

Supplemental Digital Content is available in the text.
Keywords: blood platelets, humans, pharmacogenetics, signal transduction, thrombin
Abstract
Objective—
Platelet activation after stimulation of PAR (protease-activated receptor) 4 is heightened in platelets from blacks compared with those from whites. The difference in PAR4 signaling by race is partially explained by a single-nucleotide variant in PAR4 encoding for either an alanine or threonine at amino acid 120 in the second transmembrane domain. The current study sought to determine whether the difference in PAR4 signaling by this PAR4 variant is because of biased Gq signaling and whether the difference in PAR4 activity results in resistance to traditional antiplatelet intervention.
Approach and Results—
Membranes expressing human PAR4-120 variants were reconstituted with either Gq or G13 to determine the kinetics of G protein activation. The kinetics of Gq and G13 activation were both increased in membranes expressing PAR4-Thr120 compared with those expressing PAR4-Ala120. Further, inhibiting PAR4-mediated platelet activation by targeting COX (cyclooxygenase) and P2Y12 receptor was less effective in platelets from subjects expressing PAR4-Thr120 compared with PAR4-Ala120. Additionally, ex vivo thrombus formation in whole blood was evaluated at high shear to determine the relationship between PAR4 variant expression and response to antiplatelet drugs. Ex vivo thrombus formation was enhanced in blood from subjects expressing PAR4-Thr120 in the presence or absence of antiplatelet therapy.
Conclusions—
Together, these data support that the signaling difference by the PAR4-120 variant results in the enhancement of both Gq and G13 activation and an increase in thrombus formation resulting in a potential resistance to traditional antiplatelet therapies targeting COX-1 and the P2Y12 receptor.
Cardiovascular disease is the leading cause of morbidity and mortality in the United States. Platelet activation plays an important role in the hemostatic and thrombotic processes, and when these vascular events become unbalanced, the result is the formation of an occlusive thrombus leading to debilitating myocardial infarction and stroke.1 Blacks have a higher incidence of cardiovascular disease compared with whites, and more importantly, mortality after a cardiovascular event is significantly higher in the black population.2,3 Heightened platelet reactivity is known to be associated with an increased risk of mortality in patients with cardiovascular diseases4,5; therefore, a more complete understanding of the racial difference in platelet activation will lead to better therapeutic options for this high-risk population.
PARs (protease-activated receptors) are a family of unconventional GPCRs (G protein–coupled receptors) that become irreversibly activated by the proteolysis of their N-terminal domain by thrombin and other proteases in circulation, unmasking a cryptic tethered ligand that binds intramolecularly.6,7 Human platelets express 2 PARs, PAR1 and PAR4, which on thrombin cleavage initiate unique signaling pathways leading to platelet activation.8–12 PAR4 is thought to be the low-affinity receptor for thrombin, requiring higher concentrations of thrombin to elicit its signal in the cell—a signal that is slower and more sustained than PAR1.8,13 Recent studies have identified that racial differences exist in the human platelet and first elucidated that PAR4 is associated with increased platelet function in blacks compared with whites.14–16 PAR4 is thought to signal through the heterotrimeric G proteins Gαq, which activates phospholipase C-β and induces calcium mobilization, and Gα13, which activates RhoA resulting in cytoskeletal arrangement and platelet shape change.9 The racial difference in PAR4-mediated platelet activation is due in part to heightened signaling in the Gαq pathway.16 Further, characterization of the racial difference in PAR4-mediated platelet activation revealed a single-nucleotide variant (rs773902) in the PAR4 gene (F2RL3), which results in an alanine/threonine polymorphism at amino acid 120 in the second transmembrane domain. The PAR4-Thr120 is more common among individuals of African ancestry, representing 57.2% of alleles, whereas the PAR4-Ala120 represents 81.0% of alleles in individuals of European (non-Finnish) ancestry (Table). This difference in allelic expression is thought to account for ≈50% of the racial difference in platelet activation by PAR4.14,15 Because of previous studies showing the potential for biased signaling through PAR1 based on the type of agonist presented (thrombin versus PAR1-AP [PAR1-activating peptide]), it is possible that the increased signaling observed in platelets from individuals who express at least 1 copy of the PAR4-Thr120 allele may be because of biased signaling toward the Gαq pathway in these subjects.17–19 To fully understand the mechanism by which PAR4 is hyperactive by 120 variant, the level of activity in the Gα13 pathway downstream of PAR4 was assessed. Finally, this study sought for the first time to determine whether the increased activity in PAR4 translates to a resistance to in vivo standard-of-care intervention by aspirin and clopidogrel (targeting COX [cyclooxygenase]-1 and the P2Y12 receptor, respectively).
Table.
Comparison of the Allelic Frequency for the PAR4 Variant (rs773902) Encoding for Either a Thr120 or Ala120 at Amino Acid Position 120 Between the Self-Identified Black and White Subjects Recruited for This Study and the Predominantly Black and White Populations in the gnomAD—an Online Repository of Data Combined From Several Large-Scale Sequencing Studies20
In the current study, the difference in PAR4 signaling by variant was assessed for PAR4-dependent activation through Gα13. The rate of Gα13 activation after PAR4 stimulation was enhanced in membranes expressing the PAR4-Thr120 variant relative to membranes expressing the PAR4-Ala120 variant. Activation of the Gα13 effector RhoA occurred at earlier time points and was elevated in PAR4-stimulated platelets from subjects expressing the PAR4-Thr120 variant compared with subjects expressing the PAR4-Ala120 variant. Further, RhoA-dependent platelet shape change was enhanced in platelets expressing the PAR4-Thr120 variant after PAR4 stimulation. To assess the effects of altered PAR4 activity on response to agonist in the presence of antiplatelet therapy, healthy subjects expressing at least 1 copy of the PAR4-Thr120 variant were placed on a 7-day regimen of either aspirin or clopidogrel (Plavix) and were shown to have high on-treatment platelet reactivity in response to PAR4 stimulation compared with subjects homozygous for PAR4-Ala120. The difference in platelet activation by PAR4 variant was additionally supported by an observed enhancement of thrombus formation in microfluidic chambers perfused with whole blood from subjects expressing at least 1 copy of PAR4-Thr120 relative to blood from individuals homozygous for PAR4-Ala120. Importantly, healthy subjects expressing at least 1 copy of the PAR4-Thr120 allele (estimated in >81% of blacks and 34% of whites based on self-identified race and ethnicity) are shown here to exhibit decreased protection from treatment with the antiplatelet drugs aspirin and clopidogrel, suggesting a potential difference in the risk for thrombosis in people expressing PAR4-Thr120. Future clinical studies focused on the relative risk for thrombosis in this population will help to determine whether alternative therapeutic approaches are warranted in people expressing PAR4-Thr120.
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Blood Collection
Research involving humans was approved by the University of Michigan and Thomas Jefferson University Institutional Review Boards and conducted in accordance with the Declaration of Helsinki. All subjects signed an institutional review board–approved consent form before being enrolled in the study. Blood was drawn into vacutainers containing sodium citrate. Blood was collected twice from healthy volunteers who took either Plavix (75 mg) or aspirin (81 mg) once daily for 7 consecutive days; 1 draw before starting treatment and a second draw on the seventh day of single-antiplatelet administration. All subjects recruited for this study were genotyped for the rs773902 SNP (single nucleotide polymorphism) in the PAR4 gene (F2RL3) encoding for either an alanine or threonine at amino acid position 120 by Taqman allelic discrimination real-time PCR (Thermo Fisher).
