

Abstract
Background:
Thyroid cancer is the most common endocrine tumor. Our previous studies have demonstrated that curcumin can induce apoptosis in human papillary thyroid carcinoma BCPAP cells. However, the underlined mechanism has not been clearly elucidated. Endoplasmic reticulum (ER) is a major organelle for synthesis, maturation, and folding proteins as well as a large store for Ca2+. Overcoming chronically activated ER stress by triggering pro-apoptotic pathways of the unfolded protein response (UPR) is a novel strategy for cancer therapeutics. Our study aimed to uncover the ER stress pathway involved in the apoptosis caused by curcumin.
Methods:
BCPAP cells were treated with different doses of curcumin (12.5–50 μM). Annexin V/PI double staining was used to determine cell apoptosis. Rhod-2/AM calcium fluorescence probe assay was performed to measure the calcium level of endoplasmic reticulum. Western blot was used to examine the expression of ER stress marker C/EBP homologous protein 10 (CHOP) and glucose-regulated protein 78 (GRP78). X-box binding protein1 (XBP-1) spliced form was examined by reverse transcriptase-polymerase chain reaction (RT-PCR).
Results:
Curcumin significantly inhibited anchorage-independent cell growth and induced apoptosis in BCPAP cells. Curcumin induced ER stress and UPR responses in a dose- and time-dependent manner, and the chemical chaperone 4-phenylbutyrate (4-PBA) partially reversed the antigrowth activity of curcumin. Moreover, curcumin significantly increased inositol-requiring enzyme 1α (IRE1α) phosphorylation and XBP-1 mRNA splicing to induce a subsets of ER chaperones. Increased cleavage of activating transcription factor 6 (ATF6), which enhances expression of its downstream target CHOP was also observed. Furthermore, curcumin induced intracellular Ca2+ influx through inhibition of the sarco-endoplasmic reticulum ATPase 2A (SERCA2) pump. The increased cytosolic Ca2+ then bound to calmodulin to activate calcium/calmodulin-dependent protein kinase II (CaMKII) signaling, leading to mitochondrial apoptosis pathway activation. Ca2+ chelator BAPTA partially reversed curcumin-induced ER stress and growth suppression, confirming the possible involvement of calcium homeostasis disruption in this response.
Conclusions:
Curcumin inhibits thyroid cancer cell growth, at least partially, through ER stress-associated apoptosis. Our observations provoked that ER stress activation may be a promising therapeutic target for thyroid cancer treatment.
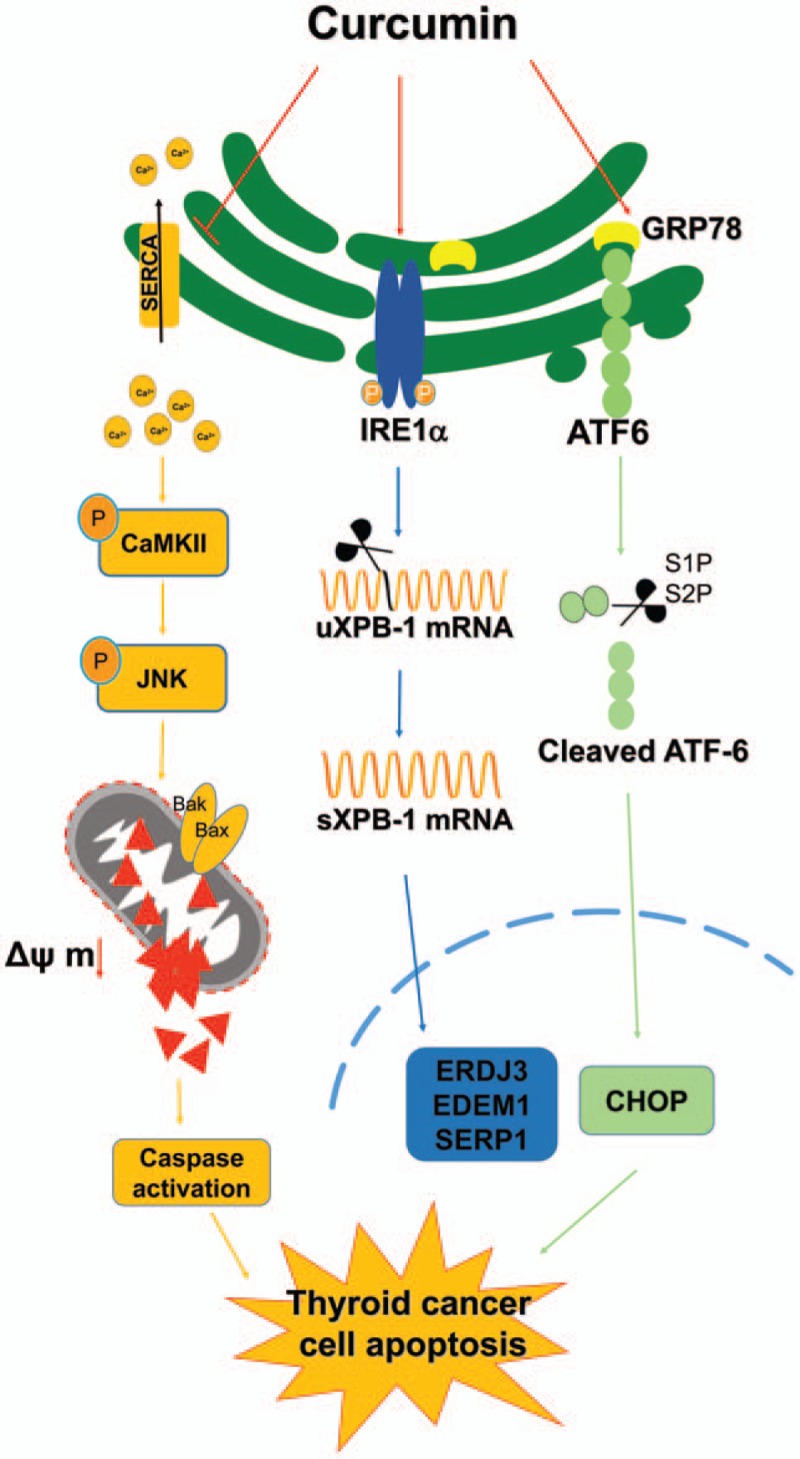
Keywords: apoptosis, curcumin, endoplasmic reticulum stress, human papillary thyroid carcinoma, intracellular calcium homeostasis
1. Introduction
Thyroid cancer is the most frequent malignancy of the endocrine system and its incidence is increasing worldwide.[1] Papillary thyroid carcinoma (PTC) is the most prevalent type, accounting for about 80% of the cases in thyroid cancer. Conventional therapies for well-differentiated thyroid cancer include surgical resection, radioactive iodine, and thyroid hormone suppression therapy.[2] Although outcomes for patients with differentiated thyroid cancer are excellent in comparison with other human cancers, advanced thyroid cancer patients such as those with radioiodine-refractory and metastatic differentiated thyroid cancer or anaplastic thyroid cancer still lack effective treatment options. Therefore, searching for an efficient method or agent is a requirement for the therapy of the cancer.
Curcumin (diferuloylmethane), a major active component of turmeric (Curcuma longa), is a natural polyphenolic compound. A large number of previous studies demonstrated that curcumin has anti-proliferative, antioxidant, anti-inflammatory, apoptosis-inducing, and anti-tumor properties in various cancer types.[3] Moreover, it has been reported that curcumin is involved in the regulation of endoplasmic reticulum (ER) stress in certain cells. However, the role of curcumin participating in ER stress regulation appears to be highly cell type and context dependent. Curcumin inhibits lipolysis via suppression of ER stress in adipose tissue and prevents hepatic insulin resistance.[4] However, in human colon cancer cells, bis-Dehydroxy-curcumin, a curcumin derivative, activates ER stress to induce cell death.[5] Another study has shown that curcumin induced apoptotic cell death via increasing ER stress and mitochondrial dysfunction in CD4+ T cells.[6] Besides, the potential benefits of targeting the endoplasmic reticulum unfolded protein response (ER-UPR) as a therapeutic strategy for thyroid cancer therapy is also explored. HNHA, a dominant HDAC inhibitor, caused apoptosis of thyroid cancer cells both in vitro and in vivo owing to its induction of ER stress.[7] Prior results in our laboratory confirmed that curcumin inhibited cell growth and induced the ER stress in the papillary thyroid carcinoma cells via activating the activating transcription factor 6 (ATF6)/X-box binding protein1 (XBP-1) pathway.[8] Nevertheless, to our knowledge, the underlying mechanism of the anticancer effect of curcumin on thyroid cancer cells needs to be further investigated.
