan B, Zhang Y. SWATH enables precise label-free quantification on proteome scale. Proteomics. 2015;15:1215–1223. doi: 10.1002/pmic.201400270. doi: 10.1002/pmic.201400270. [DOI] [PubMed] [Google Scholar]
- 11.Collins BC, Gillet LC, Rosenberger G, Röst HL, Vichalkovski A, Gstaiger M, Aebersold R. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat Methods. 2013;10:1246–1253. doi: 10.1038/nmeth.2703. doi: 10.1038/nmeth.2703. [DOI] [PubMed] [Google Scholar]
- 12.Faktor J, Michalova E, Bouchal P. [p SRM, SWATH and HRM - targeted proteomics approaches on TripleTOF 5600+ mass spectrometer and their applications in oncology research]. Klin Onkol. 2014;27(s)(uppl 1):S110–S115. doi: 10.14735/amko20141s110. [DOI] [PubMed] [Google Scholar]
- 13.McRobb LS, Lee VS, Simonian M, Zhao Z, Thomas SG, Wiedmann M, Raj JV, Grace M, Moutrie V, McKay MJ, Molloy MP, Stoodley MA. Radiosurgery alters the endothelial surface proteome: externalized intracellular molecules as potential vascular targets in irradiated brain arteriovenous malformations. Radiat Res. 2017;187:66–78. doi: 10.1667/RR14518.1. doi: 10.1667/RR14518.1. [DOI] [PubMed] [Google Scholar]
- 14.Langley SR, Willeit K, Didangelos A, et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J Clin Invest. 2017;127:1546–1560. doi: 10.1172/JCI86924. doi: 10.1172/JCI86924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horváth B, Maser-Gluth C, Greiner E, Lemmer B, Schütz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 16.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 17.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 18.Peterson SM, Liaw L, Lindner V. Ligation of the mouse common carotid artery. In: Sata M, editor. Mouse Models of Vascular Diseases. Tokyo: Springer Japan; 2016. pp. 43–68. [Google Scholar]
- 19.Kumar A, Hoover JL, Simmons CA, Lindner V, Shebuski RJ. Remodeling and neointimal formation in the carotid artery of normal and P-selectin-deficient mice. Circulation. 1997;96:4333–4342. doi: 10.1161/01.cir.96.12.4333. [DOI] [PubMed] [Google Scholar]
- 20.Myers DL, Liaw L. Improved analysis of the vascular response to arterial ligation using a multivariate approach. Am J Pathol. 2004;164:43–48. doi: 10.1016/S0002-9440(10)63094-5. doi: 10.1016/S0002-9440(10)63094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harmon KJ, Couper LL, Lindner V. Strain-dependent vascular remodeling phenotypes in inbred mice. Am J Pathol. 2000;156:1741–1748. doi: 10.1016/S0002-9440(10)65045-6. doi: 10.1016/S0002-9440(10)65045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawashima S, Yamashita T, Ozaki M, Ohashi Y, Azumi H, Inoue N, Hirata K, Hayashi Y, Itoh H, Yokoyama M. Endothelial NO synthase overexpression inhibits lesion formation in mouse model of vascular remodeling. Arterioscler Thromb Vasc Biol. 2001;21:201–207. doi: 10.1161/01.atv.21.2.201. [DOI] [PubMed] [Google Scholar]
- 23.da Cunha V, Martin-McNulty B, Vincelette J, Zhang L, Rutledge JC, Wilson DW, Vergona R, Sullivan ME, Wang YX. Interaction between mild hypercholesterolemia, HDL-cholesterol levels, and angiotensin II in intimal hyperplasia in mice. J Lipid Res. 2006;47:476–483. doi: 10.1194/jlr.M500341-JLR200. doi: 10.1194/jlr.M500341-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Ivan E, Khatri JJ, Johnson C, Magid R, Godin D, Nandi S, Lessner S, Galis ZS. Expansive arterial remodeling is associated with increased neointimal macrophage foam cell content: the murine model of macrophage-rich carotid artery lesions. Circulation. 2002;105:2686–2691. doi: 10.1161/01.cir.0000016825.17448.11. [DOI] [PubMed] [Google Scholar]
- 25.Johnson JL, Dwivedi A, Somerville M, George SJ, Newby AC. Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler Thromb Vasc Biol. 2011;31:e35–e44. doi: 10.1161/ATVBAHA.111.225623. doi: 10.1161/ATVBAHA.111.225623. [DOI] [PubMed] [Google Scholar]
- 26.Tsaousi A, Williams H, Lyon CA, Taylor V, Swain A, Johnson JL, George SJ. Wnt4/β-catenin signaling induces VSMC proliferation and is associated with intimal thickening. Circ Res. 2011;108:427–436. doi: 10.1161/CIRCRESAHA.110.233999. doi: 10.1161/CIRCRESAHA.110.233999. [DOI] [PubMed] [Google Scholar]
- 27.Williams H, Mill CA, Monk BA, Hulin-Curtis S, Johnson JL, George SJ. Wnt2 and WISP-1/CCN4 induce intimal thickening via promotion of smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2016;36:1417–1424. doi: 10.1161/ATVBAHA.116.307626. doi: 10.1161/ATVBAHA.116.307626. [DOI] [PubMed] [Google Scholar]
