

Abstract
Key points
Parkin, an E3 ubiquitin ligase encoded by the Park2 gene, has been implicated in the regulation of mitophagy, a quality control process in which defective mitochondria are degraded.
The exact physiological significance of Parkin in regulating mitochondrial function and contractility in skeletal muscle remains largely unexplored.
Using Park2−/− mice, we show that Parkin ablation causes a decrease in muscle specific force, a severe decrease in mitochondrial respiration, mitochondrial uncoupling and an increased susceptibility to opening of the permeability transition pore.
These results demonstrate that Parkin plays a protective role in the maintenance of normal mitochondrial and contractile functions in skeletal muscles.
Abstract
Parkin is an E3 ubiquitin ligase encoded by the Park2 gene. Parkin has been implicated in the regulation of mitophagy, a quality control process in which defective mitochondria are sequestered in autophagosomes and delivered to lysosomes for degradation. Although Parkin has been mainly studied for its implication in neuronal degeneration in Parkinson disease, its role in other tissues remains largely unknown. In the present study, we investigated the skeletal muscles of Park2 knockout (Park2−/−) mice to test the hypothesis that Parkin plays a physiological role in mitochondrial quality control in normal skeletal muscle, a tissue highly reliant on mitochondrial content and function. We first show that the tibialis anterior (TA) of Park2−/− mice display a slight but significant decrease in its specific force. Park2−/ − muscles also show a trend for type IIB fibre hypertrophy without alteration in muscle fibre type proportion. Compared to Park2+/+ muscles, the mitochondrial function of Park2−/− skeletal muscles was significantly impaired, as indicated by the significant decrease in ADP‐stimulated mitochondrial respiratory rates, uncoupling, reduced activities of respiratory chain complexes containing mitochondrial DNA (mtDNA)‐encoded subunits and increased susceptibility to opening of the permeability transition pore. Muscles of Park2−/− mice also displayed a decrease in the content of the mitochondrial pro‐fusion protein Mfn2 and an increase in the pro‐fission protein Drp1 suggesting an increase in mitochondrial fragmentation. Finally, Park2 ablation resulted in an increase in basal autophagic flux in skeletal muscles. Overall, the results of the present study demonstrate that Parkin plays a protective role in the maintenance of normal mitochondrial and contractile functions in normal skeletal muscles.
Keywords: respiration, reactive oxygen species, apoptosis, muscle contractility, mitophagy
Key points
Parkin, an E3 ubiquitin ligase encoded by the Park2 gene, has been implicated in the regulation of mitophagy, a quality control process in which defective mitochondria are degraded.
The exact physiological significance of Parkin in regulating mitochondrial function and contractility in skeletal muscle remains largely unexplored.
Using Park2−/− mice, we show that Parkin ablation causes a decrease in muscle specific force, a severe decrease in mitochondrial respiration, mitochondrial uncoupling and an increased susceptibility to opening of the permeability transition pore.
These results demonstrate that Parkin plays a protective role in the maintenance of normal mitochondrial and contractile functions in skeletal muscles.
Introduction
Skeletal muscle, the largest type of tissue in the human body, assumes multiple functions that are essential for general health (Wolfe, 2006). Indeed, skeletal muscles are critical for posture, mobility, thermogenesis, whole body glucose homeostasis and also act as the largest amino acid reservoir of the human body in critical catabolic conditions. It is therefore not surprising that a loss of skeletal muscle mass has been associated with impaired whole body glucose homeostasis, falls, fractures, disability and a host chronic diseases (Yang, 2014; Ebner et al. 2015; Balogun et al. 2017).
Mitochondria are intracellular organelles in charge of critical processes for skeletal muscle cells. Often referred as the powerhouse of the cell, they are also involved in the regulation of energy‐sensitive signalling pathways, reactive oxygen species (ROS) production/signalling, calcium homeostasis and the regulation of apoptosis (Brookes et al. 2004). As a result of their importance for optimal muscle cell function and performance, mitochondrial dysfunction has been implicated in a large number of adverse conditions affecting skeletal muscle health (Russell et al. 2014), including sarcopenia (Dirks & Leeuwenburgh, 2004; Chabi et al. 2008; Hepple, 2014; Gouspillou et al. 2014b), disuse‐induced muscle atrophy (Min et al. 2011) and several muscular dystrophies (Godin et al. 2012; Bernardi & Bonaldo, 2013). Maintaining an optimal mitochondrial content and function is therefore critical for skeletal muscle health (Romanello & Sandri, 2015). The maintenance of mitochondrial function relies on the subtle co‐ordination of processes involved in mitochondrial biogenesis and mitochondrial quality control processes, including the removal of dysfunctional mitochondria via autophagy (mitophagy) (Romanello & Sandri, 2015). Although the role of mitochondrial biogenesis and function has received intense research effort in the field of skeletal muscle biology (Hood et al. 2016), very little is known about the roles that mitophagy plays in skeletal muscle health.
Several pathways that regulate mitophagy have been identified (Wei et al. 2015). Amongst them, the PINk1‐Parkin‐dependent pathway has emerged as a major regulator of mitochondrial degradation in multiple biological systems (Narendra & Youle, 2011). Parkin is an E3 ubiquitin ligase encoded by the Park2 gene that translocates to depolarized mitochondria to initiate mitophagy. Parkin acts in concert with phosphatase and tensin homologue‐induced kinase 1 (PINK1), which acts upstream of Parkin. In healthy mitochondria, PINK1 is imported into the inner mitochondrial membrane and then degraded by the protease PARL (Jin et al. 2010). When mitochondria are depolarized, the import of PINK1 protein into the inner mitochondrial membrane is blocked and PINK1 is no longer degraded and becomes stabilized on the outer mitochondrial membrane (Jin et al. 2010). PINK1 then phosphorylates itself and phosphorylates ubiquitin and Parkin (Rasool et al. 2018), which leads to Parkin activation and subsequent ubiquitination of the mitochondrial outer membrane proteins such as TOMM20 and VDAC (Nardin et al. 2016). The ubiquitination of mitochondrial outer membrane proteins by Parkin triggers a broad activation of the ubiquitin‐proteasome system (Chan et al. 2011), as well as the recruitment of autophagy receptors, including LC3, SQSTM1 (p62) and NBR1 (Heo et al. 2015), comprising two steps required for the engulfing of dysfunctional mitochondria by autophagosomes.
Recent studies have shown that the biological functions of Parkin are not limited to mitophagy. Indeed, Shin et al. (2011) have shown that Parkin also plays a role in the regulation of mitochondrial biogenesis by targeting PARIS, a transcriptional repressor of PGC‐1α, for degradation. Mfn1 and Mfn2, two proteins involved in mitochondrial fusion (Chan et al. 2011; Glauser et al. 2011) and Drp1 (Wang et al. 2011), a protein involved in mitochondrial fission, have been identified as Parkin substrates, indicating that Parkin may also regulate mitochondrial dynamics. Furthermore, Parkin has recently been shown to regulate the production of subpopulations of mitochondrial‐derived vesicles, which are assumed to play a role in delivering specific cargos to lysosomes for degradation (McLelland et al. 2014) and thus contribute to mitochondrial quality control.
Although much progress has been made regarding the role of Parkin in mitochondrial recycling in cultured cells, its exact physiological significance in regulating mitochondrial function and the contractile process in vivo in skeletal muscle remains largely unexplored. To address these issues, we studied Park2−/− mice aiming to investigate the impact of Parkin ablation on skeletal muscle contractility and phenotype, as well as on muscle mitochondrial content and function. We hypothesized that skeletal muscles from Park2−/− mice would display significant impairment in mitochondrial function (i.e. respiration, ROS production, calcium retention capacity and susceptibility to trigger apoptosis), as well as in muscle contractility.
Methods
Ethic approval
The present study was carried out in strict accordance with standards established by the Canadian Council of Animal Care and the guidelines and policies of McGill University and University of Montreal. All procedures were approved by the animal ethics committees of the Université de Montréal (#13‐062). Experimental protocols were designed to minimize suffering and the number of animals used in the study. The authors declare that their work complies with the ethical principles under which The Journal of Physiology operates.