Platelet Isolation
Citrated whole blood was centrifuged (200 g for 10 minutes) to isolate platelet-rich plasma. Platelet-rich plasma was treated with acid citrate dextrose (2.5% sodium citrate, 1.5% citric acid, and 2.0% D-glucose) and apyrase (0.02 U/mL) and then centrifuged (2000 g for 10 minutes) to pellet the platelets.16 Platelets were resuspended at 3.0×108 platelets per mL in Tyrode buffer (10 mmol/L HEPES, 12 mmol/L NaHCO3, 127 mmol/L NaCl, 5 mmol/L KCl, 0.5 mmol/L NaH2PO4, 1 mmol/L MgCl2, and 5 mmol/L glucose) unless otherwise stated.
Platelet Aggregation and Shape Change
To measure aggregation, platelets were stimulated with thrombin (Enzyme Research Laboratories), PAR4-AP (PAR4-activating peptide; AYPGKF; GL Biochem), or PAR1-AP (SFLLRN; GL Biochem) in a Chrono-Log lumi-aggregometer, and light transmittance was monitored in real time for 6 minutes at 37°C under stirring conditions (1100 rpm). To quantify platelet shape change, platelets were treated with EGTA (2 mmol/L) for 2 minutes before agonist stimulation in a lumi-aggregometer.
PAR4 Membrane Preparation
High Five and Sf9 insect cells were cultured as described (Table I in the online-only Data Supplement).21 Recombinant human PAR4 baculoviruses were generated from pFastBac-1 donor plasmids using the Bac-to-Bac system (Invitrogen). Mid-log growth phase High Five cells were infected with 1:100 dilutions of secondary-amplified baculoviruses expressing PAR4-Ala120 or PAR4-Thr120. After 48 hours, cells were lysed in native buffer (20 mmol/L HEPES, pH 7.4, 2 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L DTT [dithiothreitol], protease inhibitor cocktail) by nitrogen cavitation (Parr Industries). Lysates were precleared by centrifugation at 600 g, followed by centrifugation of the supernatant at 100 000 g to pellet membranes. The membrane pellets were Dounce homogenized into native buffer containing 10% (wt/vol) sucrose. Dounced membranes were layered onto a sucrose cushion gradient made from native buffer containing 40% (wt/vol) and 25% (wt/vol) sucrose and centrifuged to equilibrium at 65 000 g. Membranes were collected from the 25% to 40% sucrose interface, diluted, and centrifuged at 100 000 g, before Dounce homogenization into 20 mmol/L HEPES, pH 7.4, 1 mmol/L EGTA, and 12% (wt/vol) sucrose.
Cell Surface Biotinylation
High Five cells infected for 48 hours with PAR4 baculoviruses were washed with PBS containing protease inhibitor cocktail.21 Intact cells were suspended in PBS containing freshly prepared 2 mmol/L Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific) for 30 minutes and then quenched by PBS with 100 mmol/L glycine, pH 8.0. Cells were lysed in 25 mmol/L HEPES, pH 7.4, 150 mmol/L NaCl, 1% Triton X-100, 2 mmol/L MgCl2, 1 mmol/L EDTA, and 2% glycerol, and cell debris was cleared by centrifugation. Biotinylated proteins were captured from lysates using streptavidin Sepharose (GE Healthcare). Cell surface biotinylated PAR4 was visualized by Western blotting with anti-PAR4 antibody, kindly provided by Dr Marvin Neiman.22
GTPγS-Binding Assay
Recombinant Gαi1, Gα13, Gαs, Gαq, and Gβ1γ2 were purified from High Five cells as described.23,24 Prepared PAR4 membranes (0.5 µg per time point) were preincubated with 100 nmol/L Gα and 500 nmol/L Gβ1γ2 in the presence or absence of PAR4-AP (500 µmol/L) for 10 minutes in preincubation buffer (50 mmol/L HEPES, pH 7.4, 1 mmol/L DTT, 1 mmol/L EDTA, and 3 µg/mL BSA) at 25°C. The kinetic assay was initiated by equal volume addition of GTPγS-binding buffer (preincubation buffer plus 10 mmol/L MgCl2, 50 mmol/L NaCl, and 2 µmol/L [35S]-GTPγS [20 000−50 000 cpm/pmol]; PerkinElmer). Triplicate samples at each time point were quenched in 20 mmol/L Tris, pH 7.7, 100 mmol/L NaCl, 10 mmol/L MgCl2, 1 mmol/L GTP, and 0.08% (wt/vol) deionized polyoxyethylene 10 lauryl ether, C12E10. Samples were filtered through Protran BA85 nitrocellulose filters (GE Healthcare) and washed with 20 mmol/L Tris, pH 7.7, 100 mmol/L NaCl, and 2 mmol/L MgCl2. Dried filters were subjected to liquid scintillation counting. Data were fitted to 1-phase monoexponential association functions or by linear regression to calculate initial GTPγS binding rates.
RhoA-GTP Pulldown
Platelets (1×109 platelets per mL) were stimulated with PAR4-AP (25 μM) for the indicated time and lysed with 2X platelet lysis buffer (100 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 2% IGEPAL [octylphenoxy poly(ethyleneoxy)ethanol, branched], 1% sodium deoxycholate, 0.05% SDS, 2 mmol/L Na3VO4, 2 mmol/L PMSF [phenylmethylsulfonyl fluoride], 2 mg/mL leupeptin, and 2 mg/mL aprotinin) and then centrifuged (10 000 g for 1 minute at 4°C). Active, GTP-bound RhoA was selectively precipitated from the supernatant using a fusion protein containing the GST (glutathione S-transferase)-tagged ρ-binding domain of rhotekin conjugated to glutathione agarose beads. After incubation with the platelet lysate, beads were washed 5× with 1X platelet lysis buffer and then resuspended in 5X Laemmli buffer (300 mmol/L Tris, pH 6.8, 10% SDS, 50% glycerol, 25% 2-mercaptoethanol, and 0.05% bromophenol blue). Platelet lysate (total RhoA) and GTP-bound RhoA were resolved by SDS polyacrylamide gel electrophoresis and immunoblotted with antibodies against RhoA (Table I in the online-only Data Supplement). The levels of active RhoA were normalized to the amount of total RhoA in each sample and reported as fold change compared with the resting sample of each donor.
Platelet Spreading
Glass coverslips were coated with fibrinogen (50 μg/mL) for 1.5 hours at ambient temperature and then blocked with 5% BSA (wt/vol) for 30 minutes at ambient temperature. Platelets (1×107 platelets per mL) treated with indomethacin (10 µmol/L) and apyrase (50 U/mL) were stimulated with PAR4-AP (25 µmol/L) and allowed to spread on coated coverslip. Platelet spreading was recorded in real time after initial platelet adhesion to fibrinogen using an inverted fluorescent microscope (Zeiss Axiovert 200 mol/L; 20X objective). Spreading was determined by platelet size as analyzed with Slidebook, program 6.0.
Clot Retraction
Citrated platelet-rich plasma adjusted to 3.0×108 platelet per mL using autologous platelet-poor plasma was recalcified (CaCl2; 2 mmol/L) and treated with thrombin at 37°C. Representative images of clots were taken at indicated times. Clot size was quantified by removing the clot from the sample, then measuring the volume of plasma eluded from the contracting clot, and subtracting that from the initial volume (500 µL).