- 28.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11:O111.016717. doi: 10.1074/mcp.O111.016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young K, Conley B, Romero D, Tweedie E, O’Neill C, Pinz I, Brogan L, Lindner V, Liaw L, Vary CP. BMP9 regulates endoglin-dependent chemokine responses in endothelial cells. Blood. 2012;120:4263–4273. doi: 10.1182/blood-2012-07-440784. doi: 10.1182/blood-2012-07-440784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young K, Tweedie E, Conley B, Ames J, FitzSimons M, Brooks P, Liaw L, Vary CP. BMP9 crosstalk with the hippo pathway regulates endothelial cell matricellular and chemokine responses. PLoS One. 2015;10:e0122892. doi: 10.1371/journal.pone.0122892. doi: 10.1371/journal.pone.0122892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivosev G, Burton L, Bonner R. Dimensionality reduction and visualization in principal component analysis. Anal Chem. 2008;80:4933–4944. doi: 10.1021/ac800110w. doi: 10.1021/ac800110w. [DOI] [PubMed] [Google Scholar]
- 33.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45:D183–D189. doi: 10.1093/nar/gkw1138. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindner V, Booth C, Prudovsky I, Small D, Maciag T, Liaw L. Members of the Jagged/Notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. Am J Pathol. 2001;159:875–883. doi: 10.1016/S0002-9440(10)61763-4. doi: 10.1016/S0002-9440(10)61763-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skórzyńska-Dziduszko KE, Olszewska A, Prendecka M, Małecka-Massalska T. Serum heat shock protein 90 alpha: a new marker of hypertension-induced endothelial injury? Adv Clin Exp Med. 2016;25:255–261. doi: 10.17219/acem/40068. [DOI] [PubMed] [Google Scholar]
- 37.Reed GW, Horr S, Young L, Clevenger J, Malik U, Ellis SG, Lincoff AM, Nissen SE, Menon V. Associations between cardiac troponin, mechanism of myocardial injury, and long-term mortality after noncardiac vascular surgery. J Am Heart Assoc. 2017;6:e005672. doi: 10.1161/JAHA.117.005672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe Y, Murdoch CE, Sano S, Ido Y, Bachschmid MM, Cohen RA, Matsui R. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1α and improve limb revascularization. Proc Natl Acad Sci USA. 2016;113:6011–6016. doi: 10.1073/pnas.1524198113. doi: 10.1073/pnas.1524198113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Jong RJ, Paulin N, Lemnitzer P, Viola JR, Winter C, Ferraro B, Grommes J, Weber C, Reutelingsperger C, Drechsler M, Soehnlein O. Protective aptitude of annexin A1 in arterial neointima formation in atherosclerosis-prone mice-brief report. Arterioscler Thromb Vasc Biol. 2017;37:312–315. doi: 10.1161/ATVBAHA.116.308744. doi: 10.1161/ATVBAHA.116.308744. [DOI] [PubMed] [Google Scholar]
- 40.Ai TJ, Sun JY, Du LJ, Shi C, Li C, Sun XN, Liu Y, Li L, Xia Z, Jia L, Liu J, Duan SZ. Inhibition of neddylation by MLN4924 improves neointimal hyperplasia and promotes apoptosis of vascular smooth muscle cells through p53 and p62. Cell Death Differ. 2018;25:319–329. doi: 10.1038/cdd.2017.160. doi: 10.1038/cdd.2017.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bowley SR, Fang C, Merrill-Skoloff G, Furie BC, Furie B. Protein disulfide isomerase secretion following vascular injury initiates a regulatory pathway for thrombus formation. Nat Commun. 2017;8:14151. doi: 10.1038/ncomms14151. doi: 10.1038/ncomms14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venugopal S, Chen M, Liao W, Er SY, Wong WS, Ge R. Isthmin is a novel vascular permeability inducer that functions through cell-surface GRP78-mediated Src activation. Cardiovasc Res. 2015;107:131–142. doi: 10.1093/cvr/cvv142. doi: 10.1093/cvr/cvv142. [DOI] [PubMed] [Google Scholar]
- 43.Noda T, Maeda K, Hayano S, Asai N, Enomoto A, Takahashi M, Murohara T. New endoplasmic reticulum stress regulator, Gipie, regulates the survival of vascular smooth muscle cells and the neointima formation after vascular injury. Arterioscler Thromb Vasc Biol. 2015;35:1246–1253. doi: 10.1161/ATVBAHA.114.304923. doi: 10.1161/ATVBAHA.114.304923. [DOI] [PubMed] [Google Scholar]