Animals
Eight‐ to 12‐week‐old male wild‐type (WT, Park2+/+) and Park2‐deficient (Park2−/−) mice (obtained from INSERM‐Sanofis‐Adventis, Gentilly, France) (Itier et al. 2003) were bred and maintained at the Institut de Recherche en Immunologie et en Cancérologie, Université de Montréal (Montréal, QC, Canada). Animals were exposed to 12:12 h light/dark cycle with free access to standard mice chow diet and water. Park2 knockout was confirmed by western blotting (Fig. 1). Animals were killed by cervical dislocation.
Figure 1. Confirmation of Park2 knockout.
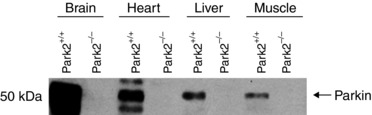
Quantification of Parkin content in brain, heart, liver and muscle in Park2−/− and Park2+/+ mice by western blotting.
In situ assessment of tibialis anterior (TA) contractility
Mice were anaesthetized with an i.p. injection of the following cocktail: ketamine (100 mg mL–1), xylazine (20 mg mL–1) and acepromazine (10 mg mL–1). Anaesthetized mice were immobilized in the supine position. The distal tendon of the TA was isolated and tied to the lever arm of a dual mode force transducer/length servomotor system mounted on a mobile micrometer stage. The exposed section of the muscle was kept moist with an isotonic saline drip (37°C) and directly stimulated with an electrode placed on the belly of the muscle. Supramaximal stimuli (pulse durations of 2 ms) were delivered using a computer‐controlled electrical stimulator (model S44; Grass Instruments, Quincy, MA, USA). Muscle force and length signals were stored on a computer using Labdat/Anadat software (RHT InfoData, Montreal, QC, Canada). Force–frequency relationships were determined at muscle optimal length by sequential supramaximal stimulations for 300 ms at 10, 30, 50, 100 and 120 Hz, with a 2 min interval between each stimulation train. At the end of this protocol, animals were killed by cervical dislocation. Muscle mass was quantified and muscle length was measured with a microcaliper. Muscle force was normalized to tissue cross‐sectional area (expressed as newtons cm–2). Muscle cross‐sectional area was estimated by dividing muscle mass by the product of muscle length and muscle density (1.056 g cm–3).
Mitochondrial functional assays in permeabilized muscle fibres
Dissections and permeabilization of fibre bundles from the gastrocnemius with saponin were performed as described previously (Picard et al. 2008). Ghost fibres were prepared by incubating permeabilized bundles in a high KCl medium that allows extraction of myosin. Permeabilized myofibres and ghost fibre bundles were kept on ice until use. All mitochondrial functional parameters were determined at minimum in duplicate and expressed per mg of dry fibre weight.
Mitochondrial respiration
Mitochondrial respiratory function was determined in permeabilized fibres as described by Picard et al. (2008) in solution B [in mm: 2.77 CaK EGTA, 7.23 K EGTA (100 nm free Ca2+), 6.56 MgCl2 (1 mm free Mg2+), 20 taurine, 0.5 DTT, 50 K‐methane sulphonate [160 mm ionic strength) 20 imidazole, pH 7.1] at 23°C under continuous stirring. Rates of O2 consumption were determined after the sequential addition of the complex I substrate glutamate–malate (5:2.5 mm; V G+M); ADP (2 mm; V ADP); the complex I blocker amytal (2 mm); the complex II substrate succinate (5 mm) (VSucc); the complex III blocker antimycin A; and p‐phenylenediamine dihydrochloride (TMPD)‐ascorbate (0.9:9 mm) (V TMPD).
Mitochondrial H2O2 emission
Net H2O2 release by respiring mitochondria was measured in permeabilized fibres incubated in buffer Z (in mm: 110 K‐MES, 35 KCl, 1 EGTA, 5 K2HPO4, 3 MgCl26H2O and 0.5 mg mL–1 BSA, 1.2 U mL–1 horseradish peroxidase, pH 7.3, 37°C) and the fluorescent probe Amplex Red (Thermo Fisher, Waltham, MA, USA) (20 μm: excitation–emission: 563–587 nm) as described previously (Picard et al. 2008). Baseline fluorescence readings were taken in the absence of any exogenous respiratory substrates. Additions were then made sequentially: glutamate (5 mm), succinate (5 mm), ADP (10 mm) and antimycin A (8 μm). Rates of H2O2 production were calculated from a standard curve established under the same experimental conditions.
Calcium retention capacity
Calcium retention capacity (CRC) was measured in ghost fibres in CRC buffer (in mm: 250 sucrose, 10 Mops, 0.005 EGTA, 10 Pi‐Tris, pH 7.3) supplemented with glutamate–malate (5:2.5 mm) and 0.5 nm oligomycin, as described previously (Picard et al. 2008). Following the addition of fibres and respiratory substrates, a single pulse of 20 nmol Ca2+ was added. CRC was defined as the total amount of Ca2+ accumulated by mitochondria prior to PTP opening‐induced Ca2+ release. The Ca2+ concentration was calculated from a standard curve relating [Ca2+] to the fluorescence of Ca–Green 5N (excitation–emission: 505–535 nm).
Enzyme activity
Activities of complex I (CI, NADH‐CoQ reductase), complex II (CII, succinate dehydrogenase), complex IV (CIV, cytochrome oxidase complex) and citrate synthase (CS) were measured spectrophotometrically with a plate reader using standard coupled enzyme assays as described previously (Marcil et al. 2006) with minor modifications (de Wit et al. 2008). Activities were expressed as mU min–1 mg–1 wet muscle weight.
Immunoblotting
Frozen muscle samples were homogenized in homogenization buffer [tris‐maleate (10 mm), EDTA (3 mm), sucrose (275 mm), DTT (0.1 mm), leupeptin (2 μg mL–1), phenylmethylsulfonyl fluoride (100 μg mL–1), aprotinin (2 μg mL–1) and pepstatin A (1 mg 100 mL–1, pH 7.2)], then centrifuged at 1000 g for 10 min in a cold room. Pellets were discarded and supernatants were designated the crude homogenate. Total muscle protein in each sample was determined using the Bradford protein assay technique. Crude homogenate (25–50 μg sample–1) was mixed with SDS sample buffer, boiled at 95°C for 8 min, loaded onto Tris‐glycine SDS‐PAGE gels and separated into proteins by electrophoresis and then transferred to nitrocellulose membranes. After 1 h of blocking in Tris‐buffered saline with 0.1% Tween 20 (TBS‐T) supplemented with 5% nonfat milk, membranes were incubated overnight at 4°C with primary antibody diluted in TBS‐T supplemented with 5% nonfat milk or 5% BSA, according to the antibody: PARK2/PARKIN (P6248; Sigma‐Aldrich, St Louis, MO, USA); SQSTM1/p62 (H00008878‐M01; Abnova, Taipei, Taiwan); LC3B (3868S; Cell Signaling, Beverly, MA, USA); BNIP3L (NIX) (39‐3300; Invitrogen, Carlsbad, CA, USA); BNIP3 (3769; Cell Signaling); GAPDH (5174; Cell Signaling); VDAC1 (4866S; Cell Signaling); SOD2 (ab16956; Abcam, Cambridge, MA, USA); OPA1 (612606; BD Transduction Laboratories, Lexington, KT, USA); HSP60 (SPA‐307; Stressgen Bioreagent, Victoria, BC, Canada); COX I (ab14705; Abcam); COX IV (4850S; Cell Signaling); 4‐HNE (HNE11S; Alpha Diagnostic International, San Antonio, TX, USA); Bax (SC526; Santa Cruz Biotechnology, Santa Cruz, CA, USA); Drp1 (8570; Cell Signaling); Mfn2 (M6319; Sigma‐Aldrich); and TOM70 (A‐8; Santa Cruz Biotechnology). After washing, membranes were incubated for 1 h at room temperature with a secondary antibody [7076 (Cell Signaling) and A0545 (Sigma‐Aldrich)] diluted in TBS‐T supplemented with 5% nonfat milk. Protein detection was performed using a chemiluminescent substrate (Amersham Biosciences Corp., Little Chalfont, UK) with film exposures ranging from 30 s to 5 min. Following the film scanning, bands were quantified using ImageJ (NIH, Bethesda, MD, USA).