ER stress is a common feature of several physiological and pathological conditions affecting the function of the protein secretory pathway. Under ER stress, the unfolded protein response (UPR), an adaptive response, was activated to reduce the misfolded or unfolded protein load. However, under irreversible ER stress, UPR shifts its signaling toward cell death by activating complex pro-apoptotic programs.[9] The mammalian UPR activates 3 principal signal transducers, RNA-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and ATF6. Under ER stress, PERK undergoes oligomerization and autophosphorylation and then induces eukaryotic initiation factor 2α (eIF2α) phosphorylation causing translational arrest. Phosphorylated eIF2α allows the selective translation of activating transcription factor 4 (ATF4). ATF4 induces C/EBP homologous protein 10 (CHOP) that in turn upregulates the expression of several pro-apoptotic proteins such as growth arrest and DNA damage protein 34 (GADD34), endoplasmic oxidoreductin 1 like protein (ERO1α), and BH3-only proteins, including Bcl-2-like Protein 11 (BIM), Bcl-2 binding component 3 (PUMA) and Phorbol-12-myristate-13-acetate-induced protein 1 (NOXA).[10] The most conserved branch of the UPR is mediated by IRE1, which is activated by autophosphorylation. Active IRE1 with endoribonuclease activity processes the mRNA encoding for X-box binding protein1 (XBP-1), producing an active transcription factor termed XBP1s. XBP1s induces the transcription of genes implicated in protein folding and quality control. ATF6 translocates to the Golgi where it is cleaved by the proteases, releasing a transcription factor that modulates the expression of chaperones and enzymes required for ER function.[11]
ER contains high concentration of Ca2+. It has been reported that a substantial burst of intracellular Ca2+ release was the primary event that triggered cell apoptosis.[12] Excessive Ca2+ accumulation in the mitochondrial matrix causes mitochondrial swelling, the dissipation of the mitochondrial membrane potential (ΔΨm), production of mitochondrial reactive oxygen species (ROS), rupture of the outer mitochondrial membrane, and subsequent release of cytochrome c (Cyt c).[13]
In the present study, we showed that curcumin induced ER stress in BCPAP cells characterized by the activation of IRE1-XBP1 axis and ATF6-CHOP pathway. Our results also provide evidences for the SERCA2-Ca2+-CaMKII-JNK pathway as a central mechanism of curcumin-induced intracellular Ca2+ homeostasis disruption and apoptosis in human papillary thyroid carcinoma BCPAP cells.
2. Materials and methods
Ethical approval was not necessary because no patients’ information was collected according to Ethics Committee of Jiangsu Institute of Nuclear Medicine.
2.1. Reagents and chemicals
Curcumin (catalog No. C7727, purity ≥94%), 4-phenylbutyrate (4-PBA, purity >98%), Hoechst 33342, Rhodamine 123 were all purchased from Sigma–Aldrich (St Louis, MO). Propidium iodide (PI) was purchased from Enzo Life Sciences (Farmingdale, NY). DMSO (dimethyl sulfoxide) and MTT (3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) were purchased from Sangon (Shanghai, China). SP600125 and Lipo 6000 were purchased from Beyotime Institute of Biotechnology (Kumamoto, Japan). Rhod-2/AM was purchased from Dojindo Molecular Technologies. ER-Tracker was purchased from Beyotime Biotechnology (China). 1,2-bis (2-aminophenoxy) ethane N,N,N′,N′-tetraacetic acid acetoxymethyl ester (BAPTA-AM) was purchased from Adooq Bioscience (Irvine, CA). Low melting point agarose was purchased from Takara (QingDao, China). Annexin V-FITC/PI apoptosis detection kit was purchased from BD Biosciences (San Jose, CA). Ca2+-ATPase kit was provided by Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The antibodies used were as follows: anti-ATF6 (sc-22799), anti-XBP-1 (sc-7160), anti-CaMKII (sc-9035), anti-CHOP (sc-7351), anti-PARP (sc-7150), anti-β-actin (sc-47778), and goat anti-mouse (sc-2005), or rabbit (sc-2004) IgG-HRP were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-SERCA2 (cst-9580), anti-p-JNK (T183/Y185, cst-4668), anti-Bax (cst-2772), anti-Bid (cst-2002), anti-caspase-8 (cst-9746), anti-caspase-7 (cst-9492), and anti-caspase-3 (cst-9665) were purchased from Cell Signaling Technology (Beverly, MA). Anti-p-CaMKII (T286, #3891) was purchased from EPITOMICS. Anti-p-IRE1α (Ser724, NB100-2323) was purchased from Novus Biologicals (Littleton, CO). Anti-IRE1α (A1601), anti-JNK (AJ518-1), anti-Bcl-2 (AB126-1), and anti-Bcl-XL (AB112-1) were purchased from Beyotime Institute of Biotechnology. Other chemicals were analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
2.2. Cell culture
BCPAP cells were obtained from the German Collection of Micro-organisms and Cell Cultures (Braunschweig, Germany) and maintained in a complete RPMI 1640 medium (Gibco, NY) with 10% calf serum (CS; Sijiqing, Hangzhou, China), 100 U/mL penicillin, and 100 U/mL streptomycin. Cells were cultured in a humid atmosphere (5% CO2, 37 °C).
2.3. Drug treatments
Curcumin was dissolved in DMSO to yield 50 mM stock solutions and stored frozen until use. Unless mentioned otherwise, BCPAP cells were treated with different concentrations of curcumin (12.5–50 μM) for 24 hours. DMSO was used as the solvent control (SC). BAPTA-AM was stored in DMSO at 40 mM and diluted to a final concentration of 0.01 to 10 μM for use. To be pointed out, the final DMSO concentration did not exceed 0.1% (v/v) in any experiment. 4-PBA was directly dissolved in culture medium at a concentration of 2.5 to 10 mM when used.
2.4. Soft agar colony formation assay
For analysis of anchorage-independent cell growth in vitro, soft agar colony formation assay was performed using a 2-layer soft agar system as described previously with some modifications.[14] Briefly, BCPAP cells pretreated with different dosages of curcumin for 6 hours were seeded into a 24-well agar plates containing an agar base layer. The cells (1 × 104 cells per well) suspended in 0.6 mL of top agar (0.6% low melting point agarose in 2 × RPMI 1640 complete cell culture medium) were overlaid onto a layer of 0.6 mL of bottom agar (1.2% low melting point agarose in the same culture medium). After 14 days incubation, the colonies were stained with 0.02% (w/v) crystal violet. The images were captured with the GIS-2019 system (Tanon, Shanghai, China) and the relative colony formation ability was calculated.
2.5. Measurement of mitochondrial membrane potential
Mitochondrial membrane was monitored using the fluorescent dye Rhodamine 123, which preferentially partitions into active mitochondria based on the highly negative mitochondrial membrane potential.[15] Depolarization of mitochondrial membrane potential results in the loss of Rhodamine 123 from the mitochondria and a decrease in intracellular fluorescence. After cultured cells were treated as indicated, cells were probed with Rhodamine 123 (10 μM) for 15 minutes at 37 °C and then washed twice with PBS for immediate detection. Fluorescence of 10,000 cells per sample was determined by flow cytometry (FACSCalibur, Becton Dickinson, CA) using the FL1 channel.
2.6. Cell apoptosis assay
For analysis of apoptosis, cells exposed to different dosages of curcumin for 24 hours were harvested. The cell pellet was then washed twice with cold PBS. An Annexin V-FITC and PI apoptosis detection kit (BD Biosciences) was used according to the manufacturer's directions. Briefly, cells were resuspended at 1 × 106 cells/mL in 100 μL of binding buffer. Annexin V-FITC (5 μL) and 10 μL of PI (20 μg/mL) were added to the cell suspension followed by incubation at room temperature for 15 minutes in the dark and then mixed with 400 μL of binding buffer. A total of 1 × 104 cells/sample was analyzed on the FACSCalibur.
2.7. Measurement of ER size
Cultured cells were treated as indicated and the medium was removed from the culture dish. Then cells were rinsed with PBS, added ER-Tracker staining solution (1 μM) and incubated for 20 minutes at 37 °C. The cells were then pelleted by centrifugation and resuspended in PBS for flow cytometry analysis.
2.8. Hoechst 33342/PI staining assay
BCPAP cells were seeded in 96-well plates at a density of 1 × 104 cells/well and cultured overnight. Then the cells were pretreated with or without 4-PBA (10 mM) for 1 hour followed by curcumin (50 μM) treatment for additional 24 hours. After incubation, the cells were stained with a combination of Hoechst 33342 (10 μg/mL) and PI (10 μg/mL) at 37 °C for 15 minutes. The cell images were captured using a fluorescence microscope (Olympus, X51, Japan). Cell were counted for five independent microscopic fields per well. Cell death rate (%) = (PI-positive staining cell number/total cell number) × 100%.
2.9. Western blotting
After BCPAP cells were exposed to different treatments, cells were collected and washed with PBS. The whole cell proteins were extracted by ice-cold lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 2 mM ethylenediaminetetraacetic acid [EDTA], 1% (w/v) Nonidet P-40, 0.02% (w/v) sodium azide) with supplemented protease inhibitors (1 mM phenylmethylsul-fonyl fluoride, PMSF). The protein concentration was measurement by Bradford assay. Equal amounts of protein were separated by 10% or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then electrotransferred onto a nitrocellulose membrane (Millipore). Then the membranes were incubated with the indicated primary antibodies overnight at 4 °C, followed by appropriate HRP-conjugated secondary antibody incubation for additional 1 hour. After that, the membranes were washed three times for 5 minutes with TBST (TBS containing 0.1% Tween 20). Finally, the membranes were incubated with ECL substrate solution (Beyotime Biotechnology, China) and visualized with autoradiography film.