- 44.McDonald RA, Pyne S, Pyne NJ, Grant A, Wainwright CL, Wadsworth RM. The sphingosine kinase inhibitor N,N-dimethylsphingosine inhibits neointimal hyperplasia. Br J Pharmacol. 2010;159:543–553. doi: 10.1111/j.1476-5381.2009.00533.x. doi: 10.1111/j.1476-5381.2009.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong CH, Bozinovski S, Hertzog PJ, Hickey MJ, Crack PJ. Absence of glutathione peroxidase-1 exacerbates cerebral ischemia-reperfusion injury by reducing post-ischemic microvascular perfusion. J Neurochem. 2008;107:241–252. doi: 10.1111/j.1471-4159.2008.05605.x. doi: 10.1111/j.1471-4159.2008.05605.x. [DOI] [PubMed] [Google Scholar]
- 46.Fedorov A, Kostareva A, Raud J, Roy J, Hedin U, Razuvaev A. Early changes of gene expression profiles in the rat model of arterial injury. J Vasc Interv Radiol. 2014;25:789–796.e7. doi: 10.1016/j.jvir.2013.11.031. doi: 10.1016/j.jvir.2013.11.031. [DOI] [PubMed] [Google Scholar]
- 47.Zhao G, Shaik RS, Zhao H, Beagle J, Kuo S, Hales CA. Low molecular weight (LMW) heparin inhibits injury-induced femoral artery remodeling in mouse via upregulating CD44 expression. J Vasc Surg. 2011;53:1359–1367.e3. doi: 10.1016/j.jvs.2010.11.048. doi: 10.1016/j.jvs.2010.11.048. [DOI] [PubMed] [Google Scholar]
- 48.Sheng K, Zhang P, Lin W, Cheng J, Li J, Chen J. Association of matrix Gla protein gene (rs1800801, rs1800802, rs4236) polymorphism with vascular calcification and atherosclerotic disease: a meta-analysis. Sci Rep. 2017;7:8713. doi: 10.1038/s41598-017-09328-5. doi: 10.1038/s41598-017-09328-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buyukterzi Z, Can U, Alpaydin S, Guzelant A, Karaarslan S, Mustu M, Kocyigit D, Gurses KM. Enhanced serum levels of matrix Gla protein and bone morphogenetic protein in acute coronary syndrome patients. J Clin Lab Anal. 2018;32:e22278. doi: 10.1002/jcla.22278. doi: 10.1002/jcla.22278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song X, Li L, Wu Y, Zhang L, Li X. Functional role of inflammation in the surgical injury induced vascular remodeling of male albino rats. Cell Mol Biol (Noisy-le-grand) 2017;63:8–12. doi: 10.14715/cmb/2017.63.9.2. [DOI] [PubMed] [Google Scholar]
- 51.Rinaldi B, Romagnoli P, Bacci S, Carnuccio R, Maiuri MC, Donniacuo M, Capuano A, Rossi F, Filippelli A. Inflammatory events in a vascular remodeling model induced by surgical injury to the rat carotid artery. Br J Pharmacol. 2006;147:175–182. doi: 10.1038/sj.bjp.0706472. doi: 10.1038/sj.bjp.0706472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sluijter JP, Smeets MB, Velema E, Pasterkamp G, de Kleijn DP. Increased collagen turnover is only partly associated with collagen fiber deposition in the arterial response to injury. Cardiovasc Res. 2004;61:186–195. doi: 10.1016/j.cardiores.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 53.Jabs A, Krämer S, Skowasch D, Welsch U, Kuhn A, Kandolf R, Lüderitz B, Bauriedel G. [Neointimal hyperplasia by luminal cell recruitment and not be transmural migration. The role of Bcl-2 and HSP47 after balloon angioplasty]. Z Kardiol. 2002;91:626–636. doi: 10.1007/s00392-002-0827-x. [DOI] [PubMed] [Google Scholar]
- 54.Yoon YW, Kwon HM, Hwang KC, Choi EY, Hong BK, Kim D, Kim HS, Cho SH, Song KS, Sangiorgi G. Upstream regulation of matrix metalloproteinase by EMMPRIN; extracellular matrix metalloproteinase inducer in advanced atherosclerotic plaque. Atherosclerosis. 2005;180:37–44. doi: 10.1016/j.atherosclerosis.2004.11.021. doi: 10.1016/j.atherosclerosis.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 55.Pyagay P, Heroult M, Wang Q, Lehnert W, Belden J, Liaw L, Friesel RE, Lindner V. Collagen triple helix repeat containing 1, a novel secreted protein in injured and diseased arteries, inhibits collagen expression and promotes cell migration. Circ Res. 2005;96:261–268. doi: 10.1161/01.RES.0000154262.07264.12. doi: 10.1161/01.RES.0000154262.07264.12. [DOI] [PubMed] [Google Scholar]
- 56.Preissner KT, Reuning U. Vitronectin in vascular context: facets of a multitalented matricellular protein. Semin Thromb Hemost. 2011;37:408–424. doi: 10.1055/s-0031-1276590. doi: 10.1055/s-0031-1276590. [DOI] [PubMed] [Google Scholar]