Autophagy flux
Park2+/+ and Park2−/− mice were injected with PBS or colchicine, a lysosomal inhibitor that prevents degradation of autophagosomes (Ju et al. 2010). To measure autophagic flux, mice received two i.p. injections, separated by a 24 h window, of PBS (control group) or colchicine (colchicine group: 0.4 mg kg–1 day–1). Twenty‐four hours after the second PBS or colchicine injection, animals were killed, muscles were excised and tissue samples were prepared for immunoblotting to detect LC3B proteins (3668S; Cell Signaling). Muscles were excised and tissue samples were prepared for immunoblotting to detect LC3B proteins. A standard curve consisting of purified LC3B was run alongside experimental gels (range 0.4–1.2 ng protein per lane) to facilitate quantification of LC3B protein levels (LC3B‐I and LC3B‐II). LC3B protein density in a given sample was converted to LC3B protein quantity by extrapolation from the standard curve using regression analysis tools. Values were then normalized to mg of total muscle protein loaded per lane. Because LC3B‐II binds tightly to autophagosomal membranes and serves as an autophagic marker protein, the difference in LC3B‐II levels in the presence and absence of colchicine represents autophagic flux.
Quantitative real‐time PCR
Frozen muscle tissue (50 mg) was homogenized in 1 ml of TRIzol reagent (15596‐026; Invitrogen) and RNA was isolated in accordance with the manufacturer's instructions. RNA concentration and integrity was verified on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) using the RNA integrity number software algorithm. Samples with a RNA integrity number score of 9 and above were used for analysis. Reverse transcription was performed in a standard fashion using a High Capacity cDNA Reverse Transcription Kit (4368814; Applied Biosystems, Foster City, CA, USA) supplemented with DNase treatment (AM2222; Ambion, Austin, TX, USA). TaqMan assays were designed for specific genes using the Roche Universal Probe Library. Map1Lc3 (Lc3), Gabarap, Bnip3, Ppargc1α, Ppargc1β, Tfam, Nrf1, Nrf2, Mt‐co1/Cox1, Mt‐co2/Cox2, Cox4i, Cox5a, Mt‐nd1/Nd1, Mt‐nd2/Nd2, TFB1 and TFB2 mRNA expressions were detected using real‐time PCR with an ABI PRISM 7000 sequence detector (Applied Biosystems) and analyzed using SDS, version 2.2.2 (Applied Biosystems). All real‐time PCR experiments were performed in triplicate. Relative mRNA quantifications of target genes were determined using the threshold cycle (ΔΔCT) method using the housekeeping genes Gapdh and Hprt1/Hprt. Data were expressed as the fold change relative to control Park2+/+ mice.
In situ determination of muscle fibre type and size
Eight micron thick serial cross‐sections were cut in a cryostat at −18°C and mounted on lysine coated slides (Superfrost; Thermo Fisher). These sections were immunolabelled for the different myosin heavy chains (MHC) as described previously (Gouspillou et al. 2014c; Leduc‐Gaudet et al. 2015). Briefly, the first cross‐sections of each animal sample were used to immunolabel MHC type I, IIa and IIb. These cross‐sections were first allowed to reach room temperature and rehydrated with PBS (pH 7.2). These sections were then blocked using goat serum (10% in PBS) and incubated for 1 h at room temperature with a primary antibody cocktail: mouse IgG2b monoclonal anti‐MHC type I (BA‐F8; dilution 1:25), mouse IgG1 monoclonal anti‐MHC type IIa (SC‐71; dilution 1:200), mouse IgM monoclonal anti‐MHC type IIb (BF‐ F3; dilution 1:200) and rabbit IgG polyclonal anti‐laminin (L9393; Sigma‐Aldrich; dilution 1:750). Muscle cross‐sections were then washed three times in PBS before being incubated for 1 h at room temperature with a secondary antibody cocktail: Alexa Fluor 350 IgG2b (y2b) goat anti‐mouse (A‐21140; Invitrogen; dilution 1:500), Alexa Fluor 594 IgG1 (y1) goat anti‐mouse (A‐21125; Invitrogen; dilution 1:100), Alexa Fluor 488 IgM goat anti‐mouse (A‐21042; Invitrogen; dilution 1:500) and Alexa Fluor 488 IgG goat anti‐rabbit (A‐11008; Invitrogen; dilution 1:500). Muscle cross‐sections were then washed three times in PBS and slides were then cover slipped using Prolong Gold (P36930; Invitrogen) as mounting medium. Identical procedures were employed for the section cross‐sections used to immunolabel for MHC type IIx, except for the primary antibody cocktail, which comprised a mouse IgM monoclonal anti‐type 2x MHC (6H1; dilution 1:25) and a rabbit IgG polyclonal anti‐laminin, and the secondary antibody cocktail that comprised Alexa Fluor 488 IgM goat anti‐mouse and Alexa Fluor 488 IgG goat anti‐rabbit. All primary antibodies selective to MHC isoforms were purchased from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA, USA).
Statistical analysis
Statistical analyses for data related to muscle specific strength, fibre size distribution, fibre cross‐sectional area, fibre type proportion, mitochondrial respiration and mitochondrial enzyme activities were performed using a two‐way ANOVA. Corrections for multiple comparisons were performed by controlling for the false discovery rate using the two‐stage method of Benjamini and Krieger and Yekutieli (with q < 0.1 and P < 0.05). All other comparisons between Park2−/− and Park2+/+ mice were performed using unpaired bilateral Student's t tests. P < 0.05 were considered statistically significant.
Results
Parkin ablation alters muscle contractility and phenotype
We first investigated the impact of Parkin ablation on skeletal muscle contractility. To this end, we quantified the in situ specific force of the TA muscle at various stimulation frequencies (Fig. 2 A). Although Park2−/− did not significantly decrease force at any given stimulation frequency, a two‐way ANOVA across stimulation frequencies revealed a significant main effect of genotype on muscle specific force. This result indicates that Park2−/− causes a mild contractile dysfunction. We then investigated the impact of Parkin ablation on muscle fibre size and type in the gastrocnemius muscle (Fig. 2 B–F). Although no change in the overall muscle fibre size was observed (Fig. 2 C), Park2−/− causes a right shift in overall fibre size distribution (Fig. 2 D), indicating a higher proportion of large fibres. Muscles from Park2−/− mice also showed a trend for higher fibre CSA of type IIb compared to muscles derived from Park+/+ mice (P = 0.051) (Fig. 2 E). No significant differences in gastrocnemius fibre type proportions were observed between Park2−/− and Park2+/+ mice (Fig. 1 F).
Figure 2. Effects of Parkin knockout on muscle contractility and phenotype.
