

Supplemental Digital Content is available in the text.
Keywords: hypoxia, inflammation, ischemia, myocardial infarction, peripheral arterial disease
Abstract
Objective—
Reduced blood flow and tissue oxygen tension conditions result from thrombotic and vascular diseases such as myocardial infarction, stroke, and peripheral vascular disease. It is largely assumed that while platelet activation is increased by an acute vascular event, chronic vascular inflammation, and ischemia, the platelet activation pathways and responses are not themselves changed by the disease process. We, therefore, sought to determine whether the platelet phenotype is altered by hypoxic and ischemic conditions.
Approach and Results—
In a cohort of patients with metabolic and peripheral artery disease, platelet activity was enhanced, and inhibition with oral antiplatelet agents was impaired compared with platelets from control subjects, suggesting a difference in platelet phenotype caused by the disease. Isolated murine and human platelets exposed to reduced oxygen (hypoxia chamber, 5% O2) had increased expression of some proteins that augment platelet activation compared with platelets in normoxic conditions (21% O2). Using a murine model of critical limb ischemia, platelet activity was increased even 2 weeks postsurgery compared with sham surgery mice. This effect was partly inhibited in platelet-specific ERK5 (extracellular regulated protein kinase 5) knockout mice.
Conclusions—
These findings suggest that ischemic disease changes the platelet phenotype and alters platelet agonist responses because of changes in the expression of signal transduction pathway proteins. Platelet phenotype and function should, therefore, be better characterized in ischemic and hypoxic diseases to understand the benefits and limitations of antiplatelet therapy.
Platelets are activated in ischemic diseases such as myocardial infarction (MI), stroke, and peripheral artery disease (PAD).1–4 Antiplatelet agents, including aspirin and clopidogrel, are recommended as part of the disease treatment. The expected antithrombotic benefits of antiplatelet agents are not observed in all patients5–7; some develop unexpected thrombosis,8 whereas others have bleeding complications.9,10 Explanations for such treatment failure includes gene polymorphisms in enzymes responsible for antiplatelet drug metabolism or in their receptors, which was reported for the P2Y12 receptor antagonist clopidogrel.11–14 The rationale behind testing for differences in metabolism of antiplatelet agents on an individual basis is that the drug type and dose may be personalized, providing a more favorable clinical outcome.15 However, reports have indicated that a personalized genetic approach to antiplatelet therapy failed to alter clinical outcomes or the progression of ischemic disease.16,17
Preclinical platelet inhibitor studies typically use platelets isolated from normal volunteers.18 There are risks associated with oversimplifying preclinical platelet studies and extrapolating findings using healthy donor platelets to studies with platelets from a diseased population.19 A prudent research approach may include determining whether the signaling processes in platelets from diseased patients are similar to healthy persons. If platelets from a diseased population have different agonist signaling properties, the design and implementation of antiplatelet agents should be tailored to adjust for these changes. For example, Jurk et al20 demonstrated that circulating platelets in patients after stroke are refractory to ex vivo stimulation, engendering an exhausted platelet phenotype, suggesting that central ischemic vascular disease may lead to the development dysfunctional platelets.
Using a murine MI model, we recently demonstrated that the circulating platelet phenotype is changed in the postinfarct environment, with a similar observation noted in patients in the peri-MI period.1,21 We now report that platelet protein expression is altered both in vitro by exposure to hypoxia and in vivo in ischemic disease, demonstrating that the altered platelet phenotype is regulated at least in part at the platelet level. Using both a murine model of critical limb ischemia and platelets from patients with metabolic disease and advanced PAD, we have revealed that platelet postreceptor signaling is altered. These findings suggest a fundamental platelet phenotype switch in ischemic disease not accounted for by current therapeutics.
Materials and Methods
Data available on request from the authors.
Antibodies and Reagents
A full list of all reagents and antibodies used are listed in the Expanded Methods in the online-only Data Supplement.
Subjects
Healthy volunteers, patients with diabetes mellitus as indicated by blood hemoglobin A1c concentration >6.5% with or without peripheral arterial disease determined objectively by the ankle brachial index were enrolled in this study. Patients with PAD were consented on the day of revascularization either by surgical bypass or by percutaneous intervention for critical limb ischemia. Venous blood was used to isolated platelet-rich plasma (PRP). Washed platelets were used for platelet stimulation studies using agonists against the P2Y12 receptor (2-methyl-ADP), PAR1 (protease-activated receptor-1; TRAP6 [thrombin receptor–activating peptide-6]), or the thromboxane receptor (U46619), with flow cytometry used to detected activated platelets by surface p-selectin expression as described previously by our group.21 This study had the approval of the Research Subjects Review Board of the University of Rochester.
Mouse Colony
All animal protocols were approved by the University Committee on Animal Resources. Eight-week-old male wild-type (WT) C57BL6/J were used in this study unless indicated. To interrogate the role of platelet ERK5 (extracellular regulated protein kinase 5) in some studies, we used ERK5flox/PF4cre(+; platelet-specific ERK5−/−) mice on a C57BL/6 background previously validated and shown to be deficient only in platelet ERK5. These platelet-specific ERK5−/− mice were matched with ERK5flox/flox mice as a control.1,22
Critical Limb Ischemia Model
Mice were anesthetized with 3% isoflurane. A skin incision was made with leg ligations made proximally and distally to the femoris profunda muscle, with 6.0 suture followed by left femoral artery dissection. The skin was closed using 4.0 coated vicryl in a subcuticular fashion. Mice were allowed to recover and returned to housing for up to 28 days. At various time points over this 28 days, mice will also be imaged under isoflurane using a laser Doppler imaging system. Expanded Methods in the online-only Data Supplement.
Mouse Hemostasis and Thrombosis Models
The tail bleeding method was used to assess the time to hemostasis. The ferric chloride-induced platelet activation and mesenteric arterial occlusion model was used to assess thrombosis. Both are described by us previously.1
Mouse Pneumonectomy Model
We performed a left pneumonectomy as described23 to create another model in which the mouse was hypoxic. Expanded Methods in the online-only Data Supplement.
Quantification of Blood Vessels
A volume of 2×250 μL ice cold Matrigel which contained all the necessary growth factors to promote angiogenesis was drawn up into prechilled 1 mL syringes and injected into the ventral surface of the mouse subcutaneously around the hindlimb area using a 27 g needle. After 7 days, the mouse was euthanized, and the solidified matrix was removed at which point blood vessels were apparent, and so hemoglobin was extracted and quantified according to the instructions using a hemoglobin colorimetric assay (Cayman Chemicals). The other injected solidified matrix was removed and fixed with 10% formalin, then sectioned for H and E staining.
Human Platelet Isolation
For human platelet function studies and for biochemical analysis, venous blood was collected into citrate plasma tubes and mixed, then isolated according to our protocol published previously.21
Mouse Platelet Isolation
Mouse platelets were collected by 2 to 3 drops of retro-orbital blood into heparinized Tyrode as described by us previously.1 Expanded Methods in the online-only Data Supplement.
Biochemistry and Protein Studies
Cell lysis and cell protein extraction, SDS PAGE, and Western blotting were conducted using buffers and techniques as described previously.1 Expanded Methods in the online-only Data Supplement.
Platelet Proteomics
Whole blood was collected into citrate plasma tubes and thoroughly mixed. The sample was centrifuged at 1100 rpm for 15 minutes using a bench top centrifuge. The supernatant was then added in a 1:1 (vol/vol) mix of supernatant/Tyrode solution with final concentration 10 μmol\L prostaglandin I2 (PG I2, Cayman Chemical) and centrifuged at 2600 rpm for 5 minutes using a bench top centrifuge. In an attempt to reduce further contaminating plasma proteins, the supernatant was discarded, and the washed platelet pellet was carefully resuspended in 1 mL fresh Tyrode solution with 10 μmol\L prostaglandin I2 and centrifuged at 2600 rpm for 5 minutes using a bench top centrifuge. The final platelet pellet was then carefully resuspended in 1 mL fresh Tyrode solution with 10 μmol\L prostaglandin I2 and centrifuged at 2600 rpm for 5 minutes using a bench top centrifuge one final time. We used CD45 and CD41 antibodies to identify leukocytes and platelets, respectively, and then gated and quantified each individual population as a proportion of all cells, not only those double positive cells. This second quality control study revealed even less leukocyte contaminants of PRP (0.12%). The resulting platelet pellet was then used for platelet protein extraction by reducing with 10 mmol/L tris (2-carboxyethyl) phosphine for 1 hour at 37°C and subsequently alkylated with 12 mmol/L iodoacetamide for 1 hour at room temperature in the dark. Samples were then diluted 1:4 with deionized water and digested with sequencing grade modified trypsin at 1:50 enzyme-to-protein ratio. After 12 hours at 37°C to promote digestion, another aliquot of the same amount of trypsin was added to the samples and further incubated at 37°C overnight. The digested samples were then acidified, cleaned up (SCX and C18) and dried as described above. An LC-MS/MS (liquid chromatography tandem mass spectrometry)-based method for quantitative proteomics using the iTRAQ (isobaric tags for relative and absolute quantitation) system reporter ion intensities as we have used previously to study the human platelet proteome and described elsewhere.24,25
Statistical Analyses
Clinical variables that are dichotomous are presented as frequencies and those that are continuous as mean with SEM unless otherwise stated. The distribution of each data set was interrogated for normality using the Shapiro–Wilk test before comparison between groups. For non-Gaussian distributed data between 2 comparative groups, data are graphically represented as median and the Mann–Whitney U test was used to assess for a difference between groups. For 3 or more groups comparisons, the Kruskal–Wallis test followed by Dunn post-test was used. For Gaussian-distributed data between 2 comparative groups, the t test was used to assess for a difference between groups. For 3 or more groups, 1-way ANOVA then the Bonferroni multiple comparisons test was used. Significance was accepted as a P value <0.05. All data were analyzed with GraphPad Prism 7 (GraphPad Software, Inc, La Jolla, CA).