2.10. Reverse transcriptase-polymerase chain reaction (RT-PCR)
Cells were collected after treated with curcumin, then whole cell RNA was extracted by the Trizol reagent (Life Technologies, Inc.). M-MLV reverse transcriptase and oligo(dT)18 primers were used to synthesized cDNA as the manufacturer's instructions. The primer sequences were as follows: XBP-1 forward: 5′- CCTTGTAGTTGAGAACCAGG-3′ and XBP-1 reverse: 5′- GGGGCTTGGTATATATGTGG-3′; ERDJ3 forward: 5′-CCCTGATGATCCACAAGC-3′, and ERDJ3 reverse: 5’′-ATTCGTCGCAGACCACCT-3′; EDEM1 forward: 5′-GCCTCCTTTCTGCTCACA-3′ and EDEM1 reverse: 5′- CACTCTGCTTTCCAACCC-3′; SERP1 forward: 5′-ATGGTCGCCAAGCAAAGG-3′ and SERP1 reverse: 5′- TCACATGCCCATCCTGAT-3′; CHOP forward: 5′-ACCAGGAAACGGAAACAG-3′ and CHOP reverse: 5′-TGCGTATGTGGGATTGAG-3′; GRP78 forward: 5′-TCAGGGCAACCGCATCAC-3′ and GRP78 reverse: 5′-CGCCACCCAGGTCAAACA-3′; ACTIN forward: 5′-GCCGGGACCTGACTGACTAC-3′ and ACTIN reverse: 5′-CGGATGTCCACGTCACACTT-3′. The PCR was performed using an initial step of denaturation at 95 °C for 5 minutes, with 30 cycles of amplification at 95 °C for 30 seconds, annealing at 55 to 60 °C (depending on the sequences of the primers) for 30 seconds, elongation at 72 °C for 30 seconds, and extension at 72 °C for 5 minutes. The PCR products were electrophoresed in 1.5% agarose gel and visualized by ethidium bromide (EB) dying. The relative expression was quantified densitometrically using the GIS-2019 system (Tanon, Shanghai, China), and calculated according to the reference bands of Actin.
2.11. Intracellular calcium measurement
Rhod 2-AM, a Ca2+-sensitive indicator, was used to detect intracellular calcium levels according to the manufacturer's protocol using calcium-free media. Briefly, BCPAP cells were preincubated in the presence or absence of a calcium chelating agent, BAPTA-AM, followed by curcumin treatment for 24 hours. The cells were subsequently loaded with Rhod-2/AM (5 μM) at 37 °C for 45 minutes in the dark and gently washed twice with D-hank's solution (calcium-free media). Rhod-2 binds cytoplasmic free calcium and emits a red fluorescence, which was analyzed by flow cytometry through FL2 channel.
2.12. Cell viability assay
Cell viability was assessed by the MTT assay which was performed as previously reported.[16]
2.13. Measurement of Ca2+-ATPase activity
The sarco-endoplasmic reticulum ATPase (SERCA) activity was measured by a Ca2+-ATPase assay kit according to the manufacturer's protocols. Inorganic phosphate (Pi), which was generated from hydrolysis of ATP by ATPase, can be measured by a simple colorimetric reaction. Ca2+-ATPase activities were expressed in units of micromolar molecular inorganic phosphate produced per milligram of the extracted cell protein per hour (μM Pi/mg protein/h).
2.14. Construction and transfection of human CHOP shRNA plasmids
pSGU6/GFP/Neo was modified to express an shRNA from the U6 promoter. Briefly, an oligonucleotide was synthesized that consisted of a sequence specific 21 nucleotide stretch designed to target the CHOP (NM_001195053.1) followed by a 2 nt overhang, a loop sequence, and finally the reverse complement of the targeting sequence. Hind III and Bbs I cloning sites were added to facilitate directional cloning immediately downstream of the U6 promoter. The shRNA sequences directed against human CHOP were as follows: 5′-GCGCATGAAGGAGAAAGAACAGG-3′ (shRNA-CHOP 1#); 5′-GAGAAAGAACAGGAGAATGAAAG-3′ (shRNA-CHOP 2#); 5′-ATGAACGGCTCAAGCAGGAAATC-3′ (shRNA-CHOP 3#). The control scrambled shRNA was constructed by the insertion of a similar structure but encoding a nonsense minigene with no homology to any known sequences in human and mouse genomes. The sequences for scramble shNC are as follows: 5′-GTTCTCCGAACGTGTCACGT-3′. Cells were transfected with plasmids by Lipo 6000 transfection reagent according to the manufacturer's instructions.
2.15. Statistical analysis
All the quantifications are expressed as mean ± S.D. from at least 3 independent biological replicates. Statistical evaluations were performed with the Student t test when 2 value sets were compared. P ≤ .05 was considered as statistically significant.
3. Results
3.1. Curcumin inhibits anchorage-independent growth of human papillary thyroid carcinoma BCPAP cells
Firstly, soft agar assays were performed to analyze the influence of curcumin on anchorage-independent growth (colony formation) of BCPAP cells. As shown in Fig. 1A, curcumin treatment resulted in a dose-dependent reduction of colony numbers of BCPAP cells after 14 days of culture, where >90% decrease in colony formation was observed with curcumin at concentrations of 25 or 50 μM. These results indicate that curcumin has a potent inhibitory effect on the growth and proliferation of BCPAP cells.
Figure 1.
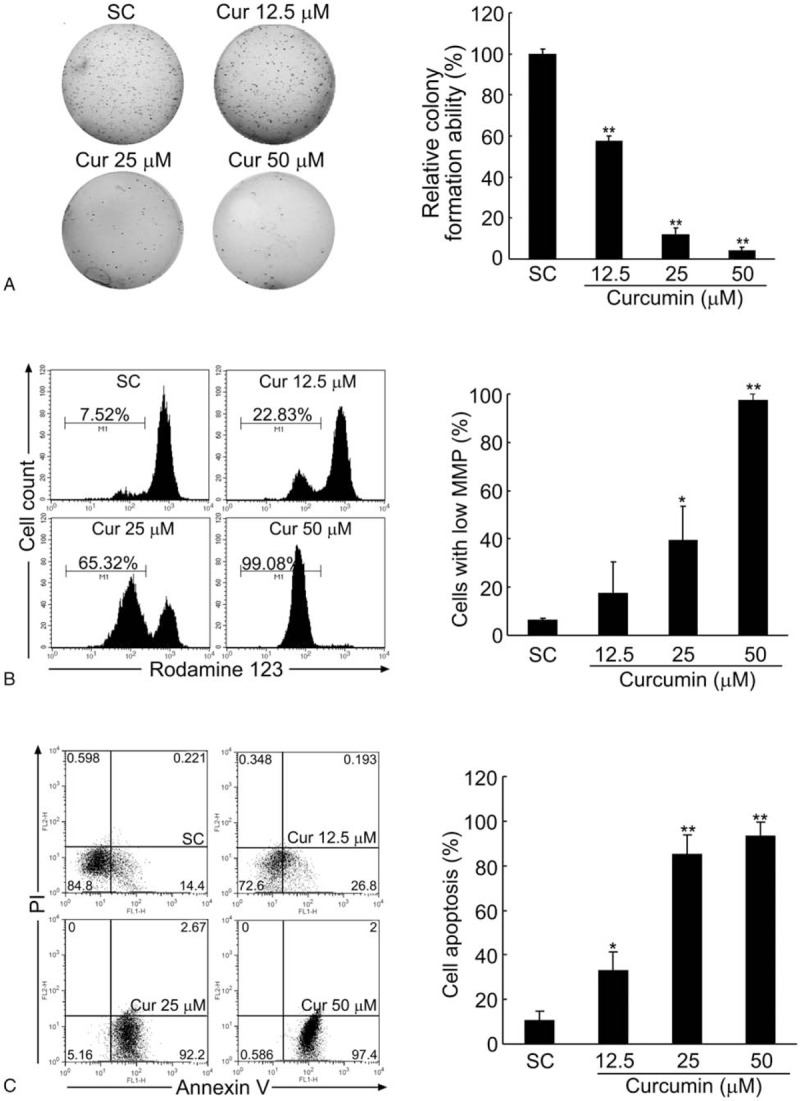
Curcumin inhibits the colony formation and induces the apoptosis of BCPAP cells. (A) Curcumin inhibits the colony formation ability of BCPAP cells in a dose-dependent manner. BCPAP cells were treated with different doses (12.5–50 μM) of curcumin, and the colony formation ability was measured by soft agar assay. (B) Curcumin decreases the mitochondrial membrane potential (MMP) of BCPAP cells. After treated by different doses (12.5–50 μM) of curcumin for 24 hours, MMP of BCPAP cells were determined by flow cytometry using Rhodamine 123 fluorescent dye. (C) Curcumin induces the apoptosis of BCPAP cells. BCPAP cells were treated with different doses (12.5–50 μM) of curcumin for 24 hours, and the apoptotic cells were determined by Annexin V-FITC/PI analysis. Percentage of living cells (lower left: Annexin V-FITC−/PI−), early apoptosis cells (lower right: Annexin V-FITC+/PI−), and late apoptosis cells (upper right: Annexin V-FITC+/PI+) were showed in the flow cytometry chart. All data shown represent the means ± S.D. of 3 independent experiments. SC = solvent control. ∗P < .05, ∗∗P < .01.
3.2. Curcumin decreases the mitochondrial membrane potential (MMP) and induces apoptosis in BCPAP cells
Reduced mitochondrial membrane potential (MMP, ΔΨm) is considered as an initial and irreversible step towards apoptosis. As shown in Fig. 1B, when BCPAP cells were treated with different doses of curcumin (12.5–50 μM) for 24 hours, the MMP was rapidly reduced, which was detected by the weakening of the fluorescence intensity of a mitochondrial specific probe, Rhodamine 123. Compared with the solvent control group which only had 6.48 ± 0.53% cells with low MMP, at 25 and 50 μM of curcumin treatment, this percent dramatically increased up to 39.47 ± 14.05% and 97.27 ± 2.72%, respectively. To determine whether the cell death was apoptotic, BCPAP cells were stained with Annexin V-FITC/PI followed by flow cytometry analysis. As illustrated in Fig. 1C, within the concentrations ranging from 12.5, 25 to 50 μM, there was a significant increase in cell apoptotic rates from 33.26 ± 7.98% to 85.33 ± 8.49% and 93.31 ± 6.05%, respectively, compared with the solvent control group (10.67 ± 3.96%). These results indicate that curcumin induces cell apoptosis in BCPAP cells.