Microfluidic Ex Vivo Flow Chamber Assays
Microfluidic perfusion chamber slides (µ-slide VI0.1; Ibidi) were coated with 100 µg/mL of type I collagen (Chrono-Log) in the presence or absence of TF (tissue factor; 250 pmol/L Innovin; Siemens) overnight at 4°C. Citrated whole blood was stained with 1 µmol/L of 3,3′-dihexyloxacarbocyanine iodide for 5 minutes at 37°C. Stained whole blood was recalcified with 5 mmol/L CaCl2 and immediately perfused at arterial shear (1500 s−1) through a coated microfluidic slide heated to 37°C. Platelet adhesion and accumulation were recorded for 4 minutes under an inverted fluorescent microscope (Zeiss Axio Observer Z1 Marianas; 20X objective). Platelet accumulation was quantified using Slidebook 6.0 (Intelligent Imaging Innovations). Whole blood was treated with inhibitors of PAR1 (RWJ-56110; 20 µmol/L), COX (acetylsalicylic acid; 100 µmol/L), or P2Y12 (2-methylthioadenosine 5′-monophosphate triethylammonium [50 µmol/L]) for 30 minutes at 37°C.
Whole-Blood Aggregation
Whole-blood impedance aggregometry was performed using a Chrono-Log lumi-aggregometer (Chrono-Log Corp). Briefly, equal volumes of saline (0.9%) and citrated whole blood (37°C) were added to an aggregation cuvette. Dilute blood samples were recalcified with 5 mmol/L CaCl2 and stimulated with either arachidonic acid (AA; Cayman), ADP (Sigma Aldrich), or PAR1-AP. Impedance was measured in real time for 3 minutes at 37°C under stirring conditions (1100 rpm).
Statistics
Unpaired, 2-tailed Student t tests and 2-way ANOVA were performed with Prism 7 (GraphPad Software) to analyze the data. Multiple statistical analyses were used in this study, and the statistical test used in each assay is reported in the figure legends.
Results
PAR4-Thr120 Variant Enhances Gαq and Gα13 Activation Compared With PAR4-Ala120
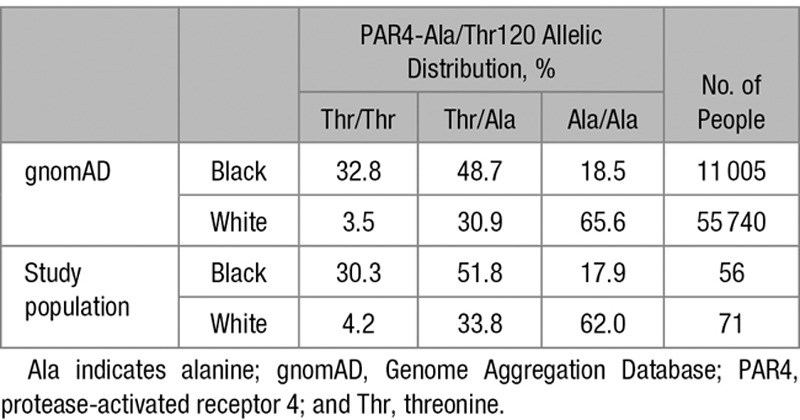
Although previous work provided indirect evidence that the PAR4-Thr120 variant facilitates increased Gαq-dependent calcium mobilization relative to the PAR4-Ala120 variant,14,16 direct evidence of a change in the rate of activation of PAR4 is lacking. Therefore, we tested whether stimulation of PAR4-Thr120 exhibits an increased rate of receptor activation compared with PAR4-Ala120. Recombinant human PAR4-Ala120 or PAR4-Thr120 were expressed in High Five insect cells, and the surface proteins from intact cells were biotinylated and isolated with Streptavidin resin. The relative amounts of PAR4-Ala120 or PAR4-Thr120 isolated from the cell surface were equivalent (Figure 1A). Native membrane homogenates were prepared from these cells and immunoblotted with PAR4 antibody to show that equivalent levels of PAR4-Ala120 or PAR4-Thr120 were present in the membrane preparations.
Figure 1.
The PAR4 (protease-activated receptor 4)-Thr120 variant enhances Gq and G13 activation compared with the PAR4-Ala120 variant. A, Left, The relative abundance of recombinant human PAR4-Ala120 and PAR4-Thr120 in prepared membranes was visualized by Western blot analysis in comparison with control noninfected membranes. Right, Intact cells expressing the recombinant human PAR4 variants were subjected to cell surface biotinylation. Biotinylated cell surface proteins were isolated, and relative PAR4 levels were compared by Western blot analysis. B–E, Prepared membranes of PAR4-Ala120 (blue) or PAR4-Thr120 (red) were preincubated with PAR4-AP (PAR4-activating peptide; closed symbols) or buffer control (open symbols) before reconstitution with the purified G protein heterotrimers: (B) G13, (C) Gq, (D) Gi1, or (E) Gsshort. The kinetics of PAR4-stimulated [35S]-GTPγS binding to G proteins (activation) are shown. B and C, (inset) Initial linear [35S]-GTPγS binding rates of (B) G13 or (C) Gq stimulated by PAR4-AP–activated PAR4-Ala120 and PAR4-Thr120. An unpaired t test, 2-tailed, was performed. Data represent mean±SEM. Ala indicates alanine; and Thr, threonine. **P<0.01, ***P<0.001, ****P<0.0001.
The PAR4 membrane preparations were treated with or without 500 µmol/L PAR4-AP before being reconstituted with purified G protein α subunits and Gβ1γ2. PAR4-stimulated G protein binding was evaluated by measuring the rates of Gα [35S]-GTPγS binding. Both PAR4 variants bound G13 and Gq in a PAR4-AP–dependent manner (Figure 1B and 1C); however, neither receptor exhibited binding to Gi or Gs over background (Figure 1D and 1E). Although the maximal binding of PAR4-Thr120 and PAR4-Ala120 was equivalent for Gq and G13 (Figure 1B and 1C, left), the initial binding kinetics were significantly faster for PAR4-Thr120 compared with PAR4-Ala120 (Figure 1B and 1C, right).