Results
To test whether the platelet phenotype is altered in human vascular and metabolic disease, we isolated platelets from patients with several cardiovascular comorbidities including PAD, diabetes mellitus, and hypertension (referred to as patients with the vascular and metabolic disease). We compared platelet function in 30 individuals: either patients or relatively healthy control subjects (Figure I in the online-only Data Supplement). We stimulated isolated platelets from healthy control subjects, healthy control subjects taking 81 mg aspirin daily, patients with vascular and metabolic comorbidities with PAD, and from patients with vascular and metabolic comorbidities without PAD (all patients were taking at least 1 antiplatelet agent). Control subjects taking 81 mg aspirin daily all showed suppression of platelet activity after surface receptor agonist stimulation compared with control subjects without aspirin therapy. However, platelet activation in response to PAR1 and thromboxane receptor stimulation (TRAP6 and U46619, respectively) was not inhibited in patients with vascular and metabolic comorbidities without PAD as we anticipated and, in fact, platelet function was enhanced in response to P2Y12 receptor stimulation (2-me-ADP) in spite of taking aspirin and clopidogrel. Platelet function in patients with vascular and metabolic comorbidities with PAD was not inhibited by antiplatelet agents in response to receptor agonists as we anticipated compared with control volunteers taking 81 mg aspirin daily (Figure 1A through 1C). These data indicate that platelets from patients with the metabolic and vascular disease have altered agonist sensitivity and apparent resistance to inhibition by antiplatelet agents compared with platelets from healthy subjects.
Figure 1.
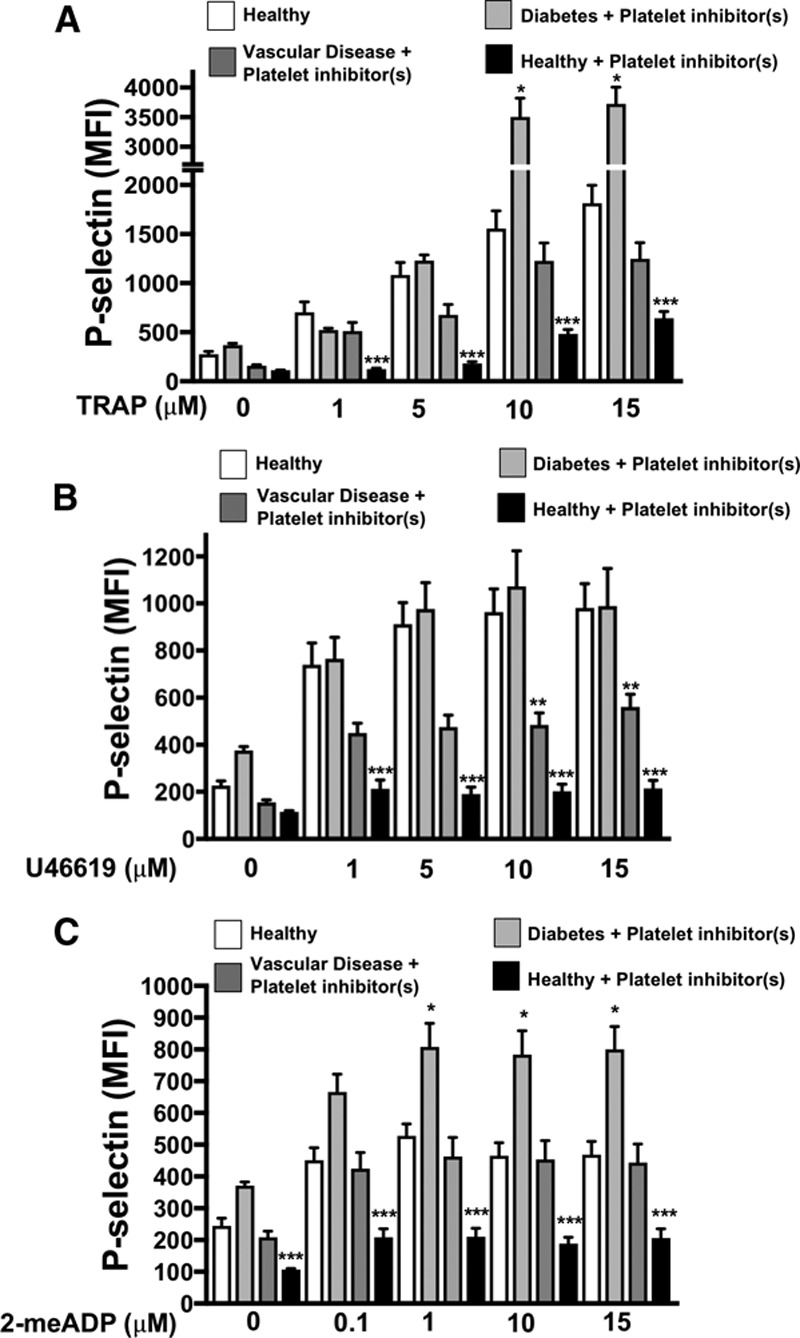
Patients with metabolic and vascular disease have dysregulated platelets. A–C, Platelets from healthy patients (4) and healthy controls taking daily 81 mg aspirin (4) were compared with patients with metabolic and vascular comorbidities including diabetes mellitus and PAD (peripheral artery disease) taking platelet inhibitors (8), and patients with diabetes mellitus without PAD taking both platelet aspirin and clopidogrel (4). Platelets were stimulated with (A) a PAR1 (protease-activated receptor-1) agonist TRAP (thrombin receptor–activating peptide), (B) a thromboxane receptor agonist U46619, or (C) a P2Y12 agonist 2-me-ADP for 15 min and activation was assessed by FACS (P-selectin expression, mean±SEM, *P<0.05 healthy vs diabetes mellitus+platelet inhibitors. **P<0.05 healthy vs vascular disease+platelet inhibitor(s). ***P<0.05 healthy vs healthy+aspirin, all by 1-way ANOVA. MFI indicates mean fluorescence intensity.
To demonstrate that these observations may in part be because of changes in the platelets themselves, platelet proteomic profiles were assessed by liquid chromatography/tandem mass spectrometry. Protein expression data in patients with cardiovascular comorbidities, including PAD, was grouped by function, showing platelet protein expression differences in processes involved in inflammation, RNA processing, protein folding and trafficking, vesicular transport, protease activity, and platelet adhesion (Figures II and III in the online-only Data Supplement). Less than one half percent leukocyte contamination was seen in PRP isolates (Figure IV in the online-only Data Supplement). These data indicate that changes in the platelet phenotype may contribute to antiplatelet drug resistance in patients with vascular and metabolic diseases.
Our prior study demonstrated that platelet protein expression is altered by ischemic disease.1 We, therefore, considered whether changes in the platelet proteome may in part be because of low tissue oxygen conditions and reactive oxygen species generated in those conditions associated with ischemic vascular diseases. Platelets were isolated from healthy subjects and incubated under normoxic (21% O2) or hypoxic (5% O2) conditions for 2 hours before stimulation with TRAP6, U46619, or 2-me-ADP. Platelet activation in response to agonist stimulation was enhanced after 2 hours in a hypoxic environment using both surface P-selectin and fibrinogen binding (activated GPIIb/IIIa) as platelet activation markers (Figure 2A through 2D), 30-minute time point showed little change (Figures V and VI in the online-only Data Supplement). We also show that activation of human platelet ERK5, which is a redox sensor, seems to be sustained after 2 hours in a hypoxic environment in vitro (Figure 2E). These data imply that hypoxia may prime platelets toward an activated state.
Figure 2.