3.3. Curcumin induces ER expansion in BCPAP cells
Perturbations in ER function, a process named ER stress, plays a crucial role in cell apoptosis.[17] Previous study reported that ER stress induced ER membrane expansion.[18] In order to determine whether curcumin can induce the ER stress-related ER expansion, the ER size was measured by flow cytometry using ER-Tracker dyes. Compared with the control cells, BCPAP cells exposed to 50 μM of curcumin exhibited a significant enhanced fluorescence intensity of ER-Tracker dye from 1.03 ± 0.64% to 66.79 ± 10.56%, indicating an expansion of ER size (Fig. 2A). As expected, pretreatment with 4-phenylbutyrate (4-PBA), which is a chemical chaperone that alleviates ER stress, ameliorated the ER expansion induced by curcumin in BCPAP cells. Compared with that treated with 50 μM of curcumin only, the percent of cells with expansive ER size in BCPAP cells treated with a combination of 50 μM curcumin and 10 mM 4-PBA reduced from 63.46% to 46.12% (Fig. 2B). These results indicate that curcumin probably induces ER stress which is characterized by ER expansion. In order to further investigate whether the death of BCPAP cells is attributed to ER stress induced by curcumin, the effect of 4-PBA on curcumin-induced cell death was measured by Hoechst/PI staining. As expected, a combination with 4-PBA significantly reduced PI-positive cell numbers compared with that of curcumin treated-alone and the cell death rate of BCPAP cells was correspondingly decreased from 55.68 ± 1.83% to 43.47 ± 2.61% (Fig. 2C). Taken together, these data demonstrate that curcumin-mediated ER stress may account, at least in part, for the cell death of BCPAP cells.
Figure 2.
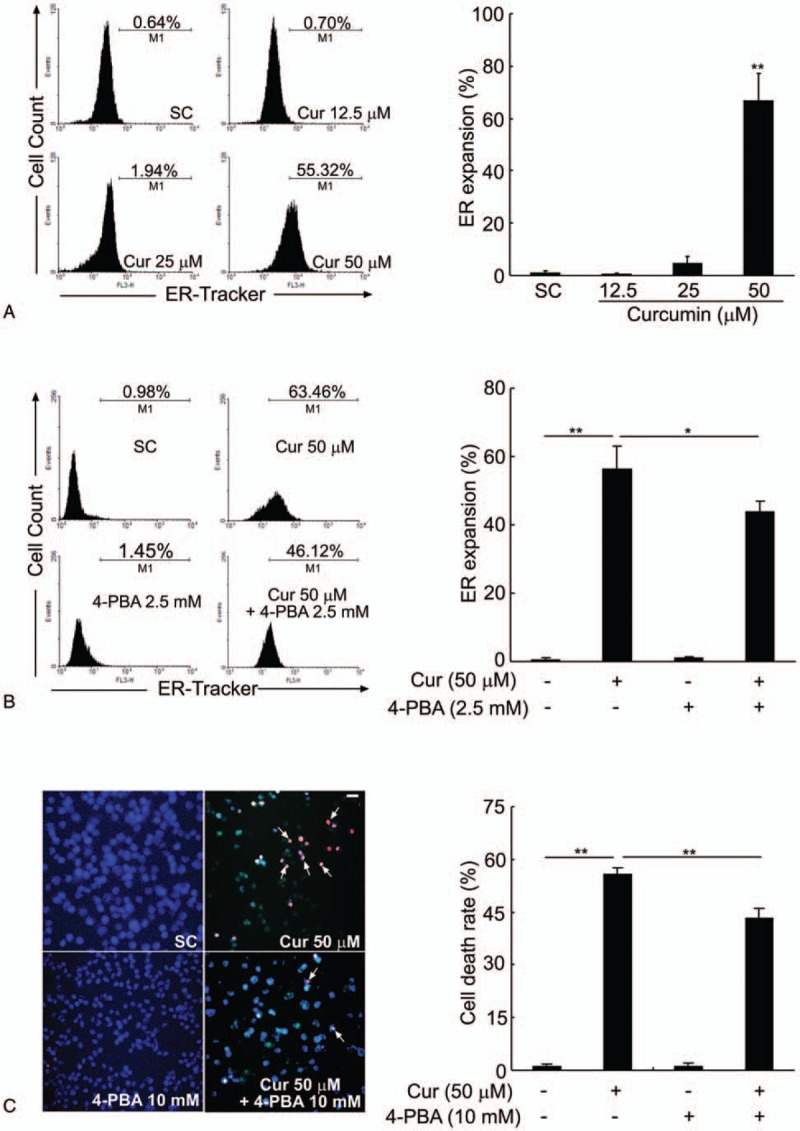
Curcumin induces ER expansion in BCPAP cells. (A) Curcumin enlarges ER size in BCPAP cells. After treatment with different doses of curcumin (12.5–50 μM), the ER size of BCPAP cells was measured by flow cytometry analysis using the ER-Tracker dyes. (B) ER stress inhibitor, 4-PBA, partially reverses the ER expansion induced by curcumin in BCPAP cells. Cells were pretreated with or without 2.5 mM of 4-PBA for 1 hour, followed by exposure to 50 μM of curcumin for another 24 hours. The degree of ER expansion was detected by flow cytometry. (C) 4-PBA ameliorates the cell death induced by curcumin. BCPAP cells were pretreated with or without 10 mM of 4-PBA for 1 hour before incubation with 50 μM of curcumin for another 24 hours. Representative images of Hoechst/PI staining were shown and white arrows indicated PI-positive cells. Scale bar, 10 μm. The cell death rate is expressed as percentage of PI-positive staining cells. All data shown represent the means ± S.D. of 3 independent experiments. SC = solvent control. ∗P < .05, ∗∗P < .01.
3.4. Curcumin increases phosphorylation of IRE1α and XBP1 mRNA splicing in BCPAP cells
ER stress triggers the UPR to recover ER function. However, a failure to adapt to ER stress results in apoptosis. UPR is orchestrated by 3 principal branches, including PERK, IRE1α, and ATF6. Recent studies indicate that hyperactivation of IRE1α branch was shown to cause cell death[19] and XBP-1, the downstream effector of IRE1α signaling, is required for ER membrane expansion.[20] As shown in Fig. 3A, the phosphorylation of IRE1α in BCPAP cells was dose-dependently enhanced by curcumin treatment without affecting the total IRE1α protein level. Curcumin treatment, most obviously at 50 μM, induced ER stress through IRE1α signaling, which resulted in activation of the RNase domain of IRE1α and removal of a 26-nt sequence from XBP-1u, producing mature XBP-1s mRNA (Fig. 3B). This, in turn, activated a translational frame-shift to generate XBP-1s, a potent transcription factor (Fig. 3C). XBP-1s subsequently binds to promoters of several genes responsible for ER-associated degradation of misfolded glycoproteins, such as ERdj3/DNAJB11 (DnaJ heat shock protein family member B11), ER degradation-enhancing mannosidase-like protein 1 (EDEM1) and stress-associated endoplasmic reticulum protein 1 (SERP1). RT-PCR analysis showed that the mRNA expression of ERDJ3 and EDEM1 increased significantly in BCPAP cells treated with 50 μM of curcumin. Whereas, among these ER chaperones, SERP1 is more susceptible to curcumin treatment as evidenced by the significant elevation of SERP1 mRNA expressions at all dose levels in BCPAP cells (Fig. 3D). Note that pretreatment with 4-PBA, a chemical chaperone, was unable to rescue the XBP-1 mRNA splicing induced by curcumin (Fig. 3E), indicating that IRE-1-mediated XBP-1 splicing is not readily reversible. These results indicate that curcumin activates the IRE1α pathway which leads to the splicing of XBP-1 mRNA in BCPAP cells.
Figure 3.
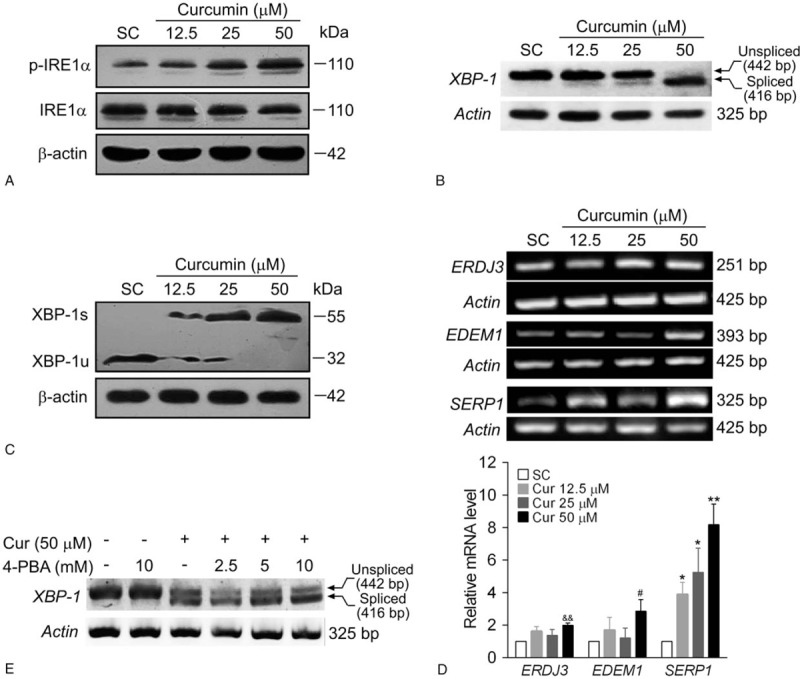
Curcumin induces phosphorylation of IRE1α and XBP-1 mRNA splicing. BCPAP cells were exposed to different dosages (12.5–50 μM) of curcumin for 24 hours. After the cells were collected, western blot or RT-PCR analysis were performed. (A) Curcumin increases the phosphorylation of IRE1α in BCPAP cells. The protein levels of phosphorylated IRE1α and total IRE1α were detected by western blot analysis. β-actin was used as a loading control. (B) Curcumin increases XBP-1 splicing in BCPAP cells. The mRNA levels of spliced and unspliced forms of XBP-1 were assessed by RT-PCR. Actin was performed as a loading control. (C) Curcumin treatment results in the conversion of inactive unspliced XBP-1 (XBP-1u) protein to an active spliced (XBP-1s) protein in BCPAP cells. The protein levels of spliced and unspliced forms of XBP-1 were detected by western blot assay. (D) Curcumin enhances the mRNA expressions of XBP-1 downstream genes. BCPAP cells were exposed to different dosages (12.5–50 μM) of curcumin for 24 hours. The mRNA expressions of ERDJ3, EDEM1, and SERP1 were determined by RT-PCR and normalized to that of Actin. The data shown represent the means ± S.D. of 3 independent experiments. &&P < .01, #P < .05, ∗P < .05, ∗∗P < .01 versus SC group. (E) 4-PBA fails to rescue the XBP-1 splicing induced by curcumin in BCPAP cells. Cells were pretreated with or without different dosages of 4-PBA (2.5–10 mM) for 1 hour. After that, cells were treated with 50 μM of curcumin for 24 hours and XBP-1 splicing was measured by RT-PCR. IRE1α = inositol-requiring enzyme 1α, RT-PCR = reverse transcriptase-polymerase chain reaction.