RhoA Is Differentially Activated by PAR4 Ala120/Thr120 Dimorphism
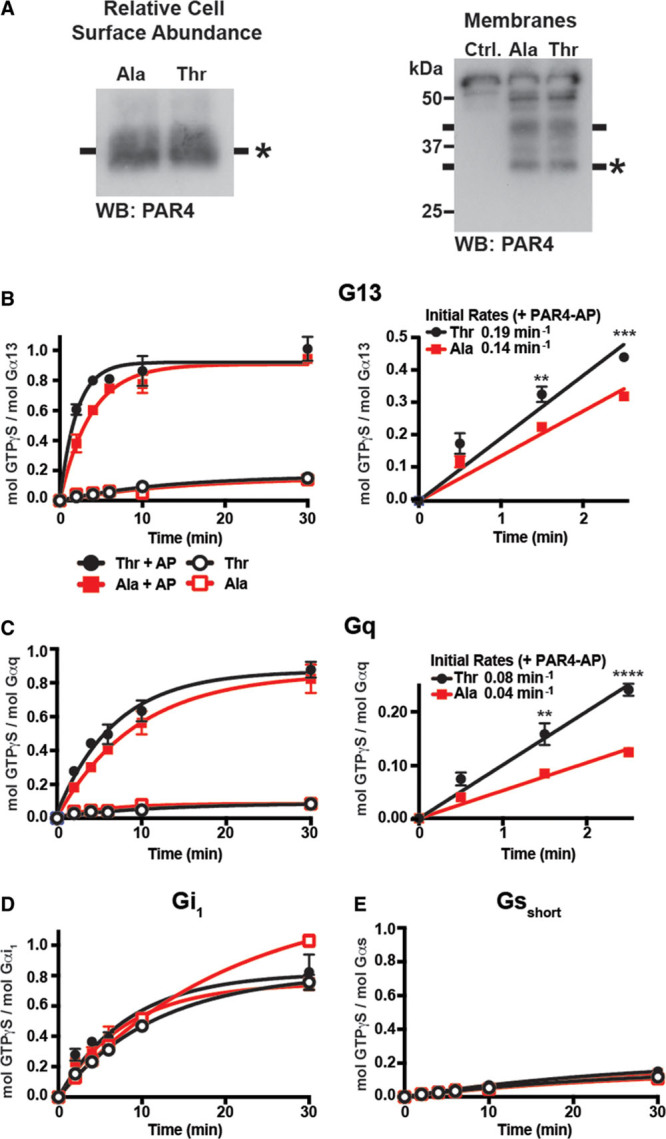
Prior studies demonstrated that because of the higher frequency of ≥1 alleles of PAR4-Thr120 in the black population relative to the white population (>80% of blacks have at least 1 copy of the PAR4-Thr120 allele compared with ≈35% of whites based on genetic database analysis; Table), signaling components of the Gq pathway are activated to a higher degree in PAR4-stimulated platelets in general from black donors relative to platelets from their white counterparts.16 To determine whether the kinetic difference observed in G13 activation by PAR4 Ala120/Thr120 dimorphism (Figure 1) results in an increase in G13 signaling in PAR4 stimulation platelets, RhoA activation was analyzed by PAR4 Ala120/Thr120 variant. Further evidence for this difference in activation is provided in Figure 1 above. To evaluate the downstream consequence of elevated Gα13 signaling in PAR4-stimulated platelets, activation of RhoA—a proximal effector dependent on Gα13 activity—was measured in PAR4-stimulated human platelets to determine whether the differences in Gα13 activity translate to potentiation of RhoA activation. In response to PAR4 stimulation (25 μM PAR4-AP), RhoA activation was enhanced in platelets from individuals containing at least 1 copy of the PAR4-Thr120 variant compared with platelets from individuals homozygous for the PAR4-Ala120 variant (Figure 2A).
Figure 2.
After PAR4 (protease-activated receptor 4) stimulation, RhoA activation and shape change are heightened in platelets from individuals expressing PAR4-T120. A, Active RhoA was selectively precipitated from the lysates of PAR4-stimulated (PAR4-AP [PAR4-activating peptide]; 25 µmol/L) platelets and normalized to the amount of total RhoA in each sample as determined by Western blot. RhoA activation data, reported as fold change relative to the unstimulated control, were analyzed by PAR4-120 variants (alanine and threonine). B, Shape change was measured in a lumi-aggregometer after PAR4 stimulation of EGTA-treated platelets, and data were analyzed by PAR4-120 variant. Two-way statistical ANOVA was performed. Data represent mean±SEM. AA indicates arachidonic acid.
PAR4-Mediated Platelet Shape Change Differs by PAR4 Ala120/Thr120 Dimorphism
Activation of RhoA downstream of Gα13 activation has been shown to induce cytoskeletal changes in platelets.25,26 These changes in the platelet are observed as a shape change from a discoid to spherical shape and are an important component of the overall activation process.27 To investigate whether the differences observed in RhoA activation by PAR4 Ala120/Thr120 dimorphism result in an enhancement of platelet shape change, platelets were stimulated with PAR4-AP, and shape change was measured as a positive inflection in an aggregometer. To measure shape change without the complication of the aggregation that follows, platelets were treated with EGTA. Platelets from subjects expressing the PAR4-T120 variant showed a significant increase in shape change in response to PAR4-AP (25 μM) compared with platelets from subjects expressing the PAR4-A120 variant (Figure 2B). To confirm that the observed differences were PAR4-specific, the experiments were repeated in the presence of PAR1-AP (0.5 μM). No difference in PAR1-AP–mediated shape change was observed between the individuals who expressed at least 1 copy of PAR4-Thr120 and individuals homozygous for PAR4-Ala120 (Figure 2B).
Gα13-Dependent Outside-In Signaling Is Not Differentially Affected by PAR4 Ala120/Thr120 Dimorphism
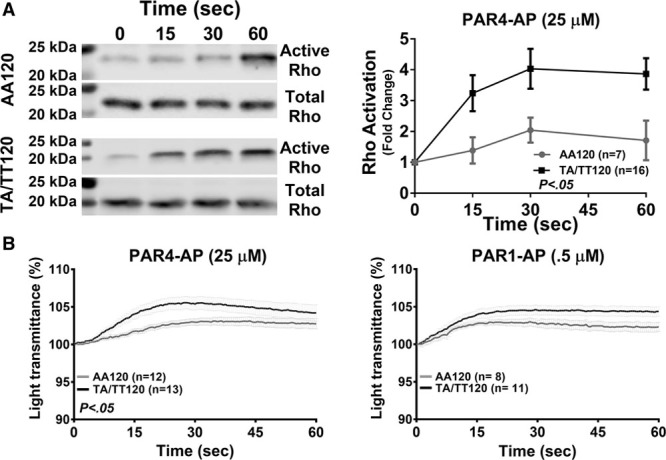
In agreement with the observations in Figure 1, activation of Gα13 proceeds primarily in a GPCR-dependent manner. However, recent reports have suggested that Gα13 may also be activated at later stages in the platelet activation through ligand engagement of the integrin αIIbβ3 (outside-in signaling) leading to GPCR-independent platelet spreading, which coincides with the inactivation of RhoA.26,28,29 Because of the difference in RhoA signaling observed in PAR4-stimulated platelets by PAR4 genotype, potential downstream effects regulated by RhoA were assessed for differences in platelet spreading. Platelets were allowed to spread on fibrinogen in the presence or absence of PAR4-AP (25 μM). The degree of spreading was measured for ≤300 seconds after stimulation. No difference in platelet spreading on fibrinogen-coated coverslips was observed irrespective of PAR4-120 variant in the presence or absence of PAR4-AP (Figure 3A).
Figure 3.
No difference in platelet spreading and clot retraction by PAR4 (protease-activated receptor 4) Ala120/Thr120 dimorphism. A, Platelets treated with indomethacin (20 μM) and apyrase (50 U/mL) were allowed to spread on fibrinogen-coated (50 μg/mL) glass cover slips in the presence or absence of PAR4-AP (PAR4-activating peptide; 25 μM). The surface area of 5 spread platelets from 5 donors was measured every 30 s for 300 s. B, Platelet-rich plasma was incubated with thrombin (2.5 or 5 nmol/L), and clot size was quantified by subtracting the volume of plasma expelled from the contracting clot from the starting/original volume (500 µL) at the indicated time. Dashed line depicts the total volume of platelet-rich plasma in samples that were not treated with thrombin after 60 min. Two-way statistical ANOVA was performed. Data represent mean±SEM. AA indicates arachidonic acid.
Clot retraction—another physiological end point that is dependent on outside-in signaling26—was also quantified to investigate potential differences in Gα13-dependent outside-in signaling in the platelet. Platelet-rich plasma was stimulated with either 2.5 or 5 nmol/L of thrombin, and the time to clot retraction was measured for ≤60 minutes. No difference in time to clot retraction was observed by PAR4-120 variant (Figure 3B). Together, these data suggest the difference in PAR4-mediated RhoA activation by PAR4 Ala120/Thr120 dimorphism does not alter outside-in–mediated spreading or clot retraction.