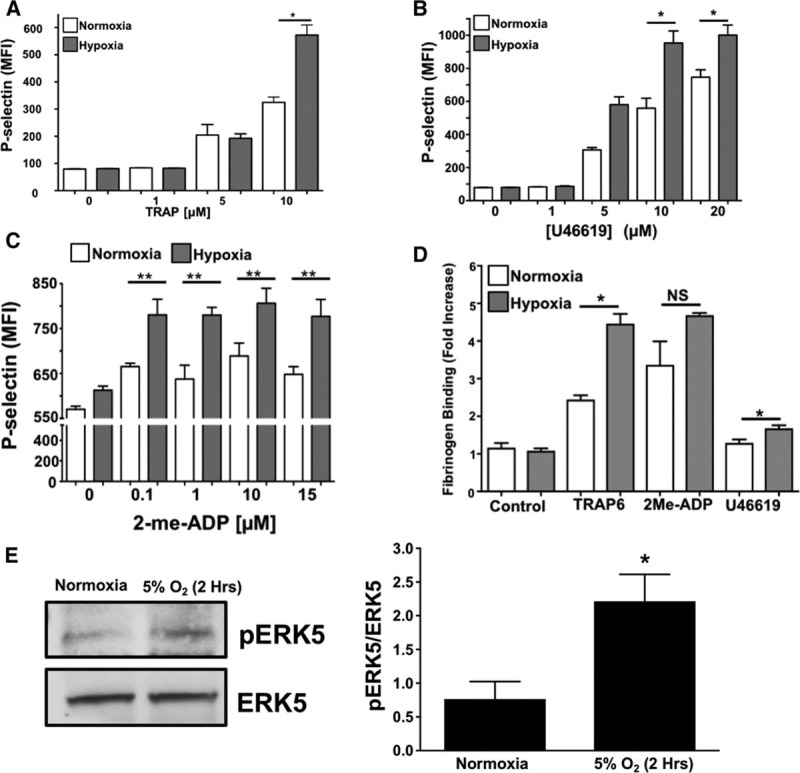
Acute hypoxia augments platelet activation. Human platelets were isolated and activation was assessed after 2 h of in vitro normoxia (21% O2) or hypoxia (5% O2). Platelets were stimulated with (A) TRAP6 (thrombin receptor–activating peptide-6), (B) U46619, or (C) 2-me-ADP for 15 min and P-selectin expression was determined by FACS (mean±SEM, n=4). *P<0.05 or **P<0.01 normoxia vs hypoxia at each agonist concentration by 1-way ANOVA. D, Human platelets were isolated and activation was assessed after 2 h of normoxia or hypoxia. Platelets were then stimulated with TRAP6 (20 µmol/L), U46619 (20 µmol/L), or 2-me-ADP (10 µmol/L) for 15 min and activation assessed by FACS for GPIIb/IIIA activation (FITC-Fibrinogen binding, mean±SEM, n=3, *P<0.01 by 1-way ANOVA, NS=not significant). E, Human platelet ERK5 (extracellular regulated protein kinase 5) is activated in normoxia or hypoxia (mean pERK5/ERK5±SEM, *P=0.02 by t test, n=5 in each group).
Our prior study using a mouse MI model demonstrated altered platelet protein expression in the post-MI environment, including ERK5, P70S6K, and RAC1.1 To assess whether platelet activation by agonists or hypoxia/ischemia alters the platelet phenotype independent of the megakaryocyte, we isolated mouse platelets and either agonist-stimulated platelets or incubated platelets in normoxic (21% O2) or reduced (5% O2) oxygen tension environments in vitro. P70S6K expression was increased by thrombin, though other agonists such as U46619, and ADP did not demonstrate the same effect (Figure 3A and 3B). Hypoxia alone, however, significantly increased the expression of ERK5, P70S6K, and RAC1 in a time-dependent manner in mouse platelets (Figure 3C). In vitro hypoxia also augmented thrombin-induced murine platelet activation (Figure 3D). Mice with either sham operation or unilateral pneumonectomy develop a chronic hypoxic state after 3 weeks as indicated by increased blood hemoglobin concentration with a coincident increased in activation of platelet redox sensor ERK5 in vivo (Figure 3E), and in vivo hypoxia enhanced thrombosis in a mouse mesenteric injury model (Figure 3F), and shortened tail bleeding time (Figure 3G). These data demonstrate that platelet function and protein expression are altered in hypoxia in a manner that may in part be because of platelet protein expression changes or changes in platelet ERK5 activation.
Figure 3.
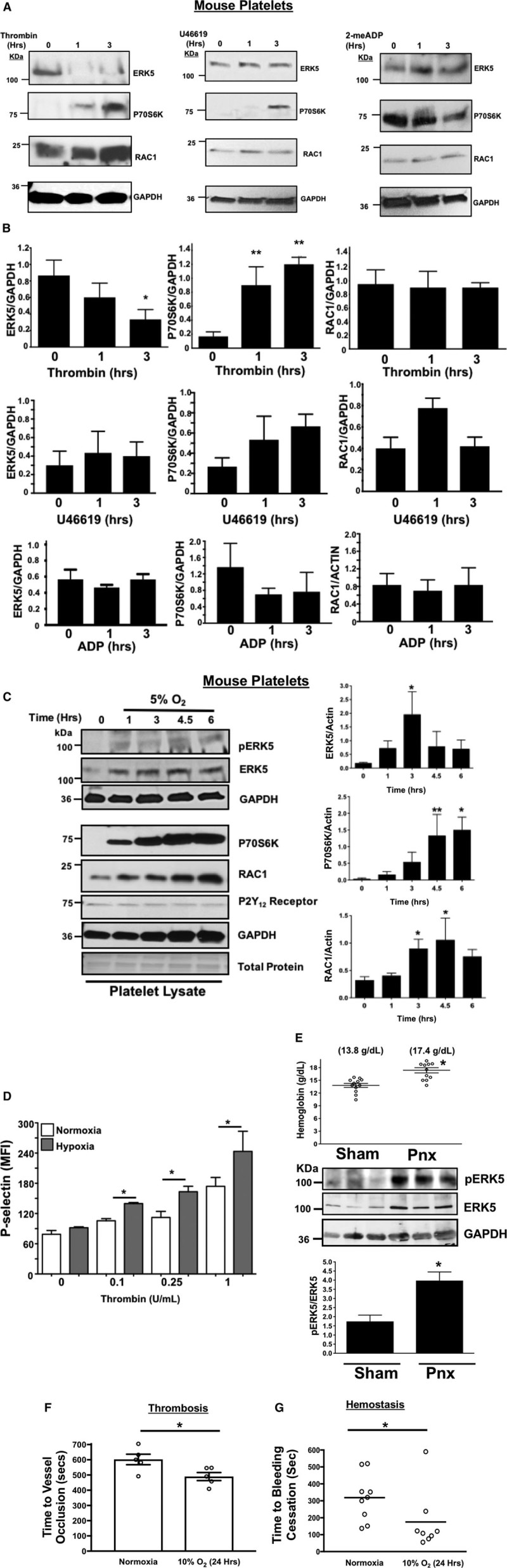
Murine platelet protein expression changes in vitro after agonist stimulation. A and B, Platelets isolated from mice were stimulated with 0.2 U/mL thrombin, 10 µmol/L U46619, or 10 µmol/L 2-me-ADP for 1 to 3 h. Protein expression was determined by (A) immunoblot (IB) and (B) quantified by densitometry (mean±SEM, *P=0.12 vs 0 for ERK5 (thrombin) and **P<0.05 vs 0 for P70S6K (thrombin) by 1-way ANOVA, n=3). Platelets isolated from mice were incubated for 0 to 6 h under hypoxic conditions (5% O2) and platelet protein expression was assessed by IB and quantified by densitometry (mean±SEM, *P<0.05 or **P=0.058 vs 0 by 1-way ANOVA, n=4–6). D, Platelets were isolated from wild-type (WT) mice and activation was assessed after 6 h of normoxia or hypoxia and stimulated with thrombin for 15 min. Activation was assessed by P-selectin expression (mean±SEM, n=4, *P<0.05 by 1-way ANOVA). E, Left pneumonectomy (Pnx) or sham surgery mice demonstrate hypoxia by a compensatory increase in circulating blood hemoglobin concentration, P=0.0002 by t test between groups without a change in platelet count (559±44 vs 604±43 between groups, P=0.48 by t test, n=12 in each group). Isolated platelets show platelet ERK5 (extracellular regulated protein kinase 5) activation (mean pERK5/ERK5±SEM, *P=0.009 by t test, n=4 in each group). Mesenteric artery thrombosis was assessed in mice after living for 24 h at ambient oxygen or in hypoxic conditions (10% O2, mean time to vessel occlusion±SEM, n=5 in each group, *P=0.026 by 1-way ANOVA for normoxia vs 10% O2). G, Tail bleeding times as an index of hemostasis were calculated as the time in seconds for bleeding to stop after surgical amputation of the tail tip, median value, n=8 to 9, *P=0.021 by Mann–Whitney U test for normoxia vs 10% O2.