3.5. Curcumin activates the ATF-CHOP pathway leading to upregulation of pro-apoptotic CHOP expression in BCPAP cells
ATF6 has been characterized as another important ER sensor. ER stress activation induces cleavage of the ER stress-regulated transcription factor p90 ATF6 into an active p50 form, which depends on a calcium-dependent serine protease S1P in association with S2P located in the Golgi apparatus.[21] As illustrated in Fig. 4A, after treatment with curcumin, the p90 ATF6 band was markedly reduced, with a concomitant dose-dependent increase in the p50 ATF6 component. The cleaved soluble transcription factor p50 ATF6 is then free to translocate to the nucleus where it subsequently activates ER stress-responsive genes, such as GRP78, GRP94, calreticulin, and CHOP.[22] These genes determine the outcome (survival or apoptosis) of the stress response. CHOP is a major pro-apoptotic transcription factor that mediates ER-stress induced apoptosis.[23] RT-PCR assay revealed that curcumin increased the CHOP mRNA expression of BCPAP cells in a dose-dependent manner (Fig. 4B). Moreover, the expression of CHOP was markedly and significantly upregulated by curcumin treatment at 50 μM following 6, 12, and 24 hours of incubations (Fig. 4C). Thus, the mRNA expression of CHOP was enhanced by curcumin both in a dose- and time-dependent manner. Unexpectedly, curcumin treatment had no effect on the mRNA expression of GRP78 at any indicated times. To be pointed out, the upregulation of CHOP mRNA induced by curcumin was abolished after pretreatment with the increasing dosage of 4-PBA. As illustrated in Fig. 4D, barely detectable level of CHOP band was observed in curcumin combined with 4-PBA at its high concentration tested (10 mM). Next, short hairpin RNAs (shRNAs) were constructed to silence the endogenous CHOP expression of BCPAP cells. Compared with the scramble shRNA, the transfection of 3 CHOP shRNA plasmids targeting different regions of human CHOP mRNA into BCPAP cells profoundly downregulated the levels of CHOP proteins as determined by western blotting (Fig. 4E). As shown in Fig. 4F, curcumin significantly decreased cell viability in scramble-transfected BCPAP cells, but the inhibitory effect of curcumin in cell growth was partly reversed in CHOP-knockdown cells. Taken together, these results show that curcumin triggers ER stress through activation of ATF-CHOP pathway in BCPAP cells.
Figure 4.
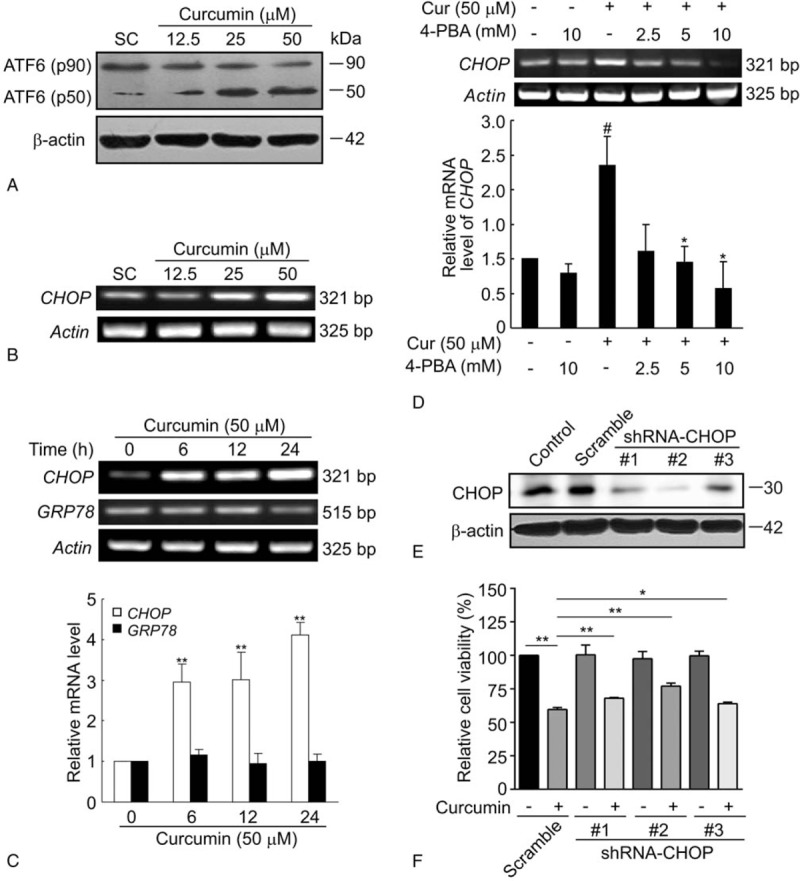
Curcumin elevates ATF6 cleavage and CHOP mRNA expression. (A) Curcumin induces the cleavage of ATF6 in BCPAP cells. Cells were treated with different dosages (12.5–50 μM) of curcumin for 24 hours and the uncleaved (90 kDa) and cleaved (50 kDa) forms of ATF6 were detected by western blot assay. (B) Curcumin dose-dependently increases the CHOP mRNA expression of BCPAP cells. BCPAP cells were exposed to different dosages (12.5–50 μM) of curcumin for 24 hours and subjected to RT-PCR analysis. (C) Curcumin increases the CHOP mRNA expression of BCPAP cells in a time-dependent manner. BCPAP cells were treated with 50 μM of curcumin for indicated times. Total RNAs were isolated and subjected to RT-PCR. CHOP and GRP78 mRNA expression were determined and normalized to that of Actin. (D) 4-PBA reverses the upregulation of CHOP mRNA induced by curcumin. BCPAP cells were pretreated with or without different dosages (2.5–10 μM) of 4-PBA before 50 μM of curcumin exposure for additional 24 hours. CHOP mRNA expression was detected by RT-PCR. All data shown represent the means ± S.D. of 3 independent experiments. #P < .05 versus untreated group; ∗P < .05, ∗∗P < .01 versus curcumin alone-treated group. (E) BCPAP cells were transfected with 3 siRNAs targeting different region of CHOP mRNA (shRNA-CHOP #1, #2, and #3) or negative control (Scramble). Cell lysates were collected 24 hours later and CHOP knockdown efficiency were determined by western blot. (F) BCPAP cells were transfected with shRNA-CHOP #1, #2, and #3 or scramble control. After 24 hours, cells were treated with or without 50 μM of curcumin for another 24 hours. The cell survival rate was determined by MTT assay. ∗P < .05, ∗∗P < .01. ATF6 = activating transcription factor 6.
3.6. Curcumin perturbs the intracellular calcium homeostasis of BCPAP cells
Apart from the classical UPR, it is well known that cellular calcium (Ca2+) signaling is another vital cellular response mechanisms to cope with ER stress.[24] ER stress frequently results in a release of Ca2+ from the ER, inducing cytosolic Ca2+ accumulation and triggering cell death.[25] Next, Ca2+ influx was measured by flow cytometry following treatment of BCPAP cells with different dosages of curcumin. As shown in Fig. 5A, the intracellular Ca2+ level in BCPAP cells significantly elevated in the presence of higher dosage of curcumin at 25 and 50 μM. However, the data in Fig. 5B showed that 4-PBA (2.5 mM)) pretreatment had only a negligible effect on curcumin-induced elevation of intracellular Ca2+ level (55.17% in curcumin treated-alone group vs 49.09% in curcumin/4-PBA combination group). BAPTA-AM is a well-known membrane permeable cytosolic Ca2+ chelator, which could block the interaction of calcium with calmodulin and thus prevent Ca2+ oscillation signals. Consistent with this point, 30-minute preincubation with 1 μM of BAPTA-AM reduced curcumin-induced Ca2+ influx from 52.82% to 42.18% (Fig. 5C). Moreover, the curcumin-mediated decrease in cell viability of BCPAP cells could also be prevented by BAPTA-AM pretreatment, further suggesting a pro-death role for cytosolic calcium in this process (Fig. 5D). Taken together, these results confirm that curcumin disturbs the steady-state of intracellular calcium in BCPAP cells.
Figure 5.