- 57.Caglayan E, Romeo GR, Kappert K, Odenthal M, Südkamp M, Body SC, Shernan SK, Hackbusch D, Vantler M, Kazlauskas A, Rosenkranz S. Profilin-1 is expressed in human atherosclerotic plaques and induces atherogenic effects on vascular smooth muscle cells. PLoS One. 2010;5:e13608. doi: 10.1371/journal.pone.0013608. doi: 10.1371/journal.pone.0013608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams HC, San Martín A, Adamo CM, Seidel-Rogol B, Pounkova L, Datla SR, Lassègue B, Bear JE, Griendling K. Role of coronin 1B in PDGF-induced migration of vascular smooth muscle cells. Circ Res. 2012;111:56–65. doi: 10.1161/CIRCRESAHA.111.255745. doi: 10.1161/CIRCRESAHA.111.255745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perisic Matic L, Rykaczewska U, Razuvaev A, et al. Phenotypic modulation of smooth muscle cells in atherosclerosis is associated with downregulation of LMOD1, SYNPO2, PDLIM7, PLN, and SYNM. Arterioscler Thromb Vasc Biol. 2016;36:1947–1961. doi: 10.1161/ATVBAHA.116.307893. doi: 10.1161/ATVBAHA.116.307893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruusalepp A, Yan ZQ, Carlsen H, Czibik G, Hansson GK, Moskaug JØ, Blomhoff R, Valen G. Gene deletion of NF-kappaB p105 enhances neointima formation in a mouse model of carotid artery injury. Cardiovasc Drugs Ther. 2006;20:103–111. doi: 10.1007/s10557-006-6755-7. doi: 10.1007/s10557-006-6755-7. [DOI] [PubMed] [Google Scholar]
- 61.Lee MY, Skoura A, Park EJ, Landskroner-Eiger S, Jozsef L, Luciano AK, Murata T, Pasula S, Dong Y, Bouaouina M, Calderwood DA, Ferguson SM, De Camilli P, Sessa WC. Dynamin 2 regulation of integrin endocytosis, but not VEGF signaling, is crucial for developmental angiogenesis. Development. 2014;141:1465–1472. doi: 10.1242/dev.104539. doi: 10.1242/dev.104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imai H, Numaguchi Y, Ishii M, Kubota R, Yokouchi K, Ogawa Y, Kondo T, Okumura K, Murohara T. Prostacyclin synthase gene transfer inhibits neointimal formation by suppressing PPAR delta expression. Atherosclerosis. 2007;195:322–332. doi: 10.1016/j.atherosclerosis.2007.01.010. doi: 10.1016/j.atherosclerosis.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 63.Hansmeier N, Buttigieg J, Kumar P, Pelle S, Choi KY, Kopriva D, Chao TC. Identification of mature atherosclerotic plaque proteome signatures using data-independent acquisition mass spectrometry. J Proteome Res. 2018;17:164–176. doi: 10.1021/acs.jproteome.7b00487. doi: 10.1021/acs.jproteome.7b00487. [DOI] [PubMed] [Google Scholar]
- 64.Hansen ML, Rasmussen LM. Associations between plasma fibulin-1, pulse wave velocity and diabetes in patients with coronary heart disease. J Diabetes Complications. 2015;29:362–366. doi: 10.1016/j.jdiacomp.2015.01.003. doi: 10.1016/j.jdiacomp.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 65.Parente JM, Pereira CA, Oliveira-Paula GH, Tanus-Santos JE, Tostes RC, Castro MM. Matrix metalloproteinase-2 activity is associated with divergent regulation of calponin-1 in conductance and resistance arteries in hypertension-induced early vascular dysfunction and remodelling. Basic Clin Pharmacol Toxicol. 2017;121:246–256. doi: 10.1111/bcpt.12787. doi: 10.1111/bcpt.12787. [DOI] [PubMed] [Google Scholar]
- 66.Belo VA, Parente JM, Tanus-Santos JE, Castro MM. Matrix metalloproteinase (MMP)-2 decreases calponin-1 levels and contributes to arterial remodeling in early hypertension. Biochem Pharmacol. 2016;118:50–58. doi: 10.1016/j.bcp.2016.08.012. doi: 10.1016/j.bcp.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 67.Ji Y, Weng Z, Fish P, Goyal N, Luo M, Myears SP, Strawn TL, Chandrasekar B, Wu J, Fay WP. Pharmacological targeting of plasminogen activator inhibitor-1 decreases vascular smooth muscle cell migration and neointima formation. Arterioscler Thromb Vasc Biol. 2016;36:2167–2175. doi: 10.1161/ATVBAHA.116.308344. doi: 10.1161/ATVBAHA.116.308344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jourd’heuil FL, Xu H, Reilly T, et al. The hemoglobin homolog cytoglobin in smooth muscle inhibits apoptosis and regulates vascular remodeling. Arterioscler Thromb Vasc Biol. 2017;37:1944–1955. doi: 10.1161/ATVBAHA.117.309410. doi: 10.1161/ATVBAHA.117.309410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fairaq A, Shawky NM, Osman I, Pichavaram P, Segar L. AdipoRon, an adiponectin receptor agonist, attenuates PDGF-induced VSMC proliferation through inhibition of mTOR signaling independent of AMPK: implications toward suppression of neointimal hyperplasia. Pharmacol Res. 2017;119:289–302. doi: 10.1016/j.phrs.2017.02.016. doi: 10.1016/j.phrs.2017.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Won KJ, Jung SH, Lee CK, Na HR, Lee KP, Lee DY, Park ES, Choi WS, Shim SB, Kim B. DJ-1/park7 protects against neointimal formation via the inhibition of vascular smooth muscle cell growth. Cardiovasc Res. 2013;97:553–561. doi: 10.1093/cvr/cvs363. doi: 10.1093/cvr/cvs363. [DOI] [PubMed] [Google Scholar]