Receptor agonist and hypoxia-induced changes in platelet protein expression were also determined using human platelets. We observed that human platelets also exhibited a similar though not identical agonist-specific protein expression change (Figure 4A; Figures VII and VIII in the online-only Data Supplement), whereas hypoxia-induced less change in human platelet proteins compared with murine platelets (Figures IX and X in the online-only Data Supplement). These data are consistent with inhibitors of P70S6K and RAC1 having little impact on human platelet posthypoxia activation (Figures XI and XII in the online-only Data Supplement), but redox-sensitive ERK5, when pharmacologically inhibited after prolonged in vitro hypoxia, demonstrated a significant attenuation of human platelet activation after hypoxia which was not as profoundly obvious in platelets in a normoxic condition (Figure XIII in the online-only Data Supplement; Figure 4B). Finally, human platelets show changes in agonist-induced activation in a hypoxic environment, possibly through additional mechanisms, including increased platelet surface receptor expression available for certain agonists after activation (Figure XIV in the online-only Data Supplement). These experiments demonstrate that platelets alter the expression of key signaling proteins independent of the bone marrow-derived precursor megakaryocyte, particularly in response to hypoxic/ischemic stress. However, the downstream mediators of increased platelet activation may be fundamentally different between murine and human platelets.
Figure 4.
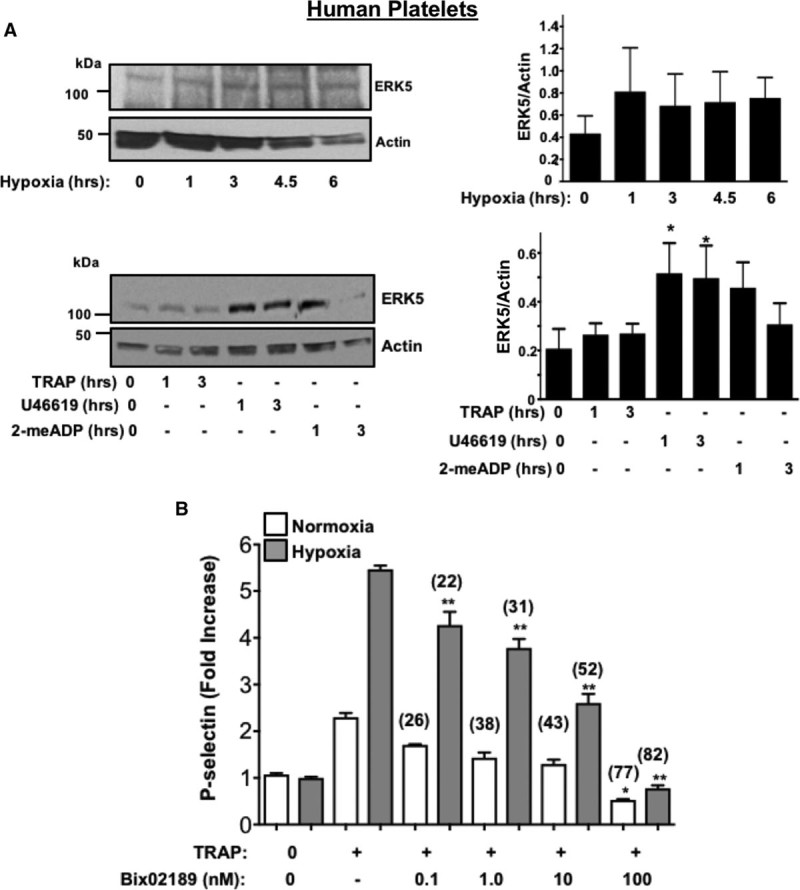
Human platelet protein expression changes in vitro after agonist stimulation. A, Human platelets were incubated in hypoxic conditions in vitro (5% O2, Top) or stimulated with TRAP (thrombin receptor–activating peptide-6; 10 µmol/L), U46619 (20 µmol/L), or 2-me-ADP (10 µmol/L). Platelet protein expression was assessed for ERK5 (extracellular regulated protein kinase 5), then quantified by densitometry and reported as mean±SEM *P<0.05 vs 0 by 1-way ANOVA, n=4. B, Human platelets were placed in normoxic or hypoxic conditions for 2 h in the presence or absence of an ERK5 inhibitor (Bix 02189, 0.1–100 nmol/L) for the last 30 min. Platelets were then stimulated with TRAP (10 µmol/L). An ERK5 inhibitor attenuated platelet activation more effectively in hypoxia (% inhibition with ERK5 inhibitor vs no ERK5 inhibitor indicated above each condition). *P=0.040 vs TRAP alone+normoxia or **P<0.0001 vs TRAP alone+hypoxia by 1-way ANOVA, n=4.
Platelet activation in response to some agonists in a hypoxic environment may be secondary to platelet activation of redox-sensitive protein kinases like ERK5.1,22,26 Murine and human platelets both increase endogenous reactive oxygen species generation in a hypoxic environment (Figure 5A) through the basal and maximal quantity of reactive oxygen species produced between species also differs. Furthermore, ERK5 activation (P-ERK5, but not total protein expression) was significantly greater in human platelets from patients with PAD (Figure 5B). ERK5 and other platelet-activating proteins including RAC1 as well as proteins well-known to promote ribosome biogenesis and support translation such as mTOR were all elevated in murine platelets in hindlimb ischemia (HLI) model (Figure 5D). This change in murine platelet protein expression coincided with dysregulated platelet activation which was not observed in sham-operated mice and coincident with lower extremity tissue remodeling and angiogenesis secondary to ischemic injury (Figure 5D; Figures XV through XVII in the online-only Data Supplement).
Figure 5.
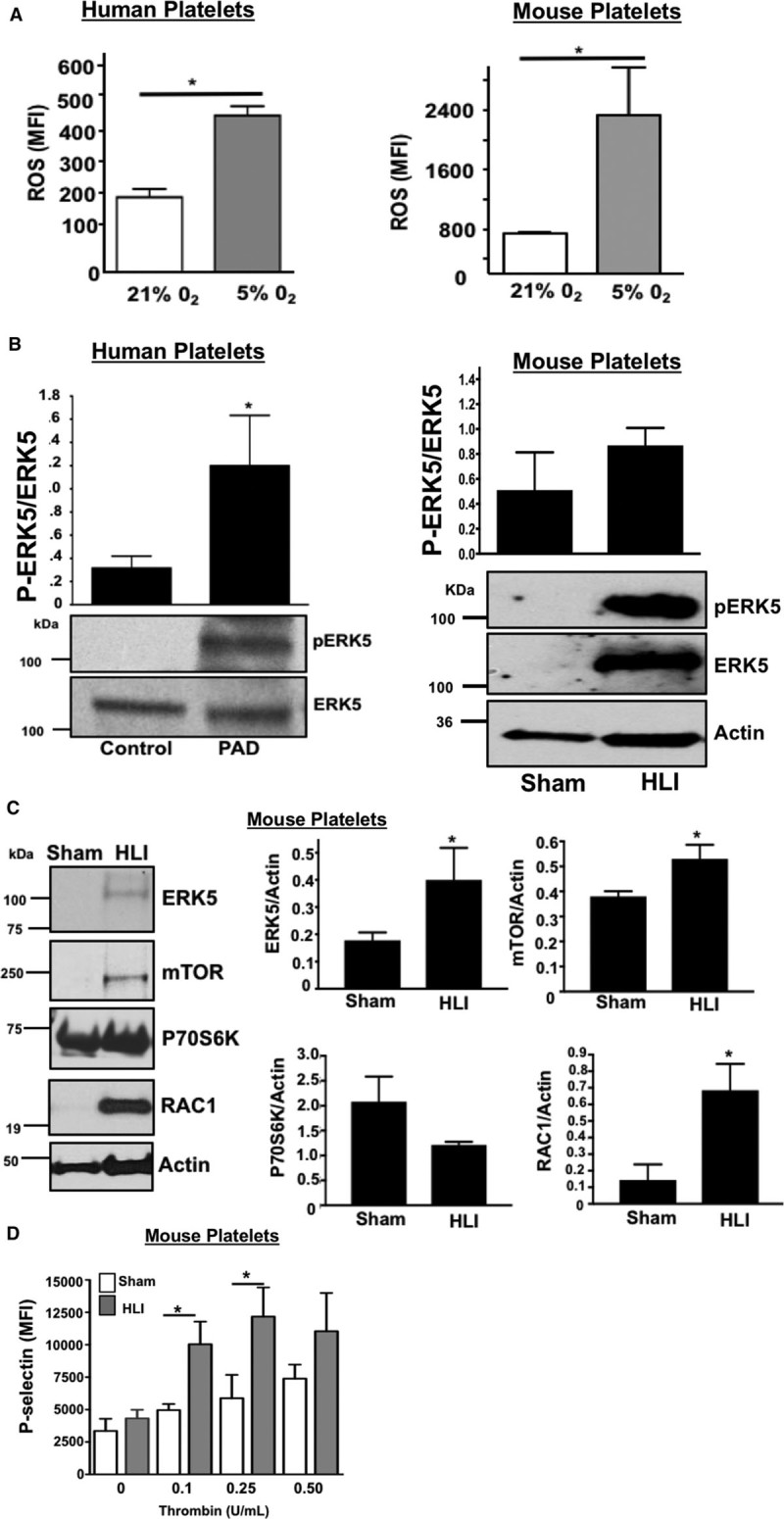
ERK5 (extracellular regulated protein kinase 5) promotes dysregulated platelet activity in critical limb ischemia. A, Platelets isolated from wild-type (WT) mice (left) or healthy humans (right) were incubated for 2 h under normoxic conditions (21% O2) or after hypoxia exposure (5% O2) and then loaded with DCFDA (2’,7’-dichlorodihydrofluorescein diacetate) to indicate reactive oxygen species (ROS) production, quantified by FACS analysis (mean±SD) *P<0.05 vs 21% O2 by t test, n=3 in each group. B, Platelets isolated from humans with peripheral artery disease (PAD) or from mice after 4 d of unilateral left leg femoral artery ligation (hindlimb ischemia [HLI]) or sham surgery were assessed for ERK5 activation using a phospho-specific antibody (p-ERK5). Actin was used as an additional loading control for ERK5 because ERK5 protein content was increased in mice with HLI. ERK5 activation was quantified by densitometry and reported as mean pERK5/ERK5±SEM. *P=0.025 for control vs PAD, N=3 to 4, and P=0.14 for sham vs HLI mice by t test, N=4. Platelets isolated from mice after 4 d of HLI or sham surgery were assessed for expression of proteins known to affect platelet activation. Protein expression was assessed by immunoblotting (IB), then quantified by densitometry and reported as mean±SEM *P=0.046 vs 0 (ERK5), P=0.034 vs 0 (mTOR) and P=0.022 vs 0 (RAC1) by t test, n=3 to 4 in each group. D, WT mice were subjected to HLI or sham surgery and platelets isolated 7 d later were stimulated with thrombin for 15 min and activation assessed by FACS (P-selectin expression, mean±SEM, n=4 in each group by 1-way ANOVA, *P<0.05 between groups).