Platelet Accumulation in Whole Blood Under Arterial Shear Differs by PAR4 Variant
Heightened platelet reactivity is associated with increased thrombotic events; however, it remains to be determined whether the heightened PAR4-mediated platelet activation observed by PAR4-Ala/Thr120 variant contributed to enhanced platelet adhesion and thrombus formation ex vivo.4 To assess whether the difference in PAR4 signaling potentiates thrombus formation, recalcified citrated whole blood was perfused through collagen-coated microfluidic chambers at arterial shear (1500 s−1). Under these conditions, platelet activation via the binding of GPVI to collagen initiates the formation of procoagulant platelets, which are capable of generating thrombin.30 Platelet accumulation was greater in collagen-coated chambers perfused with whole blood from individuals who express at least 1 copy of the PAR4-Thr120 variant and individuals homozygous for the PAR4-Ala120 variant (Figure 4A). Because chambers were coated with collagen—a known platelet activator—we performed ex vivo perfusion assays with heparinized blood to determine whether the platelet accumulation observed in our microfluidic assay was dependent on thrombin signaling. As expected, whole blood collected in heparin, the factor Xa and thrombin inhibitor(s), had a reduction in platelet accumulation independent of PAR4 variant compared with citrated blood supporting a thrombin-dependent component is essential for the platelet adhesion and thrombus formation observed in Figure 4A. Because thrombin activates platelets via PAR1 and PAR4, to determine whether the difference in thrombus formation by PAR4 variant was independent of PAR1 signaling, whole blood was treated with the PAR1 inhibitor RWJ-56110. Although the overall platelet accumulation was decreased for both groups, the difference in thrombus formation by PAR4 variant persisted in whole blood treated with a PAR1 inhibitor (Figure 4C). The formation of procoagulant platelets in vitro and thrombin generation in citrated blood under arterial shear is partially dependent on PAR4 signaling.31 Therefore, to generate thrombin independent of PAR4 signaling, perfusion chambers were coated with TF in addition to collagen. Platelet accumulation was enhanced in microfluidic chambers coated with collagen and TF when perfused with whole blood from individuals who expressed at least 1 copy of the PAR4-Thr120 variant and individuals homozygous for the PAR4-Ala120 variant (Figure 4D).
Figure 4.
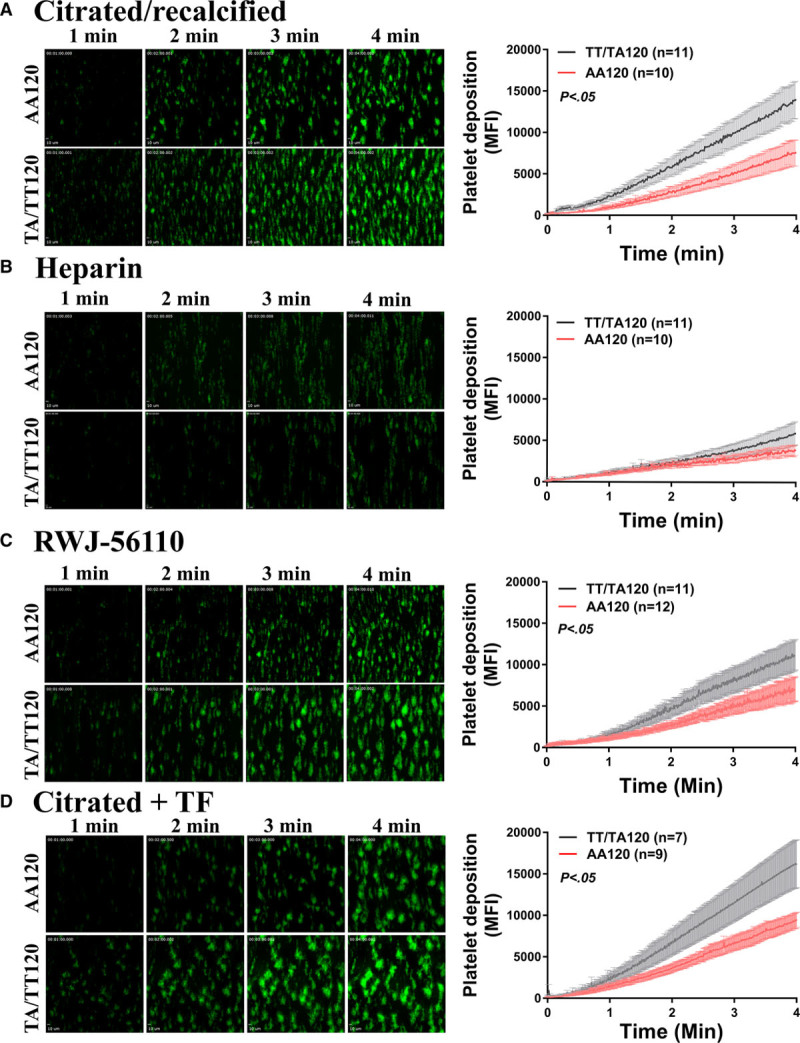
Thrombin-dependent platelet accumulation is enhanced under arterial shear ex vivo with whole blood from individuals who express PAR4 (protease-activated receptor 4)-Thr120 compared with those who are homozygous for the PAR4-Ala120 variant. A, Recalcified citrated or (B) heparinized blood was stained with 3,3′-dihexyloxacarbocyanine iodide and perfused through collagen-coated perfusion chamber at arterial shear (1500 s−1) for 4 min. Platelet accumulation as quantified by mean fluorescence intensity using an inverted fluorescent microscope (20X objective) was analyzed by PAR4-120 variants (alanine and threonine). C, Recalcified citrated blood was treated with RWJ-56110 (10 µmol/L) for 15 min before being perfused through a collagen-coated chamber. D, Recalcified citrated blood was perfused through a chamber coated with collagen and tissue factor at arterial shear (1500 s1).Two-way statistical ANOVA was performed. Data represent mean±SEM. AA indicates arachidonic acid; and MFI, mean fluorescence intensity.
Subjects Expressing the PAR4-Thr120 Variant Exhibit Elevated On-Treatment Platelet Reactivity in Response to PAR4 Stimulation
The racial difference in PAR4-mediated platelet activation between white and black individuals persists in platelets treated in vitro with aspirin and a P2Y12 antagonist.16 To determine whether subjects expressing PAR4-Thr120 on antiplatelet therapy have higher on-treatment PAR4-mediated platelet reactivity compared with subjects expressing PAR4-Ala120, PAR4-AP-induced platelet aggregation was assessed in platelets isolated from healthy individuals taking either aspirin or Plavix—2 irreversible platelet antagonists. Washed platelets isolated from healthy individuals with at least 1 copy of PAR4-Thr120 who had taken aspirin (81 mg) or Plavix (75 mg) for 7 days were hyperresponsive to PAR4-AP (0–100 µmol/L) compared with platelets from individuals homozygous for the PAR4-Ala120 variant (Figure 5A and 5B).
Figure 5.