- 71.Pilop C, Aregger F, Gorman RC, Brunisholz R, Gerrits B, Schaffner T, Gorman JH, 3rd, Matyas G, Carrel T, Frey BM. Proteomic analysis in aortic media of patients with Marfan syndrome reveals increased activity of calpain 2 in aortic aneurysms. Circulation. 2009;120:983–991. doi: 10.1161/CIRCULATIONAHA.108.843516. doi: 10.1161/CIRCULATIONAHA.108.843516. [DOI] [PubMed] [Google Scholar]
- 72.Matsui R, Watanabe Y, Murdoch CE. Redox regulation of ischemic limb neovascularization—what we have learned from animal studies. Redox Biol. 2017;12:1011–1019. doi: 10.1016/j.redox.2017.04.040. doi: 10.1016/j.redox.2017.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roy A, Basak NP, Banerjee S. Notch1 intracellular domain increases cytoplasmic EZH2 levels during early megakaryopoiesis. Cell Death Dis. 2012;3:e380. doi: 10.1038/cddis.2012.119. doi: 10.1038/cddis.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benelli D, Cialfi S, Pinzaglia M, Talora C, Londei P. The translation factor eIF6 is a Notch-dependent regulator of cell migration and invasion. PLoS One. 2012;7:e32047. doi: 10.1371/journal.pone.0032047. doi: 10.1371/journal.pone.0032047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gorantla B, Bhoopathi P, Chetty C, Gogineni VR, Sailaja GS, Gondi CS, Rao JS. Notch signaling regulates tumor-induced angiogenesis in SPARC-overexpressed neuroblastoma. Angiogenesis. 2013;16:85–100. doi: 10.1007/s10456-012-9301-1. doi: 10.1007/s10456-012-9301-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ugarte F, Ryser M, Thieme S, Fierro FA, Navratiel K, Bornhäuser M, Brenner S. Notch signaling enhances osteogenic differentiation while inhibiting adipogenesis in primary human bone marrow stromal cells. Exp Hematol. 2009;37:867–875.e1. doi: 10.1016/j.exphem.2009.03.007. doi: 10.1016/j.exphem.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 77.Kiec-Wilk B, Grzybowska-Galuszka J, Polus A, Pryjma J, Knapp A, Kristiansen K. The MAPK-dependent regulation of the Jagged/Notch gene expression by VEGF, bFGF or PPAR gamma mediated angiogenesis in HUVEC. J Physiol Pharmacol. 2010;61:217–225. [PubMed] [Google Scholar]
- 78.You C, Zhao K, Dammann P, Keyvani K, Kreitschmann-Andermahr I, Sure U, Zhu Y. EphB4 forward signalling mediates angiogenesis caused by CCM3/PDCD10-ablation. J Cell Mol Med. 2017;21:1848–1858. doi: 10.1111/jcmm.13105. doi: 10.1111/jcmm.13105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu JR, Yeh JL, Liou SF, Dai ZK, Wu BN, Hsu JH. Gamma-secretase inhibitor prevents proliferation and migration of ductus arteriosus smooth muscle cells through the Notch3-HES1/2/5 pathway. Int J Biol Sci. 2016;12:1063–1073. doi: 10.7150/ijbs.16430. doi: 10.7150/ijbs.16430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pierscianek D, Wolf S, Keyvani K, El Hindy N, Stein KP, Sandalcioglu IE, Sure U, Mueller O, Zhu Y. Study of angiogenic signaling pathways in hemangioblastoma. Neuropathology. 2017;37:3–11. doi: 10.1111/neup.12316. doi: 10.1111/neup.12316. [DOI] [PubMed] [Google Scholar]
- 81.Ramakrishnan G, Davaakhuu G, Chung WC, Zhu H, Rana A, Filipovic A, Green AR, Atfi A, Pannuti A, Miele L, Tzivion G. AKT and 14-3-3 regulate Notch4 nuclear localization. Sci Rep. 2015;5:8782. doi: 10.1038/srep08782. doi: 10.1038/srep08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang S, Da Y, Han S, He Y, Che H. Notch ligand Delta-like1 enhances degranulation and cytokine production through a novel Notch/Dok-1/MAPKs pathway in vitro. Immunol Res. 2018;66:87–96. doi: 10.1007/s12026-017-8977-0. doi: 10.1007/s12026-017-8977-0. [DOI] [PubMed] [Google Scholar]
- 83.Lai YJ, Li MY, Yang CY, Huang KH, Tsai JC, Wang TW. TRIP6 regulates neural stem cell maintenance in the postnatal mammalian subventricular zone. Dev Dyn. 2014;243:1130–1142. doi: 10.1002/dvdy.24161. doi: 10.1002/dvdy.24161. [DOI] [PubMed] [Google Scholar]