To evaluate a potential functional role for platelet ERK5 in platelet a phenotype alteration associated with ischemia, we performed HLI on WT mice and platelet-specific ERK5−/− mice which was previously characterized and shows complete absence of ERK5 protein in isolated PRP which is obviously devoid of ERK5 contaminants from other cell types (Figure XVIII in the online-only Data Supplement). Platelets were then isolated from mice on days 3, 7, and 14 post-HLI to assess activation. Platelets from platelet-specific ERK5−/− mice had attenuated platelet activation at each of the time points compared with platelets from WT mice (Figure 6A through 6C). Thermal laser Doppler imaging of the ischemic limb was also performed weekly to assess for reconstitution of blood flow as is often seen in human patients with advanced PAD. Platelet ERK5−/− mice showed more rapid recovery of limb blood flow compared with WT mice (Figure 6D and 6E), with similar platelet counts throughout the ischemic period (Figure XIX in the online-only Data Supplement). To determine whether ERK5−/− mice have improved angiogenesis in general, an in vivo Matrigel assay was used to quantify blood vessel growth in vivo in WT and platelet ERK5−/− mice. Vascular content was surprisingly similar in WT and platelet ERK5−/− mice (extracted hemoglobin concentration 1.53±0.48 g/dL versus 1.18±0.15 g/dL) implying that the more rapid reconstitution of hindlimb blood flow in platelet ERK5−/− mice is because of mechanisms other than enhanced angiogenesis, and potentially may include alterations in microvascular thrombosis. Together these data demonstrate that ischemic disease leads to a platelet phenotype that is more sensitive to agonist stimulation, and activation of platelet ERK5 may have a central role in this response (Figure 7).
Figure 6.
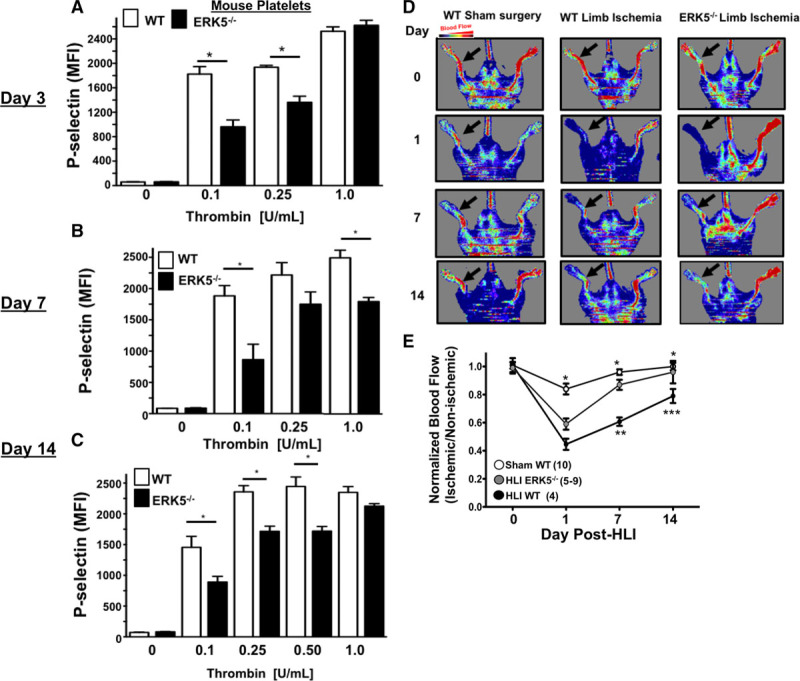
Platelet ERK5 (extracellular regulated protein kinase 5) inhibition improved blood flow in critical limb ischemia. A–C, Wild-type (WT) or ERK5−/− platelets from mice (A) 3, (B) 7, or (C) 14 d after hindlimb ischemia (HLI) were isolated and stimulated with thrombin for 15 min and activation assessed by FACS. WT platelets had more post-HLI activation compared with ERK5−/− (P-selectin expression, mean±SEM, n=4, *P<0.01 between groups by 1-way ANOVA). D and E, Thermal Doppler color imaging showed more rapid return of blood flow in ERK5−/− mouse limbs. D, Representative images, (E) quantification (mean ratio in the ischemic:nonischemic limb±SEM *P<0.001 Sham WT vs WT HLI, **P=0.013 WT HLI vs ERK5 −/− HLI, ***P=0.003 WT HLI vs ERK5 −/− HLI all by 1-way ANOVA. The group population size is indicated in parentheses.
Figure 7.
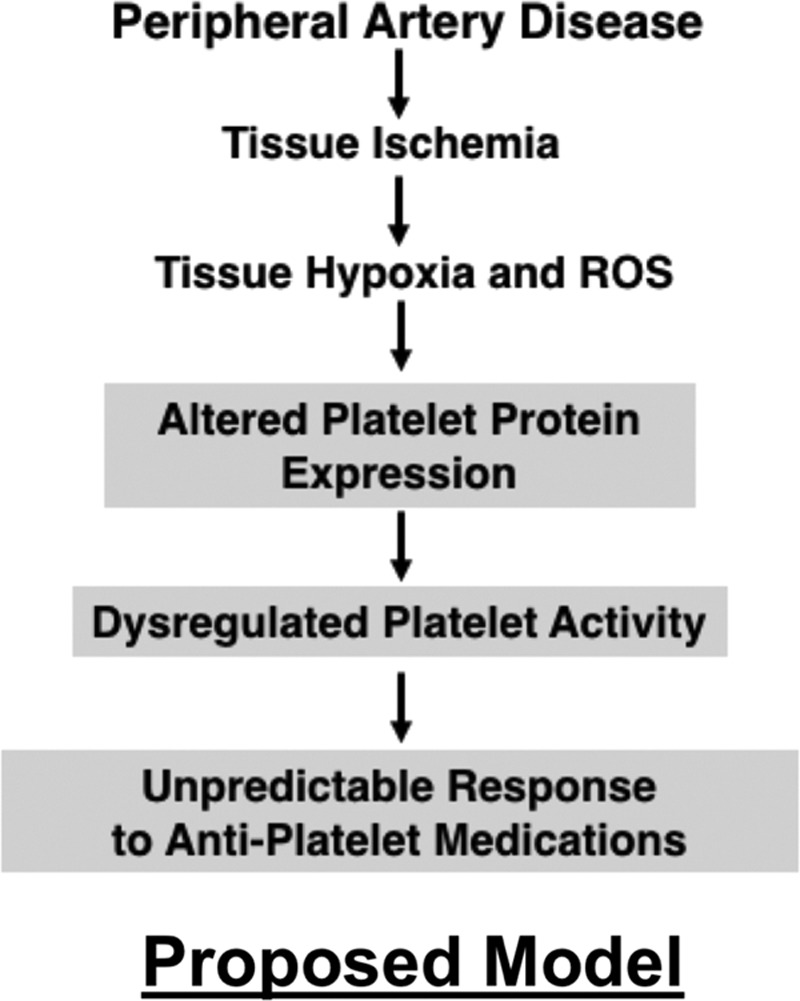
Proposed model. ROS indicates reactive oxygen species.