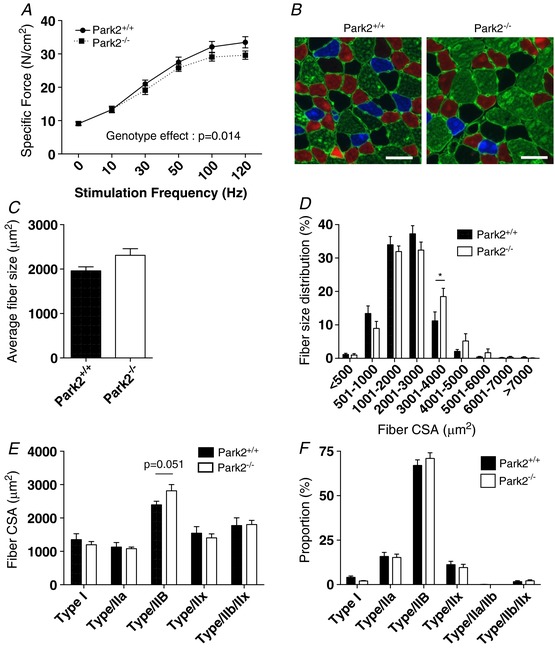
A, in situ specific force of the TA muscle of Park2−/− and Park2+/+ expressed as a function of the stimulation frequency (n = 5 or 6). B, representative immunolabeling for type I (blue), type IIa (red) and type IIb (green) MHC of a gastrocnemius cross‐section of Park2−/− (right) and Park2+/+ (left) mice. Type IIx fibre, unstained on these images, shown in black. Laminin was also immunolabelled to highlight fibre borders (green line surrounding each muscle fibre). C, quantification of the overall fibre size of the gastrocnemius muscle of Park2−/− and Park2+/+ mice (n = 6 or 7). D, quantification of the fibre size distribution of the gastrocnemius muscle of Park2−/− and Park2+/+ mice (n = 6 or 7). E, quantification of the average fibre size for each fibre type of the gastrocnemius muscle of Park2−/− and Park2+/+ mice (n = 6 or 7). F, quantification of the fibre type proportion the gastrocnemius muscle of Park2−/− and Park2+/+ mice (n = 6 or 7). Statistical analyses for data shown in (A), (D), (E) and (F) were performed using a two‐way ANOVA. Corrections for multiple comparisons were performed by controlling for the false discovery rate using the two‐stage method of Benjamini and Krieger and Yekutieli (with q < 0.1). * P < 0.05 and q < 0.1.
Parkin ablation results in mitochondrial dysfunction
Because the best‐characterized function of Parkin is its roles in mitochondrial quality control processes, we then investigated whether the contractile dysfunction observed in Park2−/− mice was associated with alteration in mitochondrial energetics. To this end, we first evaluated mitochondrial respiration in permeabilized myofibres prepared from the gastrocnemius muscle, an approach known to preserve mitochondrial structure, content and interactions with other organelles. No difference in state 2 respiration was observed when glutamate and malate were used as substrates between muscles from Park2−/− and Park2+/+ mice (Fig. 3 A). Maximal (state 3) respiration driven by complex I substrates (Column V ADP in Fig. 3 A) was 48% lower in muscles from Park2−/ − vs. Park2+/+ mice. No differences in state 3 respiration rate were observed when complexes I + II substrates were used (Column V Succ in Fig. 2 A). Maximal respiration rate driven by complex IV substrates was 34% lower in muscles from Park2−/ − vs. Park2+/+ mice (Column V TMPD in Fig. 3 A). The acceptor control ratio, an index of mitochondrial coupling efficiency calculated by dividing state 3 by state 2 respiration (Schlagowski et al. 2014), was 52% lower in muscles from Park2−/ − vs. Park2+/+ mice (Fig. 3 B). In line with the lower state 3 respiration rates driven by complex I and complex IV substrates, the individual enzymatic activity of complex I and IV were significantly lower in muscles from Park2−/ − vs. Park2+/+ mice, whereas the activity of complex II (SDH) and citrate synthase were unaltered (Fig. 3 C). Altogether, our results indicate that Parkin ablation results in severe decrease in maximal mitochondrial respiration driven by complex I and IV substrates and mitochondrial uncoupling.
Figure 3. Effects of Parkin knockout on mitochondrial respiration and enzymatic activities in skeletal muscle.
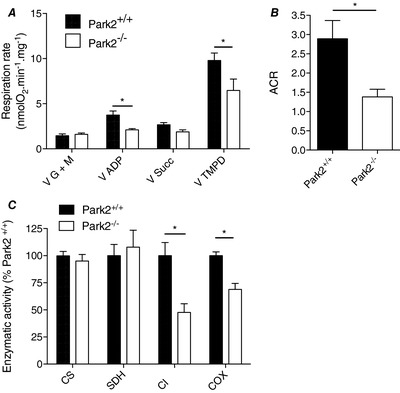
A, mitochondrial respiration rates measured in permeabilized myofibres prepared from Park2−/− and Park2+/+ mice (n = 6–8). V G + M: state II respiration driven by the complex I substrates glutamate + malate; V ADP: state III respiration rate driven by complex I substrates; VSucc: state III respiration rate driven by the complex II substrate succinate; VTMPD: state III respiration driven by the complex IV substrate TMPD. B, ACR, an index of mitochondrial coupling efficiency, assessed in permeabilized myofibres prepared from Park2−/− and Park2+/+ mice (n = 6–8). C, quantification of the activity CS, SDH, CI and COX (complex IV of the electron transfer system). Statistical analyses for data presented in (A) and (C) were performed using a two‐way ANOVA. Corrections for multiple comparisons were performed by controlling for the false discovery rate using the two‐stage method of Benjamini and Krieger and Yekutieli (with q < 0.1). * P < 0.05 and q < 0.1.
Impact of Parkin on mitochondrial biogenesis
To define whether the severe impairment in mitochondrial respiration seen in Park2−/− mice was associated with impaired mitochondrial biogenesis, we first quantified mRNA expression of key proteins involved in the regulation of the mitochondrial biogenesis program. The levels of PGC‐1α, PGC1β, NRF1, TFAM and TFB2 mRNA, but not that of NRF2 and TFB1, were all significantly lower in muscles from Park2−/ − vs. Park2+/+ mice (Fig. 4 A). Although their mRNA levels were significantly decreased in muscles from Park2−/ − mice, no difference in PGC‐1α and PGC‐1β protein contents were observed among Park2−/− and Park2+/+ mice (Fig. 4 B).
Figure 4. Effects of Parkin knockout on mitochondrial biogenesis and content in skeletal muscle.
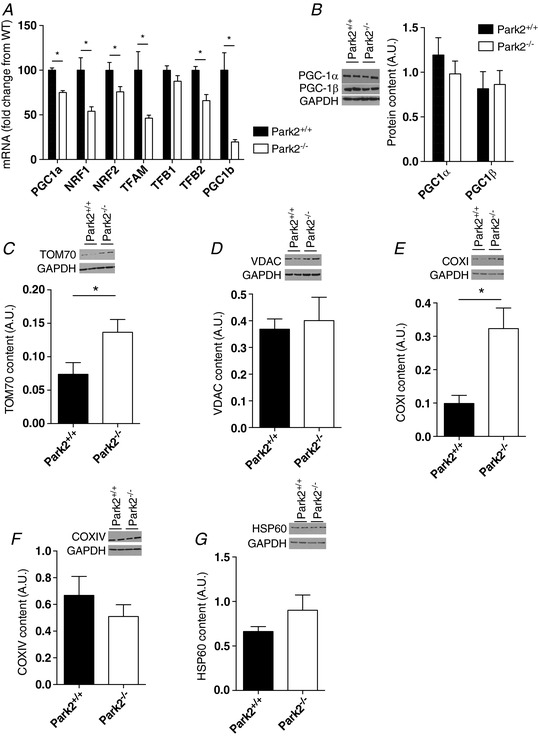
A, quantification of the mRNA content of several proteins involved in the mitochondrial biogenesis program in skeletal muscle homogenates from Park2−/− and Park2+/+ mice. B–G, quantification of the content of PGC‐1a (n = 4 per group), PGC‐1b (B) (n = 4 per group), TOMM70 (C) (n = 4 per group), VDAC (D) (n = 4 per group), COX I (E) (n = 7 or 8 per group), COX IV (F) (n = 4 per group) and HSP60 (G) (n = 4 per group) in skeletal muscle of Park2−/− and Park2+/+ mice by western blotting (n = 4 per group). * P < 0.05.