- 84.Junutula JR, De Maziére AM, Peden AA, Ervin KE, Advani RJ, van Dijk SM, Klumperman J, Scheller RH. Rab14 is involved in membrane trafficking between the Golgi complex and endosomes. Mol Biol Cell. 2004;15:2218–2229. doi: 10.1091/mbc.E03-10-0777. doi: 10.1091/mbc.E03-10-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leitch CC, Lodh S, Prieto-Echagüe V, Badano JL, Zaghloul NA. Basal body proteins regulate Notch signaling through endosomal trafficking. J Cell Sci. 2014;127(p)(t 11):2407–2419. doi: 10.1242/jcs.130344. doi: 10.1242/jcs.130344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang X, Lee SJ, Young MF, Wang MM. The small leucine-rich proteoglycan BGN accumulates in CADASIL and binds to NOTCH3. Transl Stroke Res. 2015;6:148–155. doi: 10.1007/s12975-014-0379-1. doi: 10.1007/s12975-014-0379-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mirza Z, Schulten HJ, Farsi HM, Al-Maghrabi JA, Gari MA, Chaudhary AG, Abuzenadah AM, Al-Qahtani MH, Karim S. Molecular interaction of a kinase inhibitor midostaurin with anticancer drug targets, S100A8 and EGFR: transcriptional profiling and molecular docking study for kidney cancer therapeutics. PLoS One. 2015;10:e0119765. doi: 10.1371/journal.pone.0119765. doi: 10.1371/journal.pone.0119765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fomenkov A, Huang YP, Topaloglu O, Brechman A, Osada M, Fomenkova T, Yuriditsky E, Trink B, Sidransky D, Ratovitski E. P63 alpha mutations lead to aberrant splicing of keratinocyte growth factor receptor in the Hay-Wells syndrome. J Biol Chem. 2003;278:23906–23914. doi: 10.1074/jbc.M300746200. doi: 10.1074/jbc.M300746200. [DOI] [PubMed] [Google Scholar]
- 89.Huang YP, Kim Y, Li Z, Fomenkov T, Fomenkov A, Ratovitski EA. AEC-associated p63 mutations lead to alternative splicing/protein stabilization of p63 and modulation of Notch signaling. Cell Cycle. 2005;4:1440–1447. doi: 10.4161/cc.4.10.2086. doi: 10.4161/cc.4.10.2086. [DOI] [PubMed] [Google Scholar]
- 90.Rusanescu G, Mao J. Notch3 is necessary for neuronal differentiation and maturation in the adult spinal cord. J Cell Mol Med. 2014;18:2103–2116. doi: 10.1111/jcmm.12362. doi: 10.1111/jcmm.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barnawi R, Al-Khaldi S, Majed Sleiman G, Sarkar A, Al-Dhfyan A, Al-Mohanna F, Ghebeh H, Al-Alwan M. Fascin is critical for the maintenance of breast cancer stem cell pool predominantly via the activation of the notch self-renewal pathway. Stem Cells. 2016;34:2799–2813. doi: 10.1002/stem.2473. doi: 10.1002/stem.2473. [DOI] [PubMed] [Google Scholar]
- 92.Hodkinson PS, Elliott PA, Lad Y, McHugh BJ, MacKinnon AC, Haslett C, Sethi T. Mammalian NOTCH-1 activates beta1 integrins via the small GTPase R-Ras. J Biol Chem. 2007;282:28991–29001. doi: 10.1074/jbc.M703601200. doi: 10.1074/jbc.M703601200. [DOI] [PubMed] [Google Scholar]
- 93.Jiao S, Dai W, Lu L, Liu Y, Zhou J, Li Y, Korzh V, Duan C. The conserved clusterin gene is expressed in the developing choroid plexus under the regulation of notch but not IGF signaling in zebrafish. Endocrinology. 2011;152:1860–1871. doi: 10.1210/en.2010-1183. doi: 10.1210/en.2010-1183. [DOI] [PubMed] [Google Scholar]
- 94.Arboleda-Velasquez JF, Manent J, Lee JH, et al. Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease. Proc Natl Acad Sci USA. 2011;108:E128–E135. doi: 10.1073/pnas.1101964108. doi: 10.1073/pnas.1101964108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tong M, Gonzalez-Navarrete H, Kirchberg T, Gotama B, Yalcin EB, Kay J, de la Monte SM. Ethanol-induced white matter atrophy is associated with impaired expression of aspartyl-asparaginyl-beta-hydroxylase (ASPH) and notch signaling in an experimental rat model. J Drug Alcohol Res. 2017;6:236033. doi: 10.4303/jdar/236033. doi: 10.4303/jdar/236033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cantarini MC, de la Monte SM, Pang M, Tong M, D’Errico A, Trevisani F, Wands JR. Aspartyl-asparagyl beta hydroxylase over-expression in human hepatoma is linked to activation of insulin-like growth factor and notch signaling mechanisms. Hepatology. 2006;44:446–457. doi: 10.1002/hep.21272. doi: 10.1002/hep.21272. [DOI] [PubMed] [Google Scholar]
- 97.Gundogan F, Bedoya A, Gilligan J, Lau E, Mark P, De Paepe ME, de la Monte SM. siRNA inhibition of aspartyl-asparaginyl β-hydroxylase expression impairs cell motility, Notch signaling, and fetal growth. Pathol Res Pract. 2011;207:545–553. doi: 10.1016/j.prp.2011.06.001. doi: 10.1016/j.prp.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huang PC, Chiu CC, Chang HW, Wang YS, Syue HH, Song YC, Weng ZH, Tai MH, Wu CY. Prdx1-encoded peroxiredoxin is important for vascular development in zebrafish. FEBS Lett. 2017;591:889–902. doi: 10.1002/1873-3468.12604. doi: 10.1002/1873-3468.12604. [DOI] [PubMed] [Google Scholar]