Discussion
These data demonstrate that in both humans and mouse models, metabolic and vascular disease alters the platelet phenotype. Human diseases and extreme experimental conditions of ischemia and hypoxia revealed differences in platelet surface receptor expression, agonist sensitivity, postreceptor signal transduction, and proteomic profiles which could alter the response of the platelet to environmental and pharmacological stimuli. This may provide mechanistic insight into the unpredictable patient responses to antiplatelet agents in hypoxic and ischemic diseases.21,27,28 The expectation that the platelet phenotype in a diseased state closely resembles healthy conditions may be incorrect. Preclinical studies evaluating antiplatelet agents, therefore, ought to include both healthy donors and donors with the metabolic and vascular disease because platelet antagonists presently available do not account for an evolving, disease-dependent platelet phenotype. The platelet phenotypic switch observed in diseased conditions may in part explain unpredictable platelet responses previously ascribed to changes in antiplatelet drug metabolism or platelet receptor variants.17,29,30 Resistance to antiplatelet therapeutic agents has been described in diabetics, in MI, and in patients with PAD.31 Explanations for such treatment failure may include metabolic comorbidities which alter the inflammatory environment, the metabolism of antiplatelet agents, and interactions of antiplatelet agents with other drugs.32–36 Comparing differences in the platelet phenotype between clinical groups is challenging given the difficulty in exactly matching control human populations in a complex disease group such as PAD, where the clinical pathology leading to the vascular injury is multifactorial.
A few studies showed that platelets from diseased populations might have altered surface receptor expression, implying a change in the mature platelet proteome.37–39 Our data offer some mechanistic explanations for these observations because we demonstrate an adaptive platelet phenotype in models of ischemia. This includes changes in platelet receptor agonist sensitivity and the expression and activation of postreceptor signal transduction proteins. Although we, in fact, observe some important differences in human compared with murine responses to hypoxia, platelet ERK5 seems to be a common mediator of dysregulated platelet activation in both species.
In as little as a few hours, we found changes in the expression of platelet proteins in vitro in a hypoxic environment or after a few days in vivo after limb ischemia that coincide with enhanced sensitivity to multiple platelet surface receptor agonists. We also show that the platelet proteome in patients with advanced vascular disease is not the same as in relatively healthy subjects, with the former demonstrating more signaling proteins involved in protein synthesis, inflammation, and thrombosis (Figures II and III in the online-only Data Supplement) These data extend prior observations in experimental models of extreme hypoxia in vitro and in models of venous thrombosis and sickle cell disease which all show increased platelet activation.40–42 The change in platelet function observed in just a few hours in vitro may be sufficient to tip the balance of platelet protein synthesis and degradation toward an activated phenotype.43,44 Previous studies indicate that P70S6K and RAC1 are involved in platelet cytoskeletal rearrangement and activation.45–47 We previously showed that platelet ERK5 is a regulator of protein stability and platelet function in the inflammatory post-MI environment and that platelet-specific ERK5−/− mice have attenuated platelet activation post-MI with reduced expression of P70S6K and RAC1.1 Our current study supports these prior observations by demonstrating markedly increased murine platelet P70S6K protein expression after in vitro hypoxia, independent of the megakaryocyte. Ribosomal protein S6 promotes protein translation efficiency may be especially important in ischemic disease.48,49 There is also the tantalizing possibility that platelet mRNA stability is markedly altered in different diseases with consequences on the final platelet translatome and subsequent proteome. It is tempting to speculate P70S6K is an ERK5 downstream mediator of dysregulated platelet activity in murine platelets under hypoxic conditions. An important observation in our investigation is that human and murine platelets, although similar in many ways, do in fact show clear differences in their responses to environmental cues that drive postreceptor signaling pathways and translation efficiency in vitro. These observations serve as a gentle reminder to investigators that experimental models, even when conducted rigorously, sometimes lack important features of human pathophysiology, which limits their ability to reveal a therapeutic solution for diseases.
Patients with vascular diseases such as PAD have more on-treatment thrombotic events compared with other thrombotic diseases.50,51 A recent report indicates diabetic platelets have dysregulated P2Y12 receptor signaling which was independently replicated in the present study of isolated platelets from humans with metabolic and vascular comorbidities including diabetes mellitus and advanced PAD.52 PAD is common in diabetic patients, and two-thirds of the patients in our study had diabetes mellitus. In support of other studies, despite taking daily platelet inhibitors, platelets from patients with PAD could be activated through several platelet receptor signaling pathways, and especially through the P2Y12 receptor. In the analogous murine HLI model, we show that platelets are markedly more activated by agonists compared with sham-operated animals and this phenotype is partly reversed in platelet ERK5−/− mice. We were quite surprised to observe that platelet ERK5−/− mice also showed enhanced blood flow in the weeks after HLI compared with WT mice. These findings imply ERK5 inhibition may decrease small vessel thrombotic burden in the early ischemic limb, promoting blood flow.
Our investigation has some limitations. Although we show that patients with the metabolic and vascular disease have a different platelet phenotype and seem to be somewhat resistant to antiplatelet medications, our control subjects had fewer comorbidities than those patients with diabetes mellitus and PAD, and there was an imbalance in the representation of sex among some groups. These present as potential confounding variables in data interpretation. A better and more direct correlation between these human and mouse data will require a larger cohort, ideally compared with control subjects with exactly matched comorbidities. However, these human disease-based data are only intended to highlight the basic conclusion of our study that the platelet phenotype is changed in vascular disease processes and that this may in part be because of changes in the platelet itself as well as in the vascular compartment.
In summary, the present study confirms that metabolic and ischemic stressors alter platelet signal transduction pathways and subsequent agonist responsiveness. This may in part be regulated at the level of the platelet itself, independent of the megakaryocyte. A concerted effort should be made to personalize antiplatelet therapy, not only with respect to race and sex but also to the thrombotic disease in question.
Acknowledgments
Drs Cameron and Morrell performed experiments, analyzed data, and wrote article. S.K. Ture, R.A. Schmidt, A. Mohan, D. Pariser, Dr Shah, Dr Chen, Dr Zhang, D.J. Field, Dr Modjeski performed experiments and analyzed data. Drs Mix, Stoner, and Toth were involved in study conception and assisted in acquiring platelets from human donors as well as analyzing clinical data.
Sources of Funding
National Institutes of Health (NIH) grants 5T32HL066988-1, NIH 2K08HL128856, and HL12020 to Dr Cameron; the Sable Heart Fund (Independent Order of Oddfellows) and the University of Rochester Department of Medicine Pilot Grant to Dr Cameron; NIH 5R01HL124018 and American Heart Association 13EIA14250023 to Dr Morrell.
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- ERK5
- extracellular regulated protein kinase 5
- HLI
- hindlimb ischemia
- MI
- myocardial infarction
- PAD
- peripheral artery disease
- WT
- wild-type
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.118.311186/-/DC1.
Highlights
Platelets regulate protein expression in response to environmental cues which changes their sensitivity to activation and threshold for inhibition by antiplatelet agents.
ERK5 (extracellular regulated protein kinase 5) is a mediator of ischemic platelet activation.
Platelet dysfunction is dependent on ERK5 in ischemic disease models.