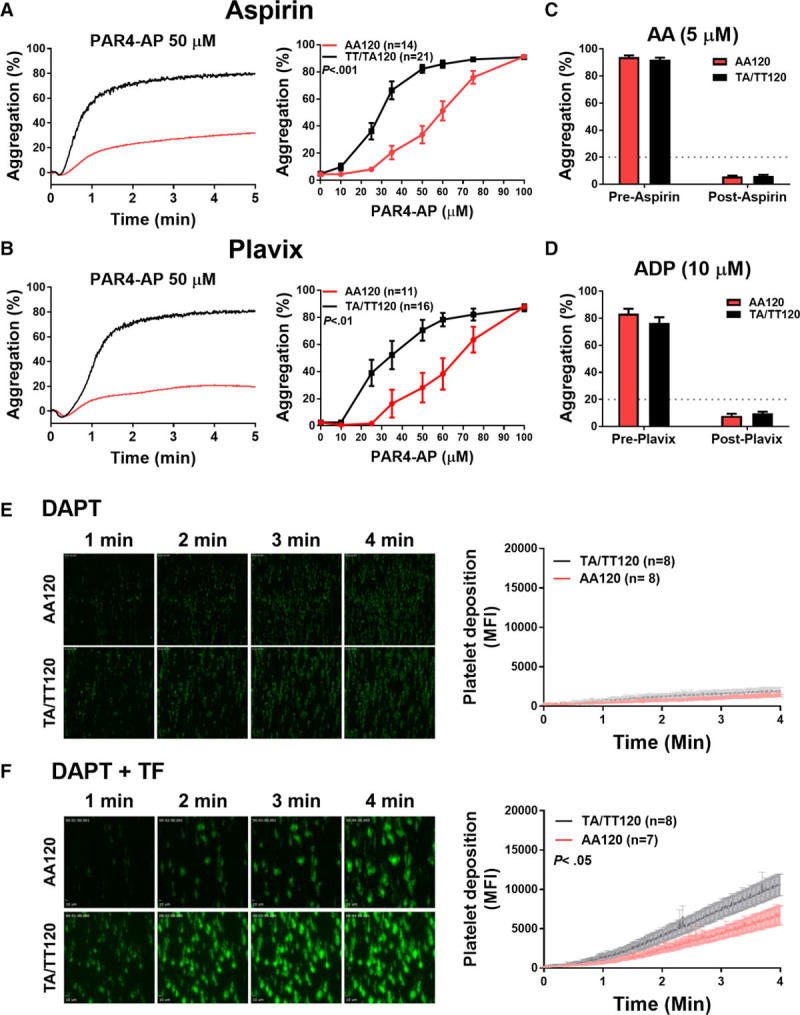
The difference in PAR4 (protease-activated receptor 4)-mediated platelet activation by PAR4 Ala120/Thr120 dimorphism persists in individuals treated with either aspirin or Plavix. Representative aggregation tracings of platelets from individuals who are homozygous for the PAR4-Ala120 variant or individuals who express at least 1 copy of the PAR4-Thr120 variant on aspirin (A) or Plavix (B) in response to 50 µmol/L PAR4-AP (PAR4-activating peptide). Quantification of the maximum aggregation by PAR4 variant of platelets isolated from individuals on aspirin (A) or Plavix (B) in response to increasing concentrations of PAR4-AP. Platelets isolated from healthy individuals on aspirin (81 mg) or Plavix (75 mg) were stimulated in an aggregometer with (C) arachidonic acid (AA) or (D) ADP, respectively. Donors on single-antiplatelet therapy who had an aggregation response ≥20% (dashed line) to either AA or ADP, respectively, were excluded from the study. Recalcified citrated whole blood was treated with dual-antiplatelet therapy (DAPT), aspirin (100 µmol/L), and 2-methylthioadenosine 5′-monophosphate triethylammonium (50 µmol/L), before being perfused through a collagen-coated chamber at arterial shear (1500 s−1) in the (E) presence or (F) absence of TF (tissue factor). Two-way statistical ANOVA was performed. Data represent mean±SEM. MFI indicates mean fluorescence intensity.
TXA2 and ADP potentiate the aggregation of platelets stimulated with low doses of PAR4-AP16; therefore, platelets from individuals on single-antiplatelet therapy were stimulated with either AA or ADP to determine whether they responded to antiplatelet therapy. Clinically, an aggregation of <20% in response to platelet stimulation with AA or ADP is a commonly used cutoff to determine whether an individual is responsive to aspirin or P2Y12 inhibition, respectively.32 To exclude data variability because of noncompliance, data from donors on single-antiplatelet therapy whose platelets aggregated ≥20% in response to AA (5 µmol/L) or ADP (10 µmol/L), respectively, for aspirin and Plavix treatment, were excluded from the study (Figure 5C and 5D). Whether a donor’s failure to respond to single-antiplatelet therapy was because of variations in pharmacokinetics or noncompliance was not further investigated.
To determine whether a difference exists in thrombus formation by PAR4 variant in the presence of dual-antiplatelet therapy, whole blood was treated ex vivo with aspirin (COX-1 inhibitor; 100 µmol/L) and 2-methylthioadenosine 5′-monophosphate triethylammonium (P2Y12 receptor inhibitor; 50 µmol/L) before being perfused through a collagen-coated perfusion channel. No difference in thrombus formation by PAR4 variant was observed in the presence of dual-antiplatelet therapy when the blood was perfused over collagen-coated channels (Figure 5E). To confirm that the COX-1 and P2Y12 receptors on the platelets had been sufficiently inhibited, whole-blood impedance aggregrometry was performed on blood treated with aspirin and 2-methylthioadenosine 5′-monophosphate triethylammonium. Whole blood treated with vehicle control aggregated after stimulation with AA or ADP, whereas treatment with aspirin and 2-methylthioadenosine 5′-monophosphate triethylammonium resulted in a significantly attenuated response to either AA or ADP (Figure I in the online-only Data Supplement). The formation of procoagulant platelets requires prolonged calcium release, which is inhibited by dual-antiplatelet therapy. Whole blood treated with dual-antiplatelet therapy was perfused through chambers coated with TF in addition to collagen to initiate coagulation independent of procoagulant platelet formation. Thrombus formation was enhanced in chambers perfused with dual-antiplatelet therapy–treated whole blood from individuals with at least 1 copy of PAR4-Thr120. Platelets from these individuals exhibited an increase in thrombus formation compared with individuals homozygous for PAR4-Ala120 (Figure 5F).
Discussion
The racial difference in cardiovascular disease risk has been observed for many decades, and recently, the genetic underpinnings for this difference have been partially identified.15 Subsequent studies have identified that the significant difference in platelet activation by race is selectively regulated by PAR4 in human platelets and that PAR4 expresses several single-nucleotide variants, including the PAR4 Thr120/Ala120 dimorphism, that shift platelet reactivity.9,14,16 Because PAR4 signaling is required for the procoagulant activity of platelets,31 enhanced PAR4-mediated signaling observed in platelets in vitro from individuals who express at least 1 copy of the PAR4-Thr120 variant could potentiate thrombus formation or minimize bleeding via enhanced platelet activation and thrombin generation. Further, although PAR4 signaling in platelets is required for thrombus growth, it is unclear whether the differences in PAR4 activation by PAR4 variant observed in vitro increase thrombus formation. The PAR4-120 variant distribution of the white and black subjects recruited for this study mirror those in the Genome Aggregation Database—the largest sequencing database (Table). In the current study, platelet accumulation was shown to be greater in TF-coated chambers perfused with blood from individuals with at least 1 copy of the PAR4-Thr120 variant compared with individuals homozygous for the PAR4-Ala120 variant. Interestingly, the difference in platelet accumulation by PAR4 variant was ablated in whole blood treated with heparin—a thrombin inhibitor suggesting the difference in platelet accumulation was because of thrombin-dependent signaling.