- 99.Hatch E, Morrow D, Liu W, Cahill PA, Redmond EM. Differential effects of alcohol and its metabolite acetaldehyde on vascular smooth muscle cell notch signaling and growth. Am J Physiol Heart Circ Physiol. 2018;314:H131–H137. doi: 10.1152/ajpheart.00586.2017. doi: 10.1152/ajpheart.00586.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Khadilkar RJ, Rodrigues D, Mote RD, Sinha AR, Kulkarni V, Magadi SS, Inamdar MS. ARF1-GTP regulates Asrij to provide endocytic control of Drosophila blood cell homeostasis. Proc Natl Acad Sci USA. 2014;111:4898–4903. doi: 10.1073/pnas.1303559111. doi: 10.1073/pnas.1303559111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Williams SE, Beronja S, Pasolli HA, Fuchs E. Asymmetric cell divisions promote Notch-dependent epidermal differentiation. Nature. 2011;470:353–358. doi: 10.1038/nature09793. doi: 10.1038/nature09793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen K, Chen QJ, Wang LJ, Liu ZH, Zhang Q, Yang K, Wang HB, Yan XX, Zhu ZB, Du R, Zhang RY, Shen WF, Lu L. Increment of HFABP level in coronary artery in-stent restenosis segments in diabetic and nondiabetic minipigs: HFABP overexpression promotes multiple pathway-related inflammation, growth and migration in human vascular smooth muscle cells. J Vasc Res. 2016;53:27–38. doi: 10.1159/000446652. doi: 10.1159/000446652. [DOI] [PubMed] [Google Scholar]
- 103.Lee B, Lee TH, Shim J. Emerin suppresses Notch signaling by restricting the Notch intracellular domain to the nuclear membrane. Biochim Biophys Acta. 2017;1864:303–313. doi: 10.1016/j.bbamcr.2016.11.013. doi: 10.1016/j.bbamcr.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 104.Koch AJ, Holaska JM. Loss of emerin alters myogenic signaling and miRNA expression in mouse myogenic progenitors. PLoS One. 2012;7:e37262. doi: 10.1371/journal.pone.0037262. doi: 10.1371/journal.pone.0037262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fuss B, Josten F, Feix M, Hoch M. Cell movements controlled by the Notch signalling cascade during foregut development in Drosophila. Development. 2004;131:1587–1595. doi: 10.1242/dev.01057. doi: 10.1242/dev.01057. [DOI] [PubMed] [Google Scholar]
- 106.Lu L, Wang J, Wu Y, Wan P, Yang G. Rap1A promotes ovarian cancer metastasis via activation of ERK/p38 and notch signaling. Cancer Med. 2016;5:3544–3554. doi: 10.1002/cam4.946. doi: 10.1002/cam4.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin L, Vu T, Yuan G, Datta PK. STRAP promotes stemness of human colorectal cancer via epigenetic regulation of the NOTCH pathway. Cancer Res. 2017;77:5464–5478. doi: 10.1158/0008-5472.CAN-17-0286. doi: 10.1158/0008-5472.CAN-17-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weng MT, Luo J. The enigmatic ERH protein: its role in cell cycle, RNA splicing and cancer. Protein Cell. 2013;4:807–812. doi: 10.1007/s13238-013-3056-3. doi: 10.1007/s13238-013-3056-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Osmanagic-Myers S, Rus S, Wolfram M, Brunner D, Goldmann WH, Bonakdar N, Fischer I, Reipert S, Zuzuarregui A, Walko G, Wiche G. Plectin reinforces vascular integrity by mediating crosstalk between the vimentin and the actin networks. J Cell Sci. 2015;128:4138–4150. doi: 10.1242/jcs.172056. doi: 10.1242/jcs.172056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gerke V. Annexins A2 and A8 in endothelial cell exocytosis and the control of vascular homeostasis. Biol Chem. 2016;397:995–1003. doi: 10.1515/hsz-2016-0207. doi: 10.1515/hsz-2016-0207. [DOI] [PubMed] [Google Scholar]
- 111.Liao M, Zhou J, Wang F, Ali YH, Chan KL, Zou F, Offermanns S, Jiang Z, Jiang Z. An x-linked myh11-creer(t2) mouse line resulting from y to x chromosome-translocation of the cre allele. Genesis. 2017;55:e23054. doi: 10.1002/dvg.23054. doi: 10.1002/dvg.23054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lindner V, Collins T. Expression of NF-kappa B and I kappa B-alpha by aortic endothelium in an arterial injury model. Am J Pathol. 1996;148:427–438. [PMC free article] [PubMed] [Google Scholar]