References
- 1.Cameron SJ, Ture SK, Mickelsen D, Chakrabarti E, Modjeski KL, McNitt S, Seaberry M, Field DJ, Le NT, Abe J, Morrell CN. Platelet extracellular regulated protein kinase 5 is a redox switch and triggers maladaptive platelet responses and myocardial infarct expansion. Circulation. 2015;132:47–58. doi: 10.1161/CIRCULATIONAHA.115.015656. doi: 10.1161/CIRCULATIONAHA.115.015656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aronow H, Hiatt WR. The burden of peripheral artery disease and the role of antiplatelet therapy. Postgrad Med. 2009;121:123–135. doi: 10.3810/pgm.2009.07.2038. doi: 10.3810/pgm.2009.07.2038. [DOI] [PubMed] [Google Scholar]
- 3.Curzen N, Gurbel PA, Myat A, Bhatt DL, Redwood SR. What is the optimum adjunctive reperfusion strategy for primary percutaneous coronary intervention? Lancet. 2013;382:633–643. doi: 10.1016/S0140-6736(13)61453-1. doi: 10.1016/S0140-6736(13)61453-1. [DOI] [PubMed] [Google Scholar]
- 4.Cherian P, Hankey GJ, Eikelboom JW, Thom J, Baker RI, McQuillan A, Staton J, Yi Q. Endothelial and platelet activation in acute ischemic stroke and its etiological subtypes. Stroke. 2003;34:2132–2137. doi: 10.1161/01.STR.0000086466.32421.F4. doi: 10.1161/01.STR.0000086466.32421.F4. [DOI] [PubMed] [Google Scholar]
- 5.van Geffen JP, Kleinegris MC, Verdoold R, Baaten CC, Cosemans JM, Clemetson KJ, Ten Cate H, Roest M, de Laat B, Heemskerk JW. Normal platelet activation profile in patients with peripheral arterial disease on aspirin. Thromb Res. 2015;135:513–520. doi: 10.1016/j.thromres.2014.12.029. doi: 10.1016/j.thromres.2014.12.029. [DOI] [PubMed] [Google Scholar]
- 6.Petzold T, Thienel M, Konrad I, et al. Oral thrombin inhibitor aggravates platelet adhesion and aggregation during arterial thrombosis. Sci Transl Med. 2016;8:367ra168. doi: 10.1126/scitranslmed.aad6712. doi: 10.1126/scitranslmed.aad6712. [DOI] [PubMed] [Google Scholar]
- 7.Ault KA, Cannon CP, Mitchell J, McCahan J, Tracy RP, Novotny WF, Reimann JD, Braunwald E. Platelet activation in patients after an acute coronary syndrome: results from the TIMI-12 trial. Thrombolysis In Myocardial Infarction. J Am Coll Cardiol. 1999;33:634–639. doi: 10.1016/s0735-1097(98)00635-4. [DOI] [PubMed] [Google Scholar]
- 8.Park DW, Yun SC, Lee SW, Kim YH, Lee CW, Hong MK, Cheong SS, Kim JJ, Park SW, Park SJ. Stent thrombosis, clinical events, and influence of prolonged clopidogrel use after placement of drug-eluting stent data from an observational cohort study of drug-eluting versus bare-metal stents. JACC Cardiovasc Interv. 2008;1:494–503. doi: 10.1016/j.jcin.2008.06.011. doi: 10.1016/j.jcin.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 9.Carrabba N, Parodi G, Marcucci R, Valenti R, Gori AM, Migliorini A, Comito V, Bellandi B, Abbate R, Gensini GF, Antoniucci D. Bleeding events and maintenance dose of prasugrel: BLESS pilot study. Open Heart. 2016;3:e000460. doi: 10.1136/openhrt-2016-000460. doi: 10.1136/openhrt-2016-000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.CAPRIE Steering Committee. A randomised, blinded, trial of Clopidogrel Versus Aspirin in Patients at Risk of Ischaemic Events (CAPRIE). CAPRIE Steering Committee. Lancet. 1996;348:1329–1339. doi: 10.1016/s0140-6736(96)09457-3. doi: 10.1016/S0140-6736(96)09457-3. [DOI] [PubMed] [Google Scholar]
- 11.Tantry US, Jeong YH, Navarese EP, Kubica J, Gurbel PA. Influence of genetic polymorphisms on platelet function, response to antiplatelet drugs and clinical outcomes in patients with coronary artery disease. Expert Rev Cardiovasc Ther. 2013;11:447–462. doi: 10.1586/erc.13.20. doi: 10.1586/erc.13.20. [DOI] [PubMed] [Google Scholar]
- 12.Zoheir N, Abd Elhamid S, Abulata N, El Sobky M, Khafagy D, Mostafa A. P2Y12 receptor gene polymorphism and antiplatelet effect of clopidogrel in patients with coronary artery disease after coronary stenting. Blood Coagul Fibrinolysis. 2013;24:525–531. doi: 10.1097/MBC.0b013e32835e98bf. doi: 10.1097/MBC.0b013e32835e98bf. [DOI] [PubMed] [Google Scholar]
- 13.Ulehlova J, Slavik L, Kucerova J, Krcova V, Vaclavik J, Indrak K. Genetic polymorphisms of platelet receptors in patients with acute myocardial infarction and resistance to antiplatelet therapy. Genet Test Mol Biomarkers. 2014;18:599–604. doi: 10.1089/gtmb.2014.0077. doi: 10.1089/gtmb.2014.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bierend A, Rau T, Maas R, Schwedhelm E, Böger RH. P2Y12 polymorphisms and antiplatelet effects of aspirin in patients with coronary artery disease. Br J Clin Pharmacol. 2008;65:540–547. doi: 10.1111/j.1365-2125.2007.03044.x. doi: 10.1111/j.1365-2125.2007.03044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sibbing D, Braun S, Morath T, Mehilli J, Vogt W, Schömig A, Kastrati A, von Beckerath N. Platelet reactivity after clopidogrel treatment assessed with point-of-care analysis and early drug-eluting stent thrombosis. J Am Coll Cardiol. 2009;53:849–856. doi: 10.1016/j.jacc.2008.11.030. doi: 10.1016/j.jacc.2008.11.030. [DOI] [PubMed] [Google Scholar]
- 16.Doll JA, Neely ML, Roe MT, Armstrong PW, White HD, Prabhakaran D, Winters KJ, Duvvuru S, Sundseth SS, Jakubowski JA, Gurbel PA, Bhatt DL, Ohman EM, Fox KA TRILOGY ACS Investigators. Impact of CYP2C19 metabolizer status on patients with ACS treated with prasugrel versus clopidogrel. J Am Coll Cardiol. 2016;67:936–947. doi: 10.1016/j.jacc.2015.12.036. doi: 10.1016/j.jacc.2015.12.036. [DOI] [PubMed] [Google Scholar]
- 17.Siller-Matula JM, Trenk D, Schrör K, Gawaz M, Kristensen SD, Storey RF, Huber K EPA (European Platelet Academy) Response variability to P2Y12 receptor inhibitors: expectations and reality. JACC Cardiovasc Interv. 2013;6:1111–1128. doi: 10.1016/j.jcin.2013.06.011. doi: 10.1016/j.jcin.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 18.Nicholson NS, Panzer-Knodle SG, Haas NF, Taite BB, Szalony JA, Page JD, Feigen LP, Lansky DM, Salyers AK. Assessment of platelet function assays. Am Heart J. 1998;135(5 pt 2 su):S170–S178. doi: 10.1016/s0002-8703(98)70245-5. [DOI] [PubMed] [Google Scholar]
- 19.Schultze AE, Walker DB, Turk JR, Tarrant JM, Brooks MB, Pettit SD. Current practices in preclinical drug development: gaps in hemostasis testing to assess risk of thromboembolic injury. Toxicol Pathol. 2013;41:445–453. doi: 10.1177/0192623312460924. doi: 10.1177/0192623312460924. [DOI] [PubMed] [Google Scholar]
- 20.Jurk K, Jahn UR, Van Aken H, Schriek C, Droste DW, Ritter MA, Bernd Ringelstein E, Kehrel BE. Platelets in patients with acute ischemic stroke are exhausted and refractory to thrombin, due to cleavage of the seven-transmembrane thrombin receptor (PAR-1). Thromb Haemost. 2004;91:334–344. doi: 10.1160/TH03-01-0044. doi: 10.1160/TH03-01-0044. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt RA, Morrell CN, Ling FS, Simlote P, Fernandez G, Rich DQ, Adler D, Gervase J, Cameron SJ. The platelet phenotype in patients with ST-segment elevation myocardial infarction is different from non-ST-segment elevation myocardial infarction. Transl Res. 2018;195:1–12. doi: 10.1016/j.trsl.2017.11.006. doi: 10.1016/j.trsl.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang M, Cooley BC, Li W, Chen Y, Vasquez-Vivar J, Scoggins NO, Cameron SJ, Morrell CN, Silverstein RL. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood. 2017;129:2917–2927. doi: 10.1182/blood-2016-11-750133. doi: 10.1182/blood-2016-11-750133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakurai MK, Greene AK, Wilson J, Fauza D, Puder M. Pneumonectomy in the mouse: technique and perioperative management. J Invest Surg. 2005;18:201–205. doi: 10.1080/08941930591004485. doi: 10.1080/08941930591004485. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Liu T, Zhang Z, et al. CPTAC Investigators. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166:755–765. doi: 10.1016/j.cell.2016.05.069. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah P, Yang W, Sun S, Pasay J, Faraday N, Zhang H. Platelet glycoproteins associated with aspirin-treatment upon platelet activation. Proteomics. 2017;17 doi: 10.1002/pmic.201600199. doi: 10.1002/pmic.201600199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Z, Gao W, Fan X, Chen X, Mei H, Liu J, Luo X, Hu Y. Extracellular signal-regulated kinase 5 associates with casein kinase II to regulate GPIb-IX-mediated platelet activation via the PTEN/PI3K/Akt pathway. J Thromb Haemost. 2017;15:1679–1688. doi: 10.1111/jth.13755. [DOI] [PubMed] [Google Scholar]