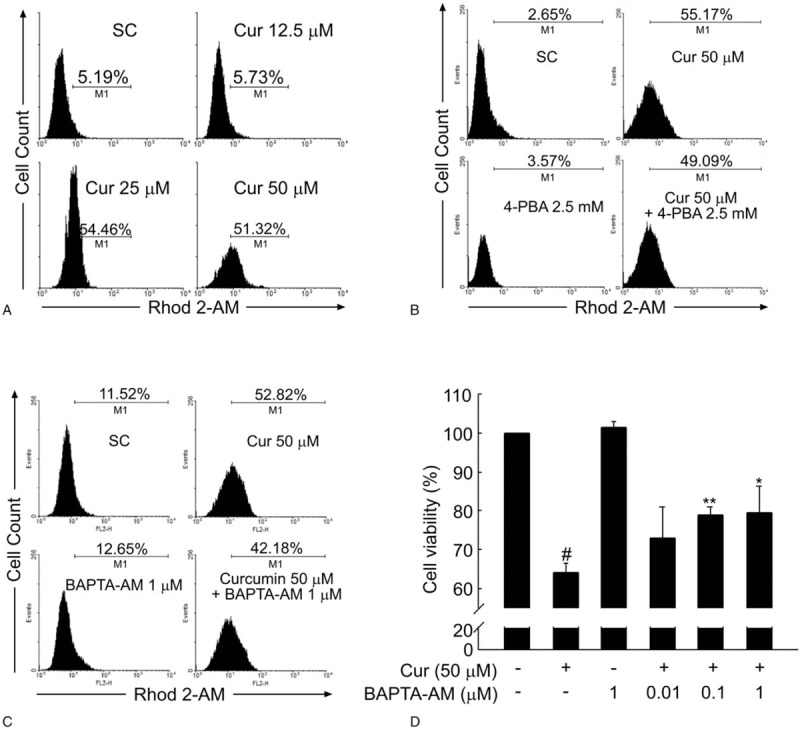
Curcumin increases the intracellular Ca2+ level of BCPAP cells. (A) Curcumin increases the intracellular Ca2+ content in BCPAP cells. After BCPAP cells were exposed to different dosages (12.5–50 μM) of curcumin for 24 hours, intracellular Ca2+ level was detected by flow cytometry analysis using Rhod 2-AM. (B) 4-PBA fails to rescue the increased intracellular Ca2+ content induced by curcumin. After treatment with 50 μM curcumin, 2.5 mM 4-PBA, or the combination of 50 μM curcumin and 2.5 mM 4-PBA, the intracellular Ca2+ content of BCPAP cells was determined by Flow cytometry. (C) BAPTA-AM decreases the elevated intracellular Ca2+ level induced by curcumin. BCPAP cells were preincubated in the presence or absence of BAPTA-AM (1 μM) for 0.5 hour followed by curcumin (50 μM) treatment for 24 hours. After incubation, the intracellular Ca2+ content of BCPAP cells was determined by flow cytometry. (D) BAPTA-AM protects BCPAP cells from curcumin-induced cytotoxicity. Cells were treated as in (C) and the cell viability of BCPAP cells was measured by MTT assay. All data shown represent the means ± S.D. of 3 independent experiments. #P < .05 versus untreated group; ∗P < .05, ∗∗P < .01 versus curcumin alone-treated group.
3.7. Curcumin promotes apoptosis by activating Ca2+-CaMKII-JNK signaling pathway in BCPAP cells
High concentrations of Ca2+ in ER lumen are essential for ER folding capacity. SERCA, resided in the ER membrane, acts as a Ca2+-ATPase that transfers Ca2+ from the cytosol to the lumen of the sarcoplasmic reticulum at the expense of ATP hydrolysis.[26] It has been reported that inhibition of SERCA activity impairs ER Ca2+ homeostasis and causes severe ER stress.[27] First, the effect of curcumin on the protein expression of SERCA2 was detected. As shown in Fig. 6A, the protein levels of SERCA2 were only slightly affected by curcumin, as there was approximate 90% of SERCA2 remained even though higher dosage of curcumin was used at 25 and 50 μM. Next, the activity of Ca2+-ATPase in BCPAP cells was measured after incubation with various concentrations of curcumin for 24 hours. As shown in Fig. 6B, curcumin significantly inhibited the activity of Ca2+-ATPase in a dose-dependent manner. Compared with solvent control cells possessing Ca2+-ATPase activity at 0.091 ± 0.011 unit/mg protein, BCPAP cells exposed to curcumin at 12.5, 25, and 50 μM showed a sharp decrease in Ca2+-ATPase activity, with only 0.054 ± 0.018, 0.048 ± 0.017, and 0.028 ± 0.006 unit/mg of the cells, respectively. Thus, the inhibition of SERCA2 activity by curcumin could explain, in part, the increased cytosolic Ca2+ in BCPAP cells. Accumulated cytosolic Ca2+ then binds to calmodulin and this complex subsequently initiates the autophosphorylation and activation of calcium/calmodulin-dependent protein kinase II (CaMKII).[28] Then CaMKII in turn, can lead to activation of c-Jun N-terminal Kinase (JNK) and subsequent mitochondrial-dependent apoptosis.[29] As expected from the data in Fig. 6C, there was an increase in phospho-Thr287 CaMKII at relatively high doses of curcumin loading (25 and 50 μM), consistent with the significant increased intracellular Ca2+ level at these 2 dosage of curcumin treatment (Fig. 5A). As CaMKII was upstream of JNK activation, curcumin treatment was also associated with an increase in the phosphorylation of JNK (Fig. 6D). SP600125, an anthrapyrazolone inhibitor of JNK was also used. As shown in Fig. 6E and F, pretreatment with 10 μM of SP60012510 inhibited curcumin-induced phosphorylation of JNK and decreased the cytotoxicity of curcumin. Furthermore, to characterize the apoptotic process, the expressions of apoptosis-related proteins were examined by western blotting. As illustrated in Fig. 6G, curcumin treatment led to a dose-dependent increased expression of pro-apoptotic Bax with a concomitant decrease in the level of anti-apoptotic proteins Bcl-XL, Bcl-2, and Bid. In addition, curcumin treatment induced the cleavage of caspase-3, -7, and -8 (Fig. 6H), as well as the proteolysis of poly ADP-ribose polymerase (PARP) (Fig. 6I). Collectively, these results demonstrate that curcumin promotes apoptosis by activating Ca2+-CaMKII-JNK pathway in BCPAP cells.
Figure 6.
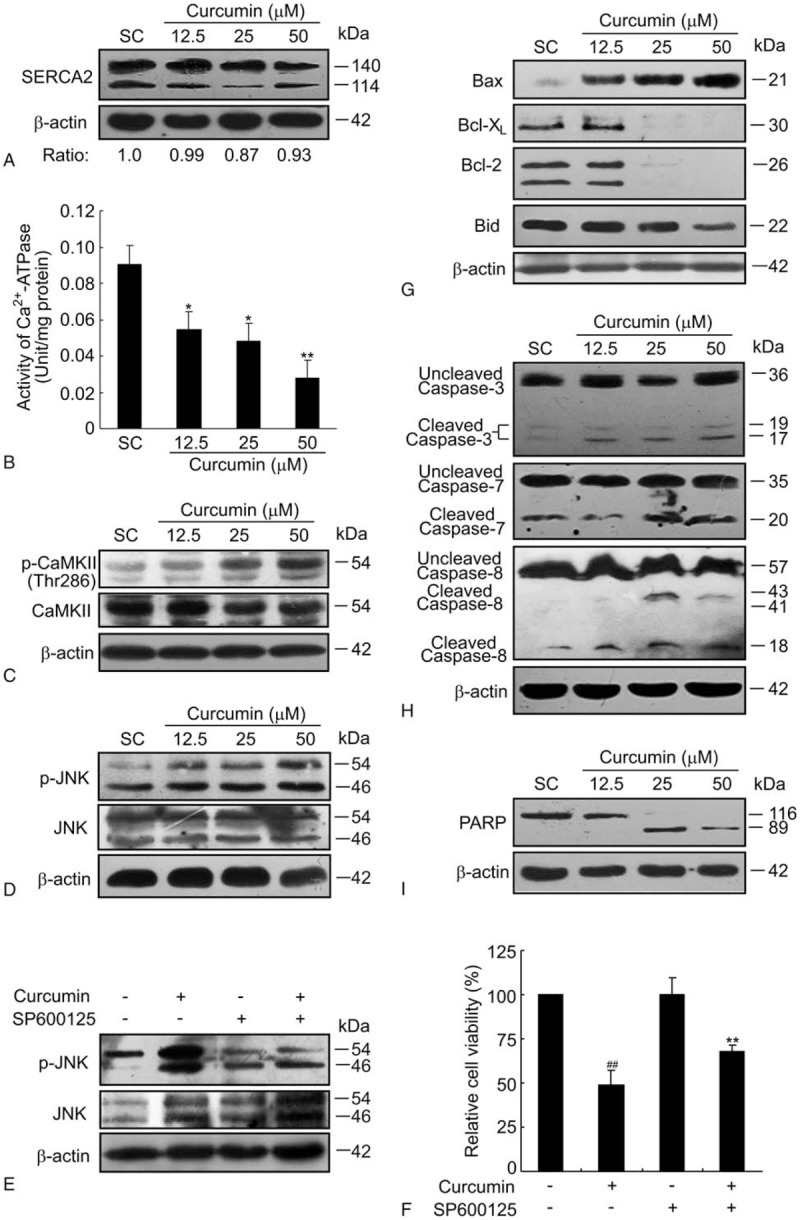
Curcumin promotes apoptosis by activating Ca2+-CaMKII-JNK pathway. (A) Curcumin slightly decreases the protein level of SERCA2 in BCPAP cells. BCPAP cells were exposed to different dosages (12.5–50 μM) of curcumin for 24 hours and the protein level of SERCA2 was detected by western blotting. β-actin was used as a loading control. Relative protein levels of SERCA2 were shown at the bottom of the graph. (B) Curcumin inhibits SERCA2 activity of BCPAP cells. Cells were incubated with various concentrations of curcumin for 24 hours. Then the cells were harvested and the Ca2+-ATPase activity was measured according to the instructions of the Ca2+-ATPase detection kit. Error bars are means ± S.D. of 3 independent experiments. ∗P < .05, ∗∗P < .01 versus SC group. (C) Curcumin treatment increases the phosphorylation of CaMKII in BCPAP cells. (D) Curcumin increases the phosphorylation of JNK of BCPAP cells. (E) BCPAP cells were either treated with 50 μM of curcumin for 24 hours or pre-treated with 10 μM of SP600125 for 1 hour before curcumin treatment. The phosphorylated and total JNK protein levels were detected by western blot assay. β-actin was used as a loading control. (F) BCPAP cells were treated as in (E). MTT assay was used to determine the cell viability. ##P < .01 versus untreated group; ∗∗P < .01 versus curcumin alone-treated group. (G) Curcumin induces apoptosis of BCPAP cells. BCPAP cells were treated with curcumin at different dosages for 24 hours and then harvested for apoptosis analysis. The pro-apoptotic protein Bax and anti-apoptotic protein Bcl-XL, Bcl2, Bid were measured by western blotting. (H) Curcumin activates caspase-3, -7, and -8 pathway in BCPAP cells. (I) Curcumin treatment leads to an increased cleavage of PARP. These apoptosis-related proteins were determined at least 3 times and representative data were shown. PARP = poly ADP-ribose polymerase; SC = solvent control.