We next quantified the protein content of representative proteins located in the outer mitochondrial membrane, the inner mitochondrial membrane and the mitochondrial matrix. Muscles from Park2−/− mice contained higher TOMM70 protein levels but similar VDAC protein levels to those detected in muscles from Park+/+ mice (Fig. 4 C and D). Although the content of the inner mitochondrial membrane protein COX I was higher in muscles from Park2−/− animals, the content of COX IV levels was similar to that of Park+/+ mice (Fig. 4 E and F). Finally, no difference in the content of the mitochondrial matrix protein HSP60 was observed (Fig. 4 G). Overall, our results suggest that Parkin ablation significantly downregulates pathways controlling mitochondrial biogenesis and that Parkin might be involved in the selective degradation of outer and inner mitochondrial membrane proteins. Finally, because none of the proteins we probed for showed a lower abundance, our results suggest that mitochondrial content is not decreased by Parkin ablation.
Parkin ablation does not impact mitochondrial H2O2 emission but result in the accumulation of a marker of oxidative stress
Because oxidative stress can decrease muscle force production (Carnio et al. 2014), we investigated the impact of Parkin ablation on mitochondrial ROS production. To this end, we measured in permeabilized fibres the efflux of H2O2 using the Amplex Red system. For all of the conditions we tested (under state II and state III conditions, with various substrates and inhibitors), no difference in mitochondrial H2O2 emission and SOD2 content were observed between muscles from Park2−/− and Park+/+ animals (Fig. 5 A and B); however, 4‐HNE content (a marker of lipid peroxidation) was significantly higher in muscles from Park2−/− vs. Park2+/+ mice, indicating that Parkin ablation favours oxidative stress (Fig. 5 C).
Figure 5. Effects of Parkin knockout on mitochondrial H2O2 emission, anti‐oxidant enzyme expression and oxidative stress.
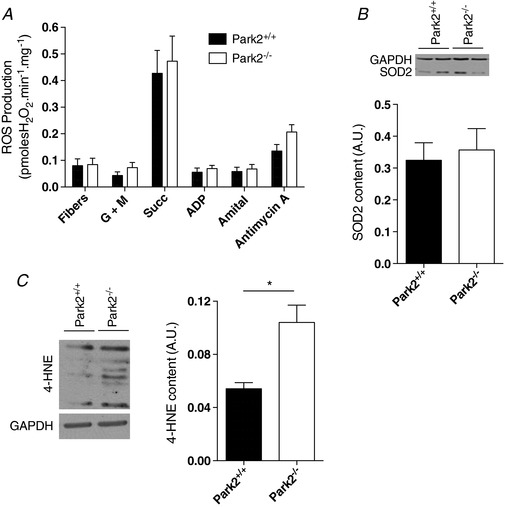
A, quantification of the mitochondrial H2O2 emission performed using various substrates and inhibitors in permeabilized myofibres from Park2−/− and Park2+/+ mice (n = 6–8). Fibre: basal H2O2 emission in the absence of mitochondrial substrates. G + M: state II H2O2 emission with the complex I substrates glutamate + malate. Succ: H2O2 emission with both complex I and complex II substrates (glutamate + malate + succinate present in the incubation medium). ADP: state III H2O2 emission with complex I and II substrates. Amytal (complex I inhibitor): state III H2O2 emission with the complex II substrate succinate. Antymycin A: H2O2 emission with the complex II substrate succinate in the presence of both the complex I inhibitor amytal and the complex III inhibitor antiycin A. B, quantification of the SOD2 content in skeletal muscle of Park2−/− and Park2+/+ mice by western blotting (n = 4 per group). C, quantification of the 4HNE content in skeletal muscle of Park2−/− and Park2+/+ mice by western blotting (n = 4 per group). * P < 0.05.
Parkin ablation results in a sensitization of the mitochondrial permeability transition pore (mPTP)
In addition to their roles in ATP synthesis and ROS production, mitochondria also play a key role in the regulation of apoptosis. For example, the opening of the mitochondrial permeability transition pore, which can be triggered by ROS overproduction or calcium overload, can release pro‐apoptotic factor normally sequestered in the intermembrane space to the cytosol (Halestrap, 2009). We therefore investigated the impact of Parkin loss on mitochondrial mediated apoptosis. We first assessed mitochondrial CRC and time to mPTP opening. Although no significant difference in CRC was observed between muscles from Park2−/− and Park2+/+ mice (Fig. 6 A), muscles from Park2−/− mice had a significantly lower time to mPTP opening compared to Park2+/+ animals (Fig. 6 B). No difference in the activities of Caspase‐3 and Caspase‐9 (Fig. 6 C), as well as in Bax protein level content, was observed among muscles from Park2−/− and Park2+/+ (Fig. 6 D).
Figure 6. Effects of Parkin knockout on the kinetics of the mitochondrial permeability transition pore and on mitochondrial‐mediated apoptosis in skeletal muscle.
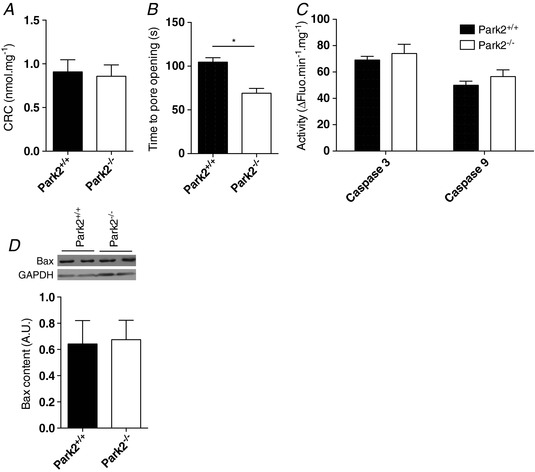
Quantification of the mitochondrial calcium retention capacity (A) and the time to pore opening (B) performed in permeabilized myofibres from Park2−/− and Park2+/+ mice (n = 6–8). Quantification of the caspase 3 and 9 activities (C) performed on muscle homogenates from Park2−/− and Park2+/+ mice (n = 6–8). Quantification of Bax content (D) performed on muscle homogenates from Park2−/− and Park2+/+ mice by western blotting (n = 4). * P < 0.05.
Parkin ablation alters mitochondrial morphology and fusion/fission processes in skeletal muscle
Because of the tight link existing between mitochondrial dynamics and function, we next investigated the impact of Parkin ablation on the content of proteins regulating mitochondrial fusion and fission. Muscles from Park2−/‐ mice displayed significantly lower levels of the pro mitochondrial fusion protein Mfn2 and significantly higher levels of the pro fission protein Drp1 compared to Park2+/+ animals (Fig. 7). The association of the higher Drp1 and lower Mfn2 protein contents observed in Park2−/− mice suggests that Parkin ablation results in the fragmentation of skeletal muscle mitochondria.
Figure 7. Effects of Parkin knockout on the content of proteins regulating mitochondrial fusion and fission in skeletal muscle.
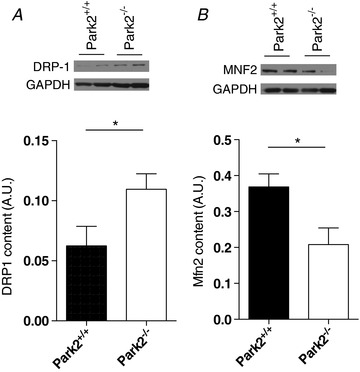
Quantification of DRP1 (A) and Mfn2 (B) content performed on muscle homogenates from Park2−/− and Park2+/+ mice by western blotting (n = 4 per group). * P < 0.05.
Parkin ablation increases global autophagy
Given the central role of Parkin in mitochondrial autophagy, we aimed to determine whether overall autophagy was perturbed in muscles from Park2−/− mice. Muscles from Park2−/− animals expressed significantly higher mRNA levels of three key autophagy proteins: LC3, Gabaparl1 and Bnip3 (Fig. 8 A). Although no significant difference in the levels of SQSTM1 (p62) was observed between muscles from Park2−/− and Park2+/+ mice (Fig. 8 B), a significantly higher autophagic flux (assessed as the evolution of LC3B‐II levels in the presence and absence of colchicine) was found in muscles from Park2−/− vs. Park2+/+ mice (Fig. 8 C).
