

Abstract
Aberrant insulin-like growth factor I receptor (IGF1R) signaling pathway serves as a well-established target for cancer drug therapy. The intragenic antisense long noncoding RNA (lncRNA) IRAIN, a putative tumor suppressor, is downregulated in breast cancer cells, while IGF1R is overexpressed, leading to an abnormal IGF1R/IRAIN ratio that promotes tumor growth. To precisely target this pathway, we developed an “antisense lncRNA-mediated intragenic cis competition” (ALIC) approach to therapeutically correct the elevated IGF1R/IRAIN bias in breast cancer cells. We used CRISPR-Cas9 gene editing to target the weak promoter of IRAIN antisense lncRNA and showed that in targeted clones, intragenic activation of the antisense lncRNA potently competed in cis with the promoter of the IGF1R sense mRNA. Notably, the normalization of IGF1R/IRAIN transcription inhibited the IGF1R signaling pathway in breast cancer cells, decreasing cell proliferation, tumor sphere formation, migration, and invasion. Using “nuclear RNA reverse transcription-associated trap” sequencing, we uncovered an IRAIN lncRNA-specific interactome containing gene targets involved in cell metastasis, signaling pathways, and cell immortalization. These data suggest that aberrantly upregulated IGF1R in breast cancer cells can be precisely targeted by cis transcription competition, thus providing a useful strategy to target disease genes in the development of novel precision medicine therapies.
Keywords: IGF1R signaling pathway, antisense cis competition, long noncoding RNA, IRAIN, breast cancer
Graphical Abstract
Introduction
The insulin-like growth factor (IGF) signaling axis plays a critical role in development, growth, and maintenance of many tissues.1, 2 The mitogenic ligands IGF-I and IGF-II interact with the cell membrane IGF-I receptor (IGF1R), which belongs to the tyrosine kinase receptor family. Increased activity of the IGF axis has long been recognized as a critical factor in tumor growth and progression.3, 4, 5 IGF1R is dysregulated in a variety of human malignancies, including breast cancer.6, 7, 8 Activation of this pathway leads to stimulation of downstream mitogen-activated protein kinase (MAPK) and/or phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling cascades,9 resulting in increases in cell proliferation, antiapoptosis, and drug resistance through autocrine, paracrine, and endocrine pathways.10, 11, 12, 13 As a result, IGF1R has been recognized as a promising target for the development of precision tumor therapy.14, 15
In the past decade, numerous extensive cancer trials have been performed using a variety of agents that are specifically directed against the IGF1R signaling pathway.16, 17, 18 Unfortunately, the vast majority of therapies using monoclonal antibodies and tyrosine kinase inhibitors to target IGF1R failed in late clinical trials.17, 19 Thus, other novel approaches are urgently needed to target this pathway in tumors.
Approximately 50% of breast tumors show increased transcription of IGF1R,14 but little is known about how IGFIR becomes dysregulated in tumors. Using a novel R3C (RNA-guided chromatin conformation capture) method, we recently identified IRAIN, an intragenic antisense long noncoding RNA (lncRNA), that arises within the IGF1R promoter complex.20 IRAIN was expressed in a monoallelic manner, with the expression of the lncRNA exclusively from the paternal chromosome, and it appeared to serve as a tumor suppressor in hematopoietic tumors20. IRAIN was also aberrantly regulated in breast cancer, exhibiting a pattern of allele-switch: the allele expressed in normal tissues was suppressed, while the normally silenced allele was expressed.21 Recent studies have shown that this lncRNA is also dysregulated in non-small-cell lung cancer22 and pancreatic cancer.23
IRAIN is transcribed in an antisense orientation using a promoter located in intron 1 of IGF1R. By overlapping with the IGF1R promoter in antisense, IRAIN lncRNA directly competes with IGF1R in cis for transcriptional machinery.20 In cancer cells, however, IRAIN is downregulated, and the decrease in this cis competition control leads to upregulation of IGF1R, increasing the activation of the IGF signaling pathway.20, 21 In this communication, we attempted to target the IGF1R pathway in tumors by increasing the transcription of the downregulated IRAIN antisense suppressor lncRNA, thereby enhancing the cis competition mechanism. The rebalanced production of the oncogenic IGF1R and tumor suppressor IRAIN should decrease the signaling cascades that stimulate the growth of breast cancer cells.