4. Discussion
Curcumin, derived from the rhizome of Curcuma longa L., has been reported to exhibit therapeutic efficacy in patients with several types of progressive advanced cancers.[30] Several phase I and phase II clinical trials indicate that curcumin is clinically safe and tolerable.[31] Recent studies from our group have also proved that curcumin inhibits metastasis[32] and induces ROS-independent DNA damage in thyroid cancer cells,[33] indicating a potential anti-cancer ability of curcumin against thyroid cancer. In the present study, we found curcumin significantly inhibited anchorage-independent cell growth and induced apoptosis in BCPAP cells, a popularly used human papillary thyroid cancer cell line (Fig. 1).
ER is a major organelle for synthesis, maturation, and folding proteins. To some extent, the cell's fate is decided by the balance between survival and apoptotic signaling, and the specific ER stressor plays a crucial role in tuning cell homeostasis.[17] Aimed to balance the extracellular stimulation, unfolded protein response (UPR) is activated to reduces unfolded/misfolded protein loads, however, prolonged ER stress initiates the apoptotic cell death which may dependent or independent of mitochondrial signaling.[34] UPR is triggered in cells when ER transmembrane protein sensors (PERK, IRE, and ATF6) detect the accumulation of unfolded proteins. It has been reported that each components of UPR sensors exert fundamental roles in ER-mediated cell death.[17] In the case of IRE1, its endoribonuclease activity catalyzes the unconventional splicing of the mRNA encoding XBP1, which induces genes related to protein folding. In addition, IRE1 can recruit TNF receptor-associated factor 2 (TRAF2), apoptosis-signal-regulating kinase (ASK1), and caspase-12 to target the activation of c-jun N-terminal kinase (JNK), which is known as a pro-apoptotic factor.[35] Activation of the transcription factor, ATF6, and its downstream targets, that is, CHOP, has also been demonstrated as a regulator of ER-associated cell survival or death.[36] Indeed, CHOP plays a pro-apoptotic role and is the most well recognized indicator of ER stress-induced apoptosis. It has been reported that CHOP suppressed the expression of anti-apoptotic Bcl-2 to upregulate the ratio of Bax/Bcl-2 and it also can be upregulated by TRAIL receptors to promote the cellular apoptotic pathway.[37] In the present study, curcumin triggered ER stress in BCPAP cells as evidenced by the expansion of ER membrane (Fig. 2). Moreover, curcumin significantly increased the key components of the UPR pathway via phosphorylation of IRE1α, XBP-1 mRNA splicing (Fig. 3) as well as activation of ATF6 and CHOP expression (Fig. 4).
ER participates in the initiation of apoptosis by at least 2 different mechanisms. One is the classical UPR, the other is the cellular Ca2+ signaling. Intracellular Ca2+ signals mediate various cellular processes, including mitochondrial homeostasis, cell survival, and cell death.[38] ER is a critical reservoir of Ca2+ and depletion of the ER Ca2+ store inhibits proliferation and induces cell death in transformed cells.[27] SERCA and inositol trisphosphate receptors (IP3Rs), which located on the ER membrane, regulate the Ca2+ flux by pumping Ca2+ from cytosol and releasing Ca2+ from ER, respectively.[39] SERCA has 3 subfamily members (SERCA1, SERCA2, and SERCA3) and their relative expression is cell type-specific. SERCA2 is one of the family members and its aberrant increase in the expression level has been observed in certain cancers than in normal tissues. For example, it has been reported that SERCA2 mRNA is overexpressed in ovarian cancer.[40] In another case, the overexpression of SERCA2 mRNA was detected in 90% of human colorectal cancer tissues.[41] These studies suggest a possible role of SERCA2 in the development and progression of human cancers and SERCA may be a novel therapeutic target in cancer treatment. Consistent with the previous study that curcumin inhibits SERCA activity in various cell types through binding with SERCA and subsequent inhibition of its structural transformation,[42] the activity of SERCA2 was dose-dependently suppressed by curcumin in BCPAP cells (Fig. 6). Accordingly, curcumin depleted ER Ca2+ and increased intracellular Ca2+ levels, and Ca2+ chelator BAPTA-AM partially reversed curcumin-induced ER stress and growth inhibition (Fig. 5). Further investigation should be focused on the possibility of SERCA2 as one of the targets in the therapy of human thyroid cancers.
The cytotoxic effects of Ca2+ released from the ER are mediated through accumulation in mitochondria (mitochondrial Ca2+ overload), which results in permeability transition and activation of apoptotic pathways.[43] Cytosolic Ca2+ resulting from ER stress induces expression of the Fas death receptor through a pathway involving Ca2+/CaMKII and JNK. Remarkably, CaMKII serves as a unifying link between ER stress and the mitochondrial-dependent apoptotic pathway by promotion of mitochondrial calcium uptake.[29] Consistent with this view, under Ca2+ influx stimulation, curcumin induced the activation of CaMKII which phosphorylated JNK, resulting in the activation of Bcl-2 family members and the cleavage of caspase and PARP (Fig. 6). Based on these data, we conclude that curcumin-induced disruption of Ca2+ homeostasis is involved in its induction of apoptosis in thyroid cancer cells.
Previous reports have indicated that a major signaling interaction in oncogenic transformation is that between Ras and Ca2+. Pierro et al[44] have reported that suppression of Ca2+ flux to the mitochondria is a common response to K-RasG13D in colorectal cell lines and that this contributes to a survival advantage during oncogenic transformation. Activating H-, K-, and N-ras mutations represent the most common type of abnormality of a dominant oncogene in human cancer and mutations in all 3 ras genes were found both in the benign (micro-) follicular thyroid adenomas and in the follicular and undifferentiated thyroid carcinomas.[45] Activated ras oncogenes has been found in approximately 80% of follicular tumors but only 20% of papillary thyroid tumors. However, both follicular and papillary carcinomas may progress to undifferentiated thyroid carcinomas.[46] Whether the presence of the oncogenic Ras mutants also modified Ca2+ signals in thyroid cancer cell lines, especially in follicular thyroid carcinoma cell lines FTC-133, remains to be investigated. In our present study, we have not monitored the extent of mitochondrial Ca2+ uptake during the increase in cytosolic Ca2+ upon curcumin treatment. Given the enhancement in curcumin-mediated Ca2+ signaling observed in BCPAP cells, we hypothesized that mitochondrial Ca2+ uptake would also be enhanced and contribute to greater sensitivity to death-inducing stimuli, such as radioiodine application or chemotherapeutic drugs treatment, in thyroid cancer cells.
In summary, when BCPAP cells are exposed to curcumin, SERCA2 activity is inhibited, impairing ER Ca2+ homeostasis, which disturbs ER folding capacity and increases unfolded and misfolded proteins, activating the signaling pathways of UPR response and causing ER stress-associated apoptosis. Our results provoked that the Ca2+-dependent cell death was induced upon curcumin treatment, which may be a promising therapeutic target for thyroid cancer treatment.
Acknowledgments
The authors thank Dr. lengxin Duan (Henan University of Science and Technology, China) for providing us with shRNA-CHOP plasmids.
Author contributions
Data curation: Xian Cheng.
Methodology: Li Zhang.
Supervision: Jiandong Bao, Huixin Yu.
Writing – original draft: Li Zhang.
Writing – review and editing: Shichen Xu.