- 113.Wang Q, Zhao N, Kennard S, Lilly B. Notch2 and Notch3 function together to regulate vascular smooth muscle development. PLoS One. 2012;7:e37365. doi: 10.1371/journal.pone.0037365. doi: 10.1371/journal.pone.0037365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Baeten JT, Jackson AR, McHugh KM, Lilly B. Loss of Notch2 and Notch3 in vascular smooth muscle causes patent ductus arteriosus. Genesis. 2015;53:738–748. doi: 10.1002/dvg.22904. doi: 10.1002/dvg.22904. [DOI] [PubMed] [Google Scholar]
- 115.Feng X, Krebs LT, Gridley T. Patent ductus arteriosus in mice with smooth muscle-specific Jag1 deletion. Development. 2010;137:4191–4199. doi: 10.1242/dev.052043. doi: 10.1242/dev.052043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Krebs LT, Norton CR, Gridley T. Notch signal reception is required in vascular smooth muscle cells for ductus arteriosus closure. Genesis. 2016;54:86–90. doi: 10.1002/dvg.22916. doi: 10.1002/dvg.22916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Deleted in proof. [Google Scholar]
- 118.Wang H, Zang C, Liu XS, Aster JC. The role of Notch receptors in transcriptional regulation. J Cell Physiol. 2015;230:982–988. doi: 10.1002/jcp.24872. doi: 10.1002/jcp.24872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pajvani UB, Shawber CJ, Samuel VT, Birkenfeld AL, Shulman GI, Kitajewski J, Accili D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat Med. 2011;17:961–967. doi: 10.1038/nm.2378. doi: 10.1038/nm.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu J, Chi F, Tsukamoto H. Notch signaling and M1 macrophage activation in obesity-alcohol synergism. Clin Res Hepatol Gastroenterol. 2015;39(s)(uppl 1):S24–S28. doi: 10.1016/j.clinre.2015.05.016. doi: 10.1016/j.clinre.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bi P, Kuang S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol Metab. 2015;26:248–255. doi: 10.1016/j.tem.2015.02.006. doi: 10.1016/j.tem.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bi P, Shan T, Liu W, Yue F, Yang X, Liang XR, Wang J, Li J, Carlesso N, Liu X, Kuang S. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat Med. 2014;20:911–918. doi: 10.1038/nm.3615. doi: 10.1038/nm.3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nakano T, Fukuda D, Koga J, Aikawa M. Delta-like ligand 4-notch signaling in macrophage activation. Arterioscler Thromb Vasc Biol. 2016;36:2038–2047. doi: 10.1161/ATVBAHA.116.306926. doi: 10.1161/ATVBAHA.116.306926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rostama B, Turner JE, Seavey GT, Norton CR, Gridley T, Vary CP, Liaw L. DLL4/Notch1 and BMP9 interdependent signaling induces human endothelial cell quiescence via P27KIP1 and thrombospondin-1. Arterioscler Thromb Vasc Biol. 2015;35:2626–2637. doi: 10.1161/ATVBAHA.115.306541. doi: 10.1161/ATVBAHA.115.306541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bhattacharyya A, Lin S, Sandig M, Mequanint K. Regulation of vascular smooth muscle cell phenotype in three-dimensional coculture system by Jagged1-selective Notch3 signaling. Tissue Eng Part A. 2014;20:1175–1187. doi: 10.1089/ten.tea.2013.0268. doi: 10.1089/ten.TEA.2013.0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu H, Kennard S, Lilly B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ Res. 2009;104:466–475. doi: 10.1161/CIRCRESAHA.108.184846. doi: 10.1161/CIRCRESAHA.108.184846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Davis-Knowlton J, Turner JE, Turner A, Damian-Loring S, Hagler N, Henderson T, Emery IF, Bond K, Duarte CW, Vary CPH, Eldrup-Jorgensen J, Liaw L. Characterization of smooth muscle cells from human atherosclerotic lesions and their responses to Notch signaling [published online ahead of print May 23, 2018] Lab Invest. doi: 10.1038/s41374-018-0072-1. doi: 10.1038/s41374-018-0072-1. https://www.nature.com/articles/s41374-018-0072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wu Y, Cain-Hom C, Choy L, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464:1052–1057. doi: 10.1038/nature08878. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- 129.Braune EB, Lendahl U. Notch—a goldilocks signaling pathway in disease and cancer therapy. Discov Med. 2016;21:189–196. [PubMed] [Google Scholar]
- 130.Rizzo P, Mele D, Caliceti C, Pannella M, Fortini C, Clementz AG, Morelli MB, Aquila G, Ameri P, Ferrari R. The role of notch in the cardiovascular system: potential adverse effects of investigational notch inhibitors. Front Oncol. 2014;4:384. doi: 10.3389/fonc.2014.00384. doi: 10.3389/fonc.2014.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]