Results
Targeted Activation of IRAIN Antisense Tumor Suppressor lncRNA
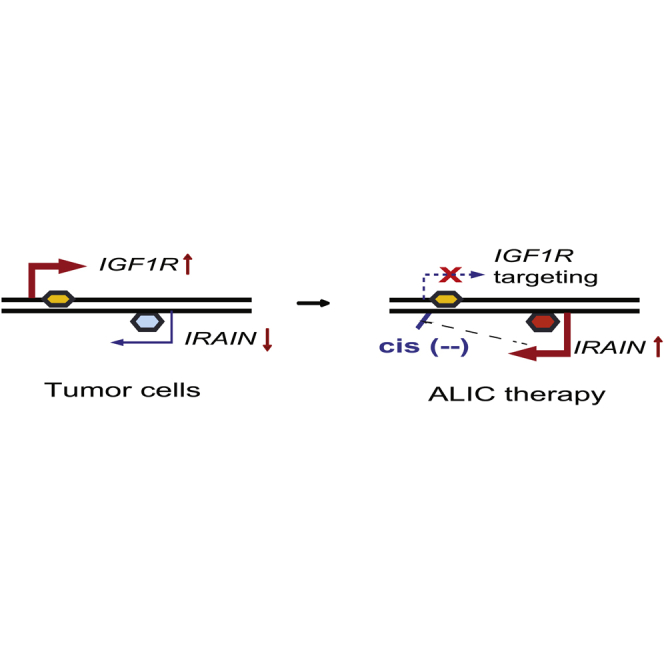
IRAIN is transcribed in an antisense direction to IGF1R from an intronic promoter (Figure 1A). In normal tissues, expression of the sense IGF1R coding mRNA and the antisense IRAIN are regulated reciprocally. Breast cancer cells, however, are characterized by upregulated IGF1R and downregulated IRAIN (Figure 1B, top). The activated IGF1R pathway in tumors is associated with tumor growth and metastasis. To precisely target the IGF1R pathway, we devised an “antisense tumor suppressor lncRNA-mediated intragenic cis competition” (ALIC) approach (Figure 1B, bottom). Specifically, the aberrant IGF1R expression in tumors was targeted by increasing the IRAIN antisense suppressor lncRNA, which competes with the IGF1R promoter in cis.
Figure 1.

Targeting the IGF1R Pathway by Antisense lncRNA IRAIN-Mediated cis Competition
(A) The orientation of IGF1R and IRAIN. The antisense IRAIN lncRNA is transcribed from an intronic promoter of the IGF1R gene. (B) Schematic diagram of the antisense lncRNA-mediated cis competition in the IGF1R signaling pathway. In normal tissues, the transcription of the IGF1R/IRAIN locus is balanced. In breast cancer cells, however, IGF1R is upregulated while IRAIN is downregulated. This unbalanced expression leads to increased activation of the IGF1R signaling pathway. An ALIC targeting approach is used to reverse this unbalance. A strong CMV promoter is inserted in front of the IRAIN lncRNA to induce increased production of IRAIN, which then competes in cis with the overlapping IGF1R promoter and dampens the IGF1R signaling pathway in tumor cells. This provides a molecular basis for the development of the precision therapy against breast cancer. (C) ALIC targeting of IGF1R by CRISPR Cas9-guided recombinant knockin. Cas9, CRISPR Cas9; gRNA, Cas9 guiding RNA; pCMV, CMV promoter; pH1, RNA polymerase III H1 promoter; Cre, Cre recombinase; pA, SV40 poly(A) signal; loxP, the locus of X-over P1 recombination site recognized by Cre; Arm 1-2, the genomic sequences used for recombination. Under the guidance of gRNAs, Cas9-mediated genomic recombination at the IRAIN locus, resulting in the insertion of the CMV promoter-puro cassette in front of the IRAIN. After puromycin selection, the cells were treated with Cre to remove the selection marker Puro+. In the selected cell clones, IRAIN is under the control of the strong promoter pCMV. The upregulated transcription of this antisense lncRNA will compete in cis with that of the sense IGF1R mRNA. (D) Initial screening of targeted cell clones by PCR. Primers were designed from the IRAIN arm, selection marker, and vector sequences. PCR was used to identify the wild-type, targeted DNA, and vector DNAs. RIC, control cells that were generated in parallel with ALIC targeting by transfecting the cells with only the donor vector DNA and selected with puromycin. As the control, RIC cells carry the randomly inserted donor vector DNA. Clones 1–12, cell clones that were generated by co-transfection with Cas9 target vector-donor vector DNAs and selected by both puromycin and ganciclovir. Targeting cell clones not only carry the IGF1R targeting allele, but also contain some amount of the randomly inserted donor vector DNA in the genome. Among them, clone 7 contains a minimum amount of randomly inserted vector DNA and was thus used for subsequent studies.