Footnotes
Abbreviations: 4-PBA = 4-phenylbutyrate, ASK1 = apoptosis-signal-regulating kinase, ATF6 = activating transcription factor 6, CaMKII = calcium/calmodulin-dependent protein kinase II, CHOP = C/EBP homologous protein 10, Cyt c = Cytochrome c, DMSO = dimethyl sulfoxide, EB = ethidium bromide, EDEM1 = ER degradation-enhancing mannosidase-like protein 1, EDTA = ethylenediaminetetraacetic acid, eIF2α = eukaryotic initiation factor 2α, ER = endoplasmic reticulum, ERdj3/DNAJB11 = DnaJ heat shock protein family member B11, ERO1α = endoplasmic oxidoreductin 1 like protein, GADD34 = Growth arrest and DNA damage protein 34, GRP78/94 = glucose-regulated protein 78/94, HDAC = histone deacetylase, IP3Rs = inositol trisphosphate receptors, IRE1α = inositol-requiring enzyme 1α, JNK = c-Jun N-terminal Kinase, MMP = mitochondrial membrane potential, PARP = poly ADP-ribose polymerase, PERK = RNA-like ER kinase, PI = propidium iodide, PMSF = phenylmethylsul-fonyl fluoride, PTC = papillary thyroid carcinoma, ROS = reactive oxygen species, SERCA2A = sarco-endoplasmic reticulum ATPase 2A, SERP1 = stress-associated endoplasmic reticulum protein 1, TRAF2 = TNF receptor-associated factor 2, UPR = unfolded protein response, XBP-1 = X-box binding protein1.
This study was funded by the Science and Research Foundation of Health Bureau of Jiangsu Province (No. H2017032), the grants from the National Natural Science Foundation of China (Nos. 81602352 and 81673436), the Natural Science Foundation of Jiangsu Province (BK20171145 and BK20151119), and Wuxi Municipal Commission of Health and Family Planning (Q201608).
The authors declare that there are no conflicts of interest.
References
- [1].Grande E, Diez JJ, Zafon C, et al. Thyroid cancer: molecular aspects and new therapeutic strategies. J Thyroid Res 2012;2012:847108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Carneiro RM, Carneiro BA, Agulnik M, et al. Targeted therapies in advanced differentiated thyroid cancer. Cancer Treat Rev 2015;41:690–8. [DOI] [PubMed] [Google Scholar]
- [3].Heger M, van Golen RF, Broekgaarden M, et al. The molecular basis for the pharmacokinetics and pharmacodynamics of curcumin and its metabolites in relation to cancer. Pharmacol Rev 2014;66:222–307. [DOI] [PubMed] [Google Scholar]
- [4].Wang L, Zhang B, Huang F, et al. Curcumin inhibits lipolysis via suppression of ER stress in adipose tissue and prevents hepatic insulin resistance. J Lipid Res 2016;57:1243–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Basile V, Belluti S, Ferrari E, et al. bis-Dehydroxy-Curcumin triggers mitochondrial-associated cell death in human colon cancer cells through ER-stress induced autophagy. PLoS One 2013;8:e53664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zheng M, Zhang Q, Joe Y, et al. Curcumin induces apoptotic cell death of activated human CD4+ T cells via increasing endoplasmic reticulum stress and mitochondrial dysfunction. Int Immunopharmacol 2013;15:517–23. [DOI] [PubMed] [Google Scholar]
- [7].Kim SM, Park KC, Jeon JY, et al. Potential anti-cancer effect of N-hydroxy-7-(2-naphthylthio) heptanomide (HNHA), a novel histone deacetylase inhibitor, for the treatment of thyroid cancer. BMC Cancer 2015;15:1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang L, Zhang L, Cheng X, et al. Curcumin induces cell death of human papillary thyroid carcinoma BCPAP cells through endoplasmic reticulum stress. RSC Adv 2016;6:52905–12. [Google Scholar]
- [9].Urra H, Dufey E, Lisbona F, et al. When ER stress reaches a dead end. Biochim Biophys Acta 2013;1833:3507–17. [DOI] [PubMed] [Google Scholar]
- [10].Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007;129:1337–49. [DOI] [PubMed] [Google Scholar]
- [11].Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007;8:519–29. [DOI] [PubMed] [Google Scholar]
- [12].Xu L, Li S, Ran X, et al. Apoptotic activity and gene responses in Drosophila melanogaster S2 cells, induced by azadirachtin A. Pest Manag Sci 2016;72:1710–7. [DOI] [PubMed] [Google Scholar]
- [13].La Rovere RM, Roest G, Bultynck G, et al. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016;60:74–87. [DOI] [PubMed] [Google Scholar]
- [14].Tanigawa N, Kern DH, Hikasa Y, et al. Rapid assay for evaluating the chemosensitivity of human tumors in soft agar culture. Cancer Res 1982;42:2159–64. [PubMed] [Google Scholar]
- [15].Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta 1986;850:436–48. [DOI] [PubMed] [Google Scholar]
- [16].Zhang L, Yu H, Sun Y, et al. Protective effects of salidroside on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells. Eur J Pharmacol 2007;564:18–25. [DOI] [PubMed] [Google Scholar]
- [17].Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta 2013;1833:3460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wu H, Wei L, Fan F, et al. Integration of Hippo signalling and the unfolded protein response to restrain liver overgrowth and tumorigenesis. Nat Commun 2015;6:6239. [DOI] [PubMed] [Google Scholar]
- [19].Ghosh R, Wang L, Wang ES, et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014;158:534–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schuck S, Prinz WA, Thorn KS, et al. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol 2009;187:525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 2000;6:1355–64. [DOI] [PubMed] [Google Scholar]
- [22].Yoshida H, Okada T, Haze K, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol 2000;20:6755–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004;11:381–9. [DOI] [PubMed] [Google Scholar]
- [24].Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 2007;14:1576–82. [DOI] [PubMed] [Google Scholar]
- [25].Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 2002;32:235–49. [DOI] [PubMed] [Google Scholar]
- [26].Monteith GR, McAndrew D, Faddy HM, et al. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer 2007;7:519–30. [DOI] [PubMed] [Google Scholar]
- [27].Li W, Ouyang Z, Zhang Q, et al. SBF-1 exerts strong anticervical cancer effect through inducing endoplasmic reticulum stress-associated cell death via targeting sarco/endoplasmic reticulum Ca(2+)-ATPase 2. Cell Death Dis 2014;5:e1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Griffith LC. Regulation of calcium/calmodulin-dependent protein kinase II activation by intramolecular and intermolecular interactions. J Neurosci 2004;24:8394–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Timmins JM, Ozcan L, Seimon TA, et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest 2009;119:2925–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shanmugam MK, Rane G, Kanchi MM, et al. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015;20:2728–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Carroll RE, Benya RV, Turgeon DK, et al. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev Res (Phila) 2011;4:354–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang L, Cheng X, Gao Y, et al. Curcumin inhibits metastasis in human papillary thyroid carcinoma BCPAP cells via down-regulation of the TGF-beta/Smad2/3 signaling pathway. Exp Cell Res 2016;341:157–65. [DOI] [PubMed] [Google Scholar]
- [33].Zhang L, Cheng X, Gao Y, et al. Induction of ROS-independent DNA damage by curcumin leads to G2/M cell cycle arrest and apoptosis in human papillary thyroid carcinoma BCPAP cells. Food Funct 2016;7:315–25. [DOI] [PubMed] [Google Scholar]
- [34].Wang WA, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. Biochim Biophys Acta 2014;1843:2143–9. [DOI] [PubMed] [Google Scholar]
- [35].Yoneda T, Imaizumi K, Oono K, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 2001;276:13935–40. [DOI] [PubMed] [Google Scholar]
- [36].Mahdi AA, Rizvi SH, Parveen A. Role of endoplasmic reticulum stress and unfolded protein responses in health and diseases. Indian J Clin Biochem 2016;31:127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rodriguez D, Rojas-Rivera D, Hetz C. Integrating stress signals at the endoplasmic reticulum: the BCL-2 protein family rheostat. Biochim Biophys Acta 2011;1813:564–74. [DOI] [PubMed] [Google Scholar]
- [38].Stewart TA, Yapa KT, Monteith GR. Altered calcium signaling in cancer cells. Biochim Biophys Acta 2015;1848:2502–11. [DOI] [PubMed] [Google Scholar]
- [39].Bittremieux M, Parys JB, Pinton P, et al. ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca(2+) signaling. Biochim Biophys Acta 2016;1863:1364–78. [DOI] [PubMed] [Google Scholar]
- [40].Bowen NJ, Walker LD, Matyunina LV, et al. Gene expression profiling supports the hypothesis that human ovarian surface epithelia are multipotent and capable of serving as ovarian cancer initiating cells. BMC Med Genomics 2009;2:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chung FY, Lin SR, Lu CY, et al. Sarco/endoplasmic reticulum calcium-ATPase 2 expression as a tumor marker in colorectal cancer. Am J Surg Pathol 2006;30:969–74. [DOI] [PubMed] [Google Scholar]
- [42].Bilmen JG, Khan SZ, Javed MH, et al. Inhibition of the SERCA Ca2+ pumps by curcumin. Curcumin putatively stabilizes the interaction between the nucleotide-binding and phosphorylation domains in the absence of ATP. Eur J Biochem 2001;268:6318–27. [DOI] [PubMed] [Google Scholar]
- [43].Rizzuto R, De Stefani D, Raffaello A, et al. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 2012;13:566–78. [DOI] [PubMed] [Google Scholar]
- [44].Pierro C, Cook SJ, Foets TC, et al. Oncogenic K-Ras suppresses IP(3)-dependent Ca(2)(+) release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca(2)(+) levels in colorectal cancer cell lines. J Cell Sci 2014;127:1607–19. [DOI] [PubMed] [Google Scholar]
- [45].Lemoine NR, Mayall ES, Wyllie FS, et al. Activated ras oncogenes in human thyroid cancers. Cancer Res 1988;48:4459–63. [PubMed] [Google Scholar]
- [46].Williams ED. The aetiology of thyroid tumours. Clin Endocrinol Metab 1979;8:193–207. [DOI] [PubMed] [Google Scholar]