Figure 8. Effects of Parkin knockout on markers of autophagy.
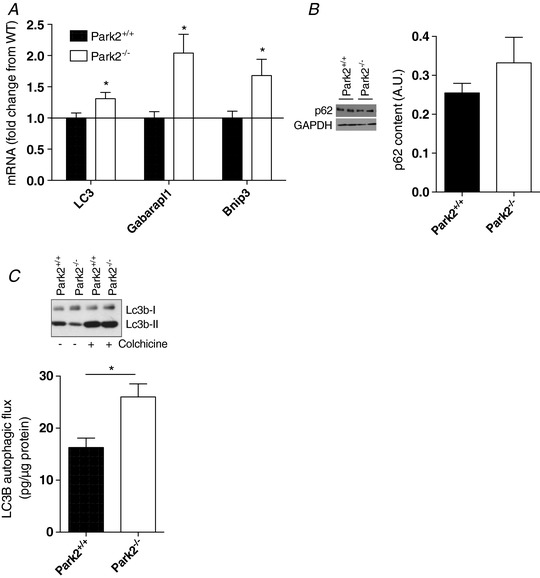
A, quantification of the mRNA expression of LC3, Gabarapl1 and Bnip3 performed using muscle homogenates from Park2−/− and Park2+/+ mice by western blotting (n = 6). B, quantification of the p62 content in skeletal muscle of Park2−/− and Park2+/+ mice by western blotting (n = 4 per group). C, quantification of the autophagic flux in skeletal muscle of Park2−/− and Park2+/+ mice (n = 6).
Discussion
Mitochondrial health is essential for optimal muscle performance, making processes involved in mitochondrial quality control critical for muscle cells. Although Parkin has been described as a major regulator of mitophagy, very little is known about its roles physiological in skeletal muscle function. In the present study, we show that Parkin ablation in mice results in: (i) mild impairment in skeletal muscle contractility; (ii) a severe decrease in mitochondrial respiration driven by complex I and IV substrates, as well as mitochondrial uncoupling; (iii) a reduction in the activities of respiratory chain complexes containing mitochondrial DNA (mtDNA) encoded subunits; and (iv) a sensitization of the mitochondrial mPTP to Ca2+. These results document a previously unappreciated role of Parkin in the normal maintenance of mitochondrial and skeletal muscle function.
It is important to note that the contractile dysfunction we observed in the TA muscle of Park2−/− mice is an intrinsic dysfunction because muscle strength was normalized to muscle cross‐sectional area (Fig. 2 A). In addition, our results obtained in the gastrocnemius suggest that the impaired contractility we observed in Park2−/− mice may occur in the absence of muscle atrophy and shift in muscle fibre type (Fig. 2). Although the mechanism linking Parkin to the regulation of skeletal muscle contractile function is currently unknown, it is possible the oxidative stress associated with Parkin ablation, as indicated by the increase in 4‐HNE content, could play a role in the depression of muscle contractility of Parkin2−/− mice. Indeed, oxidative modifications to contractile proteins, which can occur under oxidative stress, have been shown to decrease muscle force production (Carnio et al. 2014). Although oxidative stress observed in Park2−/− muscles may be attributed to an increase in mitochondrial ROS production, no significant differences in mitochondrial H2O2 emission was observed among Park2−/− and Park2+/+ muscles. Furthermore, the observation that mitochondrial anti‐oxidant SOD2 contents were also similar among these muscles strongly suggests that the sources of this increase in 4‐HNE content in Park2−/− muscles are located outside the mitochondria. Other potential ROS include NADPH oxidases, xanthine oxidases and other non‐mitochondrial ROS sources in skeletal muscle fibres. Alternatively, this increase in 4‐HNE content could also result from an impaired H2O2 handling in the cytosol, potentially through a decrease in the activity of the cytoplasmic or peroxisomal catalases. Future studies are clearly required to investigate the link between Parkin and these pathways.
The most striking impact of Parkin ablation in skeletal muscle in the present study is the significant decrease in mitochondrial respiration (Fig. 3). Interestingly, Parkin ablation in skeletal muscle appears to particularly influencing the activity of mitochondrial enzymes with subunits encoded by the mtDNA. Indeed, the activity of the two nuclear DNA encoded enzymes, complex II (SDH) and CS, were not affected by Parkin deletion, whereas the activities of complex I and complex IV, which contain seven and three mtDNA‐encoded subunits, respectively, were significantly decreased in Park2−/− muscles. Direct measurements of mitochondrial respiratory capacity provided converging evidence for this interpretation. No significant alterations in the respiration rate driven by complex II substrate was observed in permeabilized myofibres from Park2−/− mice, whereas respiration driven by complex I and complex IV substrates was severely decreased. Based on recent findings suggesting that the PINK1‐Parkin pathway is involved in the selective turnover of complexes of the respiratory chain (Vincow et al. 2013), we propose that Parkin may be involved in the turnover of complexes containing mtDNA‐encoded subunits or of the mtDNA‐encoded subunits themselves.
Our quantification of the content of two complex IV subunits support the hypothesis that Parkin may be involved in the turnover of mtDNA encoded subunits. Indeed, we found that Park2−/− muscles have significant accumulation of the mtDNA‐encoded subunit COX I, whereas no change in the nuclear DNA‐encoded subunit COX IV was observed in these animals. This selective degradation of mt‐DNA‐encoded subunits of respiratory chain complexes could be achieved through the formation of mitochondrial derived vesicles (Soubannier et al. 2012a; Soubannier et al. 2012b; McLelland et al. 2014). Recent studies have revealed that the Parkin–Pink1 pathway regulates the formation of mitochondrial derived vesicles (McLelland et al. 2014), which can contain, amongst others, the mtDNA‐encoded COX I subunit (Soubannier et al. 2012b). Interestingly, this specific impact of Parkin ablation on complexes containing mtDNA‐encoded subunits was not observed in the heart (Piquereau et al. 2013). Although mitochondrial respiration is also decreased in Park2−/− hearts (albeit to a lesser extent than in skeletal muscle), the activities of complex I and IV are similar among Park2−/− and Park2+/+ hearts (Piquereau et al. 2013). The exact mechanisms involved in the differential impact of Parkin ablation on mitochondrial respiration and mitochondrial complex activities in skeletal muscle and cardiac cells are currently unknown. It is possible that other pathways involved in mitochondrial quality control might compensate for Parkin ablation in cardiomyocytes.
The present study reveals that Parkin ablation results in a sensitization of the mPTP to Ca2+ (Fig. 6). This finding is similar to that made in the Park2−/− hearts (Piquereau et al. 2013). One of the consequences of mPTP opening is the release of pro‐apoptotic factors, such as cytochrome c, apoptosis inducing factor and EndoG, from the mitochondrial intermembrane space into the cytosol (Kroemer et al. 2007). Once released, these pro‐apoptotic factors will either translocate to the nuclease to cleave the nuclear DNA or will trigger the activation of several caspases, including the caspases 3 and 9. Interestingly, despite increased mPTP sensitivity to Ca2+, muscles of Park2−/− mice do not have significant increase in caspase‐3 and caspase‐9 activities, suggesting that mitochondria from these muscles do not trigger excessive apoptosis. This conclusion is supported by the observation that BAX protein levels was similar in muscles from Park2−/− and Park2+/+ mice. Bax is a protein able to form oligomers in the outer mitochondrial membrane to trigger the release of mitochondrial pro‐apoptotic factor (Kroemer et al. 2007). Although this may be interpreted as an absence of effect of Parkin on mitochondrial‐mediated apoptosis, the sensitized mPTP in Park2−/− muscles might render muscle mitochondria more susceptible to trigger apoptosis under stress condition.