As a proof-of-concept study to target the IGF1R pathway, we used the CRISPRCas9 gene-editing system to insert a strong cytomegalovirus (CMV) promoter in front of IRAIN that would increase the expression of the IRAIN lncRNA (Figure 1B). We hypothesized that IRAIN competes in cis with the overlapping IGF1R promoter and thus downregulates the IGF1R pathway. We co-transfected the targeting and donor vectors into MDA-MB-231 breast cancer cells (Figure 1C). With the aid of the guide RNAs (gRNAs) (Figure S1), Cas9 induced homologous recombination at the IRAIN locus. Puromycin was used to select the positive cells, and ganciclovir was then added to the cell medium to reduce cells that had random integration of the donor vector in the genome. After targeting, IRAIN would be under the control of an introduced strong promoter that drives the high expression of the lncRNA that would compete with sense IGF1R-coding mRNA (Figure 1C).
After co-transfection with the Cas9 targeting vector and the donor vector, we selected cells using puromycin and ganciclovir. We collected stable cells (clones 1–12) and confirmed the knockin targeting using a series of PCR primers located in the donor vector, arms, and genomic DNAs (Figure 1D). In selected clones, we detected the specific target DNAs (Figure 1D, top), although some cell clones also contained some random insertion of the donor vector (Figure 1D, middle). As a control, MDA-MB-231 cells were transfected with donor vector DNA and were selected with puromycin in parallel with IGF1R targeting. As expected, these random insertion cell (RIC) control cells did not carry the targeting DNA sequence. Instead, only the randomly inserted donor vector DNA was detected (Figure 1D, middle, lane 1). Because cell clone 7 contained a minimum amount of randomly inserted vector DNA in the genome, we focused on this clone for the subsequent studies.
Targeted Activation of IRAIN Inhibits IGF1R Expression
To examine the targeting of IGF1R by the ALIC approach, we first examined the expression of IGF1R and IRAIN in selected cell clones. As predicted, IGF1R was upregulated and IRAIN was downregulated in RIC control cells. In targeted clones, however, we detected a dramatic increase of the IRAIN antisense lncRNA, in parallel with a significant downregulation of IGF1R (Figure 2A).
Figure 2.

Downregulation of IGF1R by cis Competition of the Active IRAIN Antisense RNA
(A) Reciprocal transcription competition between the sense IGF1R mRNA and the antisense IRAIN lncRNA. RIC, random insertion control cells. Clones 1–12, cells that carry the IGF1R targeting allele in the genome. The abundance of IGF1R/IRAIN transcripts was measured by qPCR. The data shown are mean ± SD of three independent PCR reactions. Note the downregulated IGF1R in clone cells. (B) ALIC targeting of IGF1R in