Thrombin receptors on the human platelet are thought to signal through 2 classes of G proteins, Gq and G13, which are known to regulate calcium mobilization and cell shape change, respectively. Work in the endothelium previously showed that at least one of the thrombin receptors, PAR1, exhibits differential or biased signaling toward Gq versus G13 depending on the mechanism by which the receptor is activated.12 Recent studies demonstrated that in PAR4-stimulated platelets, components of the Gq signaling axis were elevated by both race and PAR4 genotype.14,16 Because PARs are capable of biased signaling through 1 G protein over another,19,33 this study sought to determine whether in fact differences in platelet activation by PAR4-Ala120/Thr120 dimorphism were due in part to biased activation of Gq over G13 or whether differences in PAR4 signaling by variant result in an overall activation of all pathways downstream of PAR4 activation. Quantitative evaluation of G protein activation in isolated membranes containing the PAR4-Thr120 variant exhibited a significant increase in the rate of activation of both Gq and G13 after PAR4 stimulation compared with isolated membranes containing the PAR4-Ala120 variant, suggesting the difference in PAR4 activation by PAR4 genotype was not because of biased signaling. Although biased activation is clearly not a component of the increased platelet activity in these subjects, the mechanism by which the PAR4-Thr120 variant influences its ability to activate Gq and G13 remains poorly understood. G13 activity and RhoA activity downstream of PAR4 activation were shown to be significantly elevated. RhoA activation after stimulation of PAR4 is known to facilitate inside-out signaling and possibly outside-in signaling.28 Interestingly, although the inside-out activity of G13, RhoA activation, and shape change was shown to be upregulated in platelets from subjects expressing PAR4-Thr120 variant, as expected, the outside-in signaling initiated by αIIbβ3 engagement of G13, resulting in platelet spreading and clot retraction, did not exhibit differences by PAR4 genotype suggesting that G13 activation is differentially activated by PAR4 but not by αIIbβ3 engagement.
Figures 1 through 3 identify that the increase in PAR4 signaling because of PAR4-Thr120 variant expression is not because of biased receptor activation but rather upregulation of all PAR4-dependent signaling. However, a key question that remains is whether potential differences in morbidity in patients with cardiovascular risk are due solely to hyperactivity of PAR4 or whether resistance to antiplatelet therapy because of heightened PAR4 activity may play an important role in this process. Although PAR4 activity is increased in platelets from subjects expressing the PAR4-Thr120 variant, it was expected that inhibition of COX-1 or P2Y12 receptor would significantly diminish PAR4-mediated activity. Surprisingly, healthy subjects expressing the PAR4-Thr120 variant taking a standard-of-care prevention for platelet activation, either aspirin (COX-1 inhibitor) or clopidogrel (P2Y12 receptor inhibitor), showed significant resistance to protection from PAR4-mediated platelet aggregation. Importantly, when the contribution because of thrombin was assessed ex vivo in whole-blood arterial shear conditions, it was shown that thrombin activation was necessary to elicit full activation, and under these conditions, platelets from subjects expressing the PAR4-Thr120 variant accumulated at a significantly higher rate compared with blood from subjects expressing PAR4-Ala120.
The current study found that the difference in PAR4-mediated platelet activation by PAR4 variant upregulates all PAR4-mediated signaling in the human platelet and importantly persists in healthy individuals treated with single-antiplatelet therapy. Further, the PAR4 variant difference in PAR4 signaling correlated with heightened thrombus formation in ex vivo perfusion chambers in the presence or absence of single-antiplatelet therapy. These findings were further supported through ex vivo experiments where blood spiked with dual-antiplatelet drugs targeting COX-1 and the P2Y12 receptor showed less protection in platelets expressing at least 1 allele of PAR4-Thr120. Although current antiplatelet therapy has successfully decreased morbidity and mortality because of thrombosis, a large number of individuals taking dual-antiplatelet therapy remain at risk for a thrombotic event, and its use is further limited by the increased risk for life-threatening bleeding events. Therefore, understanding interindividual variations in platelet reactivity has been a key clinical goal for improving personalized antiplatelet therapy allowing physicians to maximize antithrombotic conditions while retaining normal hemostasis. Because individuals who express the PAR4-T120 variant exhibit a higher on-treatment PAR4-mediated platelet reactivity compared with their counterparts who express the PAR4-Ala120 variant, PAR4 could represent a candidate for novel PAR4 antagonists. This work provides further evidence that PAR4 may represent a target for a precision medicine approach for prevention of platelet activation and occlusive thrombosis leading to myocardial infarction and stroke.
Acknowledgments
The PAR4 antibody was kindly provided by Marvin Neiman.
Sources of Funding
This work was funded, in part, through the National Institutes of Health grants R01 GM105671 (M. Holinstat), R01 HL114405 (M. Holinstat), R01 MD007880 (M. Holinstat and P.F. Bray), R01 GM120110 (G.G. Tall), R01 GM0088242 (G.G. Tall), K99 HL136784 (B.E. Tourdot), and K08 HL131993 (Y. Kanthi); Bo Schembechler Heart of a Champion Fund and McKay Award (Y. Kanthi); Ruth L. Kirschstein Institutional National Research Service Awards F31 HL129481 (J. Yeung) and F32 HL129491 (B.E. Tourdot); and the American Heart Association (15POST24480075; B.E. Tourdot).
Disclosures
We declare that patent 9 750 757 (M. Holinstat and P.F. Bray) relating to this work has been awarded by the US patent office.
Nonstandard Abbreviations and Acronyms
- AA
- arachidonic acid
- COX
- cyclooxygenase
- GPCR
- G protein–coupled receptor
- GST
- glutathione S-transferase
- PAR
- protease-activated receptor
- PAR1-AP
- PAR1-activating peptide
- PAR4-AP
- PAR4-activating peptide
- TF
- tissue factor
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.118.311112/-/DC1.
Highlights
The PAR (protease-activated receptor) 4-Thr120 variant enhances Gq and G13 activation compared with PAR4-Ala120.
Ex vivo thrombus formation was enhanced in blood from subjects expressing PAR4-Thr120 relative to subjects expressing PAR4-Ala120.
Expression of PAR4-Thr120 in human platelets results in resistance to dual-antiplatelet therapy.