It is important to highlight that, despite the decrease in mitochondrial respiration and the sensitization of the mPTP detected in muscles from mice Park2−/−, mitochondrial content was not decreased in muscles from mice Park2−/− mice. Indeed, several commonly used markers of mitochondrial contents, such as CS activity and VDAC content, were similar among muscles from Park2−/− and Park2+/+ mice, suggesting a similar mitochondrial content between muscles of Park2−/− and Park2+/+ mice (Fig. 4). Because of its role in mitophagy, it could also be assumed that Parkin ablation would result in an increase in mitochondrial content. However, our results suggest that Parkin ablation causes a reduction in the drive for mitochondrial biogenesis, as highlighted by the decrease in mRNA expression of multiple proteins regulating mitochondrial biogenesis (Fig. 3). This observation is in agreement with recent findings showing that Parkin plays a role in the regulation of mitochondrial biogenesis by targeting PARIS, a transcriptional repressor of PGC‐1α, for degradation (Shin et al. 2011).
An interesting observation in our study is that muscle autophagic flux is significantly higher in Park2−/− mice compared to Park2+/+ mice. A similar finding has been reported in the heart (Piquereau et al. 2013). The increase in basal autophagy may be a compensatory response designed to compensate for the lack of Parkin and the resulting reduction in mitochondrial quality and cytoplasmic oxidative stress. This increase in basal autophagy in Park2+/+ mice is in line with the findings of Chen et al. (2010) who found that Parkin in Hela cells can repress autophagy through the mono‐ubiquitinylation of Bcl‐2. Although autophagy is an important catalytic pathway involved in muscle protein degradation and the development of fibre atrophy (Sandri, 2013; Milan et al. 2015), we found that the significant increase in basal autophagy of muscles from Park2−/− mice was not associated with myofibre atrophy. The lack of fibre atrophy may be attributed to a possible activation of the hypertrophy signalling pathways, such as the AKT/ mTOR pathway in muscles lacking Parkin, resulting in increased protein synthesis. This possibility needs to be tested in future studies.
Mitophagy and mitochondrial dynamics are closely related processes. It is well established that mitochondrial fission is required for the initiation of mitophagy (Twig & Shirihai, 2011). Parkin was shown to ubiquitinylate the pro‐fusion proteins Mfn2 and Mfn1, thereby targeting these proteins for proteosomal degradation (Gegg et al. 2010). Through this action, it was suggested that Parkin can ultimately tip the fusion/fission balance towards mitochondrial fission (Gegg et al. 2010). In line with this thesis, Parkin overexpression in skeletal muscle of flies and in rat hippocampal neurons has been shown to promote mitochondrial fragmentation (Yu et al. 2011; Rana et al. 2013). Based on these observations, we anticipated that Parkin ablation would result in an increase in the content of pro‐fusion proteins and potentially in a fusion/fission balance favouring mitochondrial fusion. Our results, however, indicate that Parkin ablation in skeletal muscle results in a significant decrease in Mfn2 content and an increase in the levels of the pro‐fission protein Drp1, strongly suggesting that Parkin deletion is associated with an increase in mitochondrial fragmentation. The increase in Drp1 protein levels in muscles from Park2−/− mice in the present study may be a result of decreased degradation rather than increased transcription of this protein. This speculation is based on the recent observation that Drp1 is a Parkin substrate and that ubiquitinylation of Drp1 by Parkin targets Drp1 for gradation (Wang et al. 2011). Our results and those from the previously cited studies emphasize that the interplay between Parkin and proteins involved in mitochondrial dynamics is extremely complex and requires further investigation. Because mitochondrial fragmentation has been frequently associated with impaired mitochondrial function (Picard et al. 2011), it is also possible that the imbalance between Drp1 and Mfn2 contents caused by Parkin ablation might have contributed to the mitochondrial dysfunction that we found in muscles from Park2−/− mice.
Our current results could have important implications for the understanding of several conditions and pathologies affecting skeletal muscles and mitochondrial function. For example, a decline in Parkin expression has been reported in aged skeletal muscle (Drummond et al. 2014; Gouspillou et al. 2014b) and in skeletal muscle dysfunction induced by chemotherapy treatment (Gouspillou et al. 2015). Interestingly, in addition to depressed muscle contractility, muscles from Park2−/− mice recapitulate some of the mitochondrial phenotypes seen in aged skeletal muscles and in muscles from animals treated with chemotherapy. Similar to muscles from Park2−/− mice, aged skeletal muscle mitochondria display a sensitized mPTP and impaired mitochondrial energetics (Marcinek et al. 2005; Gouspillou et al. 2014a; Gouspillou et al. 2014b), whereas muscle from chemotherapy treated mice displays a severe decrease in mitochondrial respiration (Gouspillou et al. 2015). Although a decrease in Parkin expression is probably not the only contributor to mitochondrial and muscle dysfunction seen with normal ageing and chemotherapy treatment, our data suggest that it could represent a significant underlying mechanism.
In summary, the present study shows that Parkin is essential for the maintenance of normal mitochondrial function in skeletal muscles and that Parkin ablation leads to mild impairment in skeletal muscle contractility, as well as significant impairment of mitochondrial function. Our results thus suggest that Parkin could be targeted to improve muscle and mitochondrial health.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
GG, YB and SNAH conceived the project and designed the experiments. GG, RG, JP, MP, FMPS, MM, JM and NS performed the experiments. All authors contributed to data analyses and interpretation. GG wrote the first draft of the manuscript. All authors revised the manuscript and approved the final version submitted for publication. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by grants from the Canadian Institute of Health Research (MOP‐93760) awarded to Sabah N. A. Hussain and Yan Burelle and by grants from the Natural Sciences and Engineering Council of Canada (NSERC) awarded to Gilles Gouspillou (#RGPIN‐2014‐04668). Gilles Gouspillou is also supported by a FQRS Chercheur Boursier Junior 1 salary award (FRQS‐35184). The funders had no role in the study design, data collection and analysis, the decision to publish, or the preparation of the manuscript.
Biography
Gilles Gouspillou received a PhD in cell biology in 2010 from Bordeaux 2 University under the supervision of Drs P. Diolez and I. Bourdel‐Marchasson. He then joined Dr R. T. Hepple's lab (McGill University) in 2011 as a postdoctoral fellow. During his training, his research focused on investigating the role played by mitochondrial dysfunction in the muscle ageing process. In 2013, he was recruited by the Département des sciences de l'activité physique at UQAM as an assistant professor, where he now studies the role played by mitochondrial dynamics and mitophagy in muscle health, disease and ageing.

Edited by: Michael Hogan & Anne McArdle
This is an Editor's Choice article from the 1 July 2018 issue.