- 27.Wisman PP, Teraa M, de Borst GJ, Verhaar MC, Roest M, Moll FL. Baseline platelet activation and reactivity in patients with critical limb ischemia. PLoS One. 2015;10:e0131356. doi: 10.1371/journal.pone.0131356. doi: 10.1371/journal.pone.0131356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajagopalan S, Mckay I, Ford I, Bachoo P, Greaves M, Brittenden J. Platelet activation increases with the severity of peripheral arterial disease: implications for clinical management. J Vasc Surg. 2007;46:485–490. doi: 10.1016/j.jvs.2007.05.039. doi: 10.1016/j.jvs.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 29.Edelstein LC, Simon LM, Lindsay CR, Kong X, Teruel-Montoya R, Tourdot BE, Chen ES, Ma L, Coughlin S, Nieman M, Holinstat M, Shaw CA, Bray PF. Common variants in the human platelet PAR4 thrombin receptor alter platelet function and differ by race. Blood. 2014;124:3450–3458. doi: 10.1182/blood-2014-04-572479. doi: 10.1182/blood-2014-04-572479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong X, Simon LM, Holinstat M, Shaw CA, Bray PF, Edelstein LC. Identification of a functional genetic variant driving racially dimorphic platelet gene expression of the thrombin receptor regulator, PCTP. Thromb Haemost. 2017;117:962–970. doi: 10.1160/TH16-09-0692. doi: 10.1160/TH16-09-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cleator JH, Duvernay MT, Holinstat M, Colowick NE, Hudson WJ, Song Y, Harrell FE, Hamm HE. Racial differences in resistance to P2Y12 receptor antagonists in type 2 diabetic subjects. J Pharmacol Exp Ther. 2014;351:33–43. doi: 10.1124/jpet.114.215616. doi: 10.1124/jpet.114.215616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagihara K, Kazui M, Kurihara A, Yoshiike M, Honda K, Okazaki O, Farid NA, Ikeda T. A possible mechanism for the differences in efficiency and variability of active metabolite formation from thienopyridine antiplatelet agents, prasugrel and clopidogrel. Drug Metab Dispos. 2009;37:2145–2152. doi: 10.1124/dmd.109.028498. doi: 10.1124/dmd.109.028498. [DOI] [PubMed] [Google Scholar]
- 33.Scott SA, Sangkuhl K, Gardner EE, Stein CM, Hulot JS, Johnson JA, Roden DM, Klein TE, Shuldiner AR Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450-2C19 (CYP2C19) genotype and clopidogrel therapy. Clin Pharmacol Ther. 2011;90:328–332. doi: 10.1038/clpt.2011.132. doi: 10.1038/clpt.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hulot JS, Bura A, Villard E, Azizi M, Remones V, Goyenvalle C, Aiach M, Lechat P, Gaussem P. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108:2244–2247. doi: 10.1182/blood-2006-04-013052. doi: 10.1182/blood-2006-04-013052. [DOI] [PubMed] [Google Scholar]
- 35.Bonello L, Tantry US, Marcucci R, et al. Working Group on High On-Treatment Platelet Reactivity. Consensus and future directions on the definition of high on-treatment platelet reactivity to adenosine diphosphate. J Am Coll Cardiol. 2010;56:919–933. doi: 10.1016/j.jacc.2010.04.047. doi: 10.1016/j.jacc.2010.04.047. [DOI] [PubMed] [Google Scholar]
- 36.Łabuz-Roszak B, Pierzchała K, Tyrpień K. Resistance to acetylsalicylic acid in patients with type 2 diabetes mellitus is associated with lipid disorders and history of current smoking. J Endocrinol Invest. 2014;37:331–338. doi: 10.1007/s40618-013-0012-2. doi: 10.1007/s40618-013-0012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lanza GA, Aurigemma C, Fattorossi A, Scambia G, Crea F. Changes in platelet receptor expression and leukocyte-platelet aggregate formation following exercise in cardiac syndrome X. J Thromb Haemost. 2006;4:1623–1625. doi: 10.1111/j.1538-7836.2006.02003.x. doi: 10.1111/j.1538-7836.2006.02003.x. [DOI] [PubMed] [Google Scholar]
- 38.Floyd CN, Goodman T, Becker S, Chen N, Mustafa A, Schofield E, Campbell J, Ward M, Sharma P, Ferro A. Increased platelet expression of glycoprotein IIIa following aspirin treatment in aspirin-resistant but not aspirin-sensitive subjects. Br J Clin Pharmacol. 2014;78:320–328. doi: 10.1111/bcp.12335. doi: 10.1111/bcp.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaywin P, McDonough M, Insel PA, Shattil SJ. Platelet function in essential thrombocythemia. Decreased epinephrine responsiveness associated with a deficiency of platelet alpha-adrenergic receptors. N Engl J Med. 1978;299:505–509. doi: 10.1056/NEJM197809072991002. doi: 10.1056/NEJM197809072991002. [DOI] [PubMed] [Google Scholar]
- 40.Brill A, Suidan GL, Wagner DD. Hypoxia, such as encountered at high altitude, promotes deep vein thrombosis in mice. J Thromb Haemost. 2013;11:1773–1775. doi: 10.1111/jth.12310. doi: 10.1111/jth.12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nwankwo JO, Gremmel T, Gerrits AJ, Mithila FJ, Warburton RR, Hill NS, Lu Y, Richey LJ, Jakubowski JA, Frelinger AL, III, Chishti AH. Calpain-1 regulates platelet function in a humanized mouse model of sickle cell disease. Thromb Res. 2017;160:58–65. doi: 10.1016/j.thromres.2017.10.018. doi: 10.1016/j.thromres.2017.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubenstein DA, Yin W. Hypergravity and hypobaric hypoxic conditions promote endothelial cell and platelet activation. High Alt Med Biol. 2014;15:396–405. doi: 10.1089/ham.2013.1139. doi: 10.1089/ham.2013.1139. [DOI] [PubMed] [Google Scholar]
- 43.Gupta N, Li W, McIntyre TM. Deubiquitinases modulate platelet proteome ubiquitination, aggregation, and thrombosis. Arterioscler Thromb Vasc Biol. 2015;35:2657–2666. doi: 10.1161/ATVBAHA.115.306054. doi: 10.1161/ATVBAHA.115.306054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Srikanthan S, Li W, Silverstein RL, McIntyre TM. Exosome poly-ubiquitin inhibits platelet activation, downregulates CD36 and inhibits pro-atherothombotic cellular functions. J Thromb Haemost. 2014;12:1906–1917. doi: 10.1111/jth.12712. doi: 10.1111/jth.12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akbar H, Duan X, Saleem S, Davis AK, Zheng Y. RhoA and Rac1 GTPases differentially regulate agonist-receptor mediated reactive oxygen species generation in platelets. PLoS One. 2016;11:e0163227. doi: 10.1371/journal.pone.0163227. doi: 10.1371/journal.pone.0163227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carrizzo A, Forte M, Lembo M, Formisano L, Puca AA, Vecchione C. Rac-1 as a new therapeutic target in cerebro- and cardio-vascular diseases. Curr Drug Targets. 2014;15:1231–1246. doi: 10.2174/1389450115666141027110156. [DOI] [PubMed] [Google Scholar]
- 47.Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJ. S6K1 and mTOR regulate Rac1-driven platelet activation and aggregation. Blood. 2011;118:3129–3136. doi: 10.1182/blood-2011-02-331579. doi: 10.1182/blood-2011-02-331579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie R, Wang P, Ji X, Zhao H. Ischemic post-conditioning facilitates brain recovery after stroke by promoting Akt/mTOR activity in nude rats. J Neurochem. 2013;127:723–732. doi: 10.1111/jnc.12342. doi: 10.1111/jnc.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Medeiros C, Frederico MJ, da Luz G, Pauli JR, Silva AS, Pinho RA, Velloso LA, Ropelle ER, De Souza CT. Exercise training reduces insulin resistance and upregulates the mTOR/p70S6K pathway in cardiac muscle of diet-induced obesity rats. J Cell Physiol. 2011;226:666–674. doi: 10.1002/jcp.22387. doi: 10.1002/jcp.22387. [DOI] [PubMed] [Google Scholar]
- 50.Snoep JD, Hovens MM, Eikenboom JC, van der Bom JG, Huisman MV. Association of laboratory-defined aspirin resistance with a higher risk of recurrent cardiovascular events: a systematic review and meta-analysis. Arch Intern Med. 2007;167:1593–1599. doi: 10.1001/archinte.167.15.1593. doi: 10.1001/archinte.167.15.1593. [DOI] [PubMed] [Google Scholar]
- 51.Mueller MR, Salat A, Stangl P, Murabito M, Pulaki S, Boehm D, Koppensteiner R, Ergun E, Mittlboeck M, Schreiner W, Losert U, Wolner E. Variable platelet response to low-dose ASA and the risk of limb deterioration in patients submitted to peripheral arterial angioplasty. Thromb Haemost. 1997;78:1003–1007. [PubMed] [Google Scholar]
- 52.Hu L, Chang L, Zhang Y, Zhai L, Zhang S, Qi Z, Yan H, Yan Y, Luo X, Zhang S, Wang Y, Kunapuli SP, Ye H, Ding Z. Platelets express activated P2Y12 receptor in patients with diabetes mellitus. Circulation. 2017;136:817–833. doi: 10.1161/CIRCULATIONAHA.116.026995. doi: 10.1161/CIRCULATIONAHA.116.026995. [DOI] [PubMed] [Google Scholar]