References
- 1.Jackson SP. Arterial thrombosis–insidious, unpredictable and deadly. Nat Med. 2011;17:1423–1436. doi: 10.1038/nm.2515. doi: 10.1038/nm.2515. [DOI] [PubMed] [Google Scholar]
- 2.Berry JD, Dyer A, Cai X, Garside DB, Ning H, Thomas A, Greenland P, Van Horn L, Tracy RP, Lloyd-Jones DM. Lifetime risks of cardiovascular disease. N Engl J Med. 2012;366:321–329. doi: 10.1056/NEJMoa1012848. doi: 10.1056/NEJMoa1012848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas KL, Honeycutt E, Shaw LK, Peterson ED. Racial differences in long-term survival among patients with coronary artery disease. Am Heart J. 2010;160:744–751. doi: 10.1016/j.ahj.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 4.Tsiara S, Elisaf M, Jagroop IA, Mikhailidis DP. Platelets as predictors of vascular risk: is there a practical index of platelet activity? Clin Appl Thromb Hemost. 2003;9:177–190. doi: 10.1177/107602960300900301. [DOI] [PubMed] [Google Scholar]
- 5.Elwood PC, Renaud S, Sharp DS, Beswick AD, O’Brien JR, Yarnell JW. Ischemic heart disease and platelet aggregation. The Caerphilly Collaborative Heart Disease Study. Circulation. 1991;83:38–44. doi: 10.1161/01.cir.83.1.38. [DOI] [PubMed] [Google Scholar]
- 6.Hamilton JR, Trejo J. Challenges and opportunities in protease-activated receptor drug development. Annu Rev Pharmacol Toxicol. 2017;57:349–373. doi: 10.1146/annurev-pharmtox-011613-140016. doi: 10.1146/annurev-pharmtox-011613-140016. [DOI] [PubMed] [Google Scholar]
- 7.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 8.Sidhu TS, French SL, Hamilton JR. Differential signaling by protease-activated receptors: implications for therapeutic targeting. Int J Mol Sci. 2014;15:6169–6183. doi: 10.3390/ijms15046169. doi: 10.3390/ijms15046169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.French SL, Hamilton JR. Protease-activated receptor 4: from structure to function and back again. Br J Pharmacol. 2016;173:2952–2965. doi: 10.1111/bph.13455. doi: 10.1111/bph.13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holinstat M, Boutaud O, Apopa PL, Vesci J, Bala M, Oates JA, Hamm HE. Protease-activated receptor signaling in platelets activates cytosolic phospholipase A2α differently for cyclooxygenase-1 and 12-lipoxygenase catalysis. Arterioscler Thromb Vasc Biol. 2011;31:435–442. doi: 10.1161/ATVBAHA.110.219527. doi: 10.1161/ATVBAHA.110.219527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holinstat M, Voss B, Bilodeau ML, Hamm HE. Protease-activated receptors differentially regulate human platelet activation through a phosphatidic acid-dependent pathway. Mol Pharmacol. 2007;71:686–694. doi: 10.1124/mol.106.029371. doi: 10.1124/mol.106.029371. [DOI] [PubMed] [Google Scholar]
- 12.Holinstat M, Voss B, Bilodeau ML, McLaughlin JN, Cleator J, Hamm HE. PAR4, but not PAR1, signals human platelet aggregation via Ca2+ mobilization and synergistic P2Y12 receptor activation. J Biol Chem. 2006;281:26665–26674. doi: 10.1074/jbc.M602174200. doi: 10.1074/jbc.M602174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. 2000;39:5458–5467. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- 14.Edelstein LC, Simon LM, Lindsay CR, Kong X, Teruel Montoya R, Tourdot BE, Chen ES, Ma L, Coughlin S, Nieman M, Holinstat M, Shaw CA, Bray PF. Common variants in the human platelet PAR4 thrombin receptor alter platelet function and differ by race. Blood. 2014;124:3450–3458. doi: 10.1182/blood-2014-04-572479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edelstein LC, Simon LM, Montoya RT, Holinstat M, Chen ES, Bergeron A, Kong X, Nagalla S, Mohandas N, Cohen DE, Dong JF, Shaw C, Bray PF. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nat Med. 2013;19:1609–1616. doi: 10.1038/nm.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tourdot BE, Conaway S, Niisuke K, Edelstein LC, Bray PF, Holinstat M. Mechanism of race-dependent platelet activation through the protease-activated receptor-4 and Gq signaling axis. Arterioscler Thromb Vasc Biol. 2014;34:2644–2650. doi: 10.1161/ATVBAHA.114.304249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCoy KL, Gyoneva S, Vellano CP, Smrcka AV, Traynelis SF, Hepler JR. Protease-activated receptor 1 (PAR1) coupling to G(q/11) but not to G(i/o) or G(12/13) is mediated by discrete amino acids within the receptor second intracellular loop. Cellular Signal. 2012;24:1351–1360. doi: 10.1016/j.cellsig.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLaughlin JN, Shen L, Holinstat M, Brooks JD, DiBenedetto E, Hamm HE. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem. 2005;280:25048–25059. doi: 10.1074/jbc.M414090200. [DOI] [PubMed] [Google Scholar]
- 19.Zhao P, Metcalf M, Bunnett NW. Biased signaling of protease-activated receptors. Front Endocrinol. 2014;5:67. doi: 10.3389/fendo.2014.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lek M, Karczewski KJ, Minikel EV, et al. Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoveken HM, Hajduczok AG, Xu L, Tall GG. Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc Natl Acad Sci USA. 2015;112:6194–6199. doi: 10.1073/pnas.1421785112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mumaw MM, de la Fuente M, Arachiche A, Wahl JK, III, Nieman MT. Development and characterization of monoclonal antibodies against protease activated receptor 4 (PAR4). Thromb Res. 2015;135:1165–1171.. doi: 10.1016/j.thromres.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kozasa T, Gilman AG. Purification of recombinant G proteins from sf9 cells by hexahistidine tagging of associated subunits. Characterization of alpha 12 and inhibition of adenylyl cyclase by alpha z. J Biol Chem. 1995;270:1734–1741. doi: 10.1074/jbc.270.4.1734. [DOI] [PubMed] [Google Scholar]
- 24.Chan P, Gabay M, Wright FA, Kan W, Oner SS, Lanier SM, Smrcka AV, Blumer JB, Tall GG. Purification of heterotrimeric G protein α subunits by GST-Ric-8 association primary characterization of purified Gαolf. J Biol Chem. 2011;286:2625–2635. doi: 10.1074/jbc.M110.178897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elvers M. RhoGAPs and Rho GTPases in platelets. Hamostaseologie. 2016;36:168–177. doi: 10.5482/HAMO-14-09-0046. [DOI] [PubMed] [Google Scholar]
- 26.Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost. 2013;11:35–46. doi: 10.1111/jth.12051. doi: 10.1111/jth.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moers A, Nieswandt B, Massberg S, Wettschureck N, Grüner S, Konrad I, Schulte V, Aktas B, Gratacap MP, Simon MI, Gawaz M, Offermanns S. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9:1418–1422. doi: 10.1038/nm943. doi: 10.1038/nm943. [DOI] [PubMed] [Google Scholar]
- 28.Gong H, Shen B, Flevaris P, Chow C, Lam SC, Voyno-Yasenetskaya TA, Kozasa T, Du X. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science. 2010;327:340–343. doi: 10.1126/science.1174779. doi: 10.1126/science.1174779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flevaris P, Stojanovic A, Gong H, Chishti A, Welch E, Du X. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007;179:553–565. doi: 10.1083/jcb.200703185. doi: 10.1083/jcb.200703185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Witt SM, Swieringa F, Cavill R, et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat Commun. 2014;5:4257. doi: 10.1038/ncomms5257. doi: 10.1038/ncomms5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.French SL, Arthur JF, Lee H, Nesbitt WS, Andrews RK, Gardiner EE, Hamilton JR. Inhibition of protease-activated receptor 4 impairs platelet procoagulant activity during thrombus formation in human blood. J Thromb Haemost. 2016;14:1642–1654. doi: 10.1111/jth.13293. doi: 10.1111/jth.13293. [DOI] [PubMed] [Google Scholar]
- 32.Gurbel PA, Bliden KP, Saucedo JF, Suarez TA, DiChiara J, Antonino MJ, Mahla E, Singla A, Herzog WR, Bassi AK, Hennebry TA, Gesheff TB, Tantry US. Bivalirudin and clopidogrel with and without eptifibatide for elective stenting: effects on platelet function, thrombelastographic indexes, and their relation to periprocedural infarction results of the CLEAR PLATELETS-2 (Clopidogrel with Eptifibatide to Arrest the Reactivity of Platelets) study. J Am Coll Cardiol. 2009;53:648–657. doi: 10.1016/j.jacc.2008.10.045. doi: 10.1016/j.jacc.2008.10.045. [DOI] [PubMed] [Google Scholar]
- 33.Hollenberg MD, Mihara K, Polley D, Suen JY, Han A, Fairlie DP, Ramachandran R. Biased signalling and proteinase-activated receptors (PARs): targeting inflammatory disease. Br J Pharmacol. 2014;171:1180–1194. doi: 10.1111/bph.12544. doi: 10.1111/bph.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]