References
- Balogun S, Winzenberg T, Wills K, Scott D, Jones G, Aitken D & Callisaya ML (2017). Prospective associations of low muscle mass and function with 10‐year falls risk, incident fracture and mortality in community‐dwelling older adults. J Nutr Health Aging 21, 843–848. [DOI] [PubMed] [Google Scholar]
- Bernardi P & Bonaldo P (2013). Mitochondrial dysfunction and defective autophagy in the pathogenesis of collagen VI muscular dystrophies. Cold Spring Harbor perspectives in biology 5, a011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW & Sheu SS (2004). Calcium, ATP, and ROS: a mitochondrial love‐hate triangle. Am J Physiol Cell Physiol 287, C817–C833. [DOI] [PubMed] [Google Scholar]
- Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R & Sandri M (2014). Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep 8, 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A & Hood DA (2008). Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 7, 2–12. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S & Chan DC (2011). Broad activation of the ubiquitin‐proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Gao F, Li B, Wang H, Xu Y, Zhu C & Wang G (2010). Parkin mono‐ubiquitinates Bcl‐2 and regulates autophagy. J Biol Chem 285, 38214–38223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit LE, Scholte HR & Sluiter W (2008). Correct assay of complex I activity in human skin fibroblasts by timely addition of rotenone. Clin Chem 54, 1921–1922. [DOI] [PubMed] [Google Scholar]
- Dirks AJ & Leeuwenburgh C (2004). Aging and lifelong calorie restriction result in adaptations of skeletal muscle apoptosis repressor, apoptosis‐inducing factor, X‐linked inhibitor of apoptosis, caspase‐3, and caspase‐12. Free Radic Biol Med 36, 27–39. [DOI] [PubMed] [Google Scholar]
- Drummond MJ, Addison O, Brunker L, Hopkins PN, McClain DA, LaStayo PC & Marcus RL (2014). Downregulation of E3 ubiquitin ligases and mitophagy‐related genes in skeletal muscle of physically inactive, frail older women: a cross‐sectional comparison. J Gerontol A Biol Sci Med Sci 69, 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner N, Sliziuk V, Scherbakov N & Sandek A (2015). Muscle wasting in ageing and chronic illness. ESC Heart Fail 2, 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH & Taanman JW (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin‐dependent manner upon induction of mitophagy. Hum Mol Genet 19, 4861–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauser L, Sonnay S, Stafa K & Moore DJ (2011). Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem 118, 636–645. [DOI] [PubMed] [Google Scholar]
- Godin R, Daussin F, Matecki S, Li T, Petrof BJ & Burelle Y (2012). Peroxisome proliferator‐activated receptor γ coactivator1‐α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post‐necrotic mdx mouse skeletal muscle. J Physiol 590, 5487–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Bourdel‐Marchasson I, Rouland R, Calmettes G, Biran M, Deschodt‐Arsac V, Miraux S, Thiaudiere E, Pasdois P, Detaille D, Franconi JM, Babot M, Trezeguet V, Arsac L & Diolez P (2014a). Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell 13, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Scheede‐Bergdahl C, Spendiff S, Vuda M, Meehan B, Mlynarski H, Archer‐Lahlou E, Sgarioto N, Purves‐Smith FM, Konokhova Y, Rak J, Chevalier S, Taivassalo T, Hepple RT & Jagoe RT (2015). Anthracycline‐containing chemotherapy causes long‐term impairment of mitochondrial respiration and increased reactive oxygen species release in skeletal muscle. Sci Rep 5, 8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Kapchinsky S, Purves‐Smith F, Norris B, Pion CH, Barbat‐Artigas S, Lemieux F, Taivassalo T, Morais JA, Aubertin‐Leheudre M & Hepple RT (2014b). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J 28, 1621–1633. [DOI] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Norris B, Barbat‐Artigas S, Aubertin‐Leheudre M, Morais JA, Burelle Y, Taivassalo T & Hepple RT (2014c). The relationship between muscle fiber type‐specific PGC‐1alpha content and mitochondrial content varies between rodent models and humans. PLoS One 9, e103044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP (2009). What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46, 821–831. [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J & Harper JW (2015). The PINK1‐PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepple RT (2014). Mitochondrial involvement and impact in aging skeletal muscle. Front Aging Neurosci 6, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood DA, Tryon LD, Carter HN, Kim Y & Chen CC (2016). Unravelling the mechanisms regulating muscle mitochondrial biogenesis. Biochem J 473, 2295–2314. [DOI] [PubMed] [Google Scholar]
- Itier JM, Ibanez P, Mena MA, Abbas N, Cohen‐Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L, Ret G, Joubert C, Periquet M, Araujo F, Negroni J, Casarejos MJ, Canals S, Solano R, Serrano A, Gallego E, Sanchez M, Denefle P, Benavides J, Tremp G, Rooney TA, Brice A & Garcia de Yebenes J (2003). Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet 12, 2277–2291. [DOI] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP & Youle RJ (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191, 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Varadhachary AS, Miller SE & Weihl CC (2010). Quantitation of ‘autophagic flux’ in mature skeletal muscle. Autophagy 6, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L & Brenner C (2007). Mitochondrial membrane permeabilization in cell death. Physiol Rev 87, 99–163. [DOI] [PubMed] [Google Scholar]
- Leduc‐Gaudet JP, Picard M, St‐Jean Pelletier F, Sgarioto N, Auger MJ, Vallee J, Robitaille R, St‐Pierre DH & Gouspillou G (2015). Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6, 17923–17937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcil M, Ascah A, Matas J, Belanger S, Deschepper CF & Burelle Y (2006). Compensated volume overload increases the vulnerability of heart mitochondria without affecting their functions in the absence of stress. J Mol Cell Cardiol 41, 998–1009. [DOI] [PubMed] [Google Scholar]
- Marcinek DJ, Schenkman KA, Ciesielski WA, Lee D & Conley KE (2005). Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J Physiol 569, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM & Fon EA (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, Blaauw B, DePinho RA & Sandri M (2015). Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 6, 6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH & Powers SK (2011). Mitochondrial‐targeted antioxidants protect skeletal muscle against immobilization‐induced muscle atrophy. J Appl Physiol (1985) 111, 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardin A, Schrepfer E & Ziviani E (2016). Counteracting PINK/Parkin deficiency in the activation of mitophagy: a potential therapeutic intervention for Parkinson's disease. Curr Neuropharmacol 14, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP & Youle RJ (2011). Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal 14, 1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Csukly K, Robillard ME, Godin R, Ascah A, Bourcier‐Lucas C & Burelle Y (2008). Resistance to Ca2+‐induced opening of the permeability transition pore differs in mitochondria from glycolytic and oxidative muscles. Am J Physiol Regul Integr Comp Physiol 295, R659–R668. [DOI] [PubMed] [Google Scholar]
- Picard M, Taivassalo T, Gouspillou G & Hepple RT (2011). Mitochondria: isolation, structure and function. J Physiol 589, 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquereau J, Godin R, Deschenes S, Bessi VL, Mofarrahi M, Hussain SN & Burelle Y (2013). Protective role of PARK2/Parkin in sepsis‐induced cardiac contractile and mitochondrial dysfunction. Autophagy 9, 1837–1851. [DOI] [PubMed] [Google Scholar]
- Rana A, Rera M & Walker DW (2013). Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci U S A 110, 8638–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool S, Soya N, Truong L, Croteau N, Lukacs GL & Trempe JF (2018). PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep 19, e44981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello V & Sandri M (2015). Mitochondrial quality control and muscle mass maintenance. Front Physiol 6, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AP, Foletta VC, Snow RJ & Wadley GD (2014). Skeletal muscle mitochondria: a major player in exercise, health and disease. Biochim Biophys Acta 1840, 1276–1284. [DOI] [PubMed] [Google Scholar]
- Sandri M (2013). Protein breakdown in muscle wasting: role of autophagy‐lysosome and ubiquitin‐proteasome. Int J Biochem Cell Biol 45, 2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlagowski AI, Singh F, Charles AL, Gali Ramamoorthy T, Favret F, Piquard F, Geny B & Zoll J (2014). Mitochondrial uncoupling reduces exercise capacity despite several skeletal muscle metabolic adaptations. J Appl Physiol (1985) 116, 364–375. [DOI] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL & Dawson TM (2011). PARIS (ZNF746) repression of PGC‐1alpha contributes to neurodegeneration in Parkinson's disease. Cell 144, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA & McBride HM (2012a). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22, 135–141. [DOI] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA & McBride HM (2012b). Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 7, e52830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G & Shirihai OS (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal 14, 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ & Pallanck LJ (2013). The PINK1‐Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A 110, 6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J & Chen Q (2011). Parkin ubiquitinates Drp1 for proteasome‐dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 286, 11649–11658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Liu L & Chen Q (2015). Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim Biophys Acta 1853, 2784–2790. [DOI] [PubMed] [Google Scholar]
- Wolfe RR (2006). The underappreciated role of muscle in health and disease. Am J Clin Nutr 84, 475–482. [DOI] [PubMed] [Google Scholar]
- Yang J (2014). Enhanced skeletal muscle for effective glucose homeostasis. Prog Mol Biol Transl Sci 121, 133–163. [DOI] [PubMed] [Google Scholar]
- Yu W, Sun Y, Guo S & Lu B (2011). The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet 20, 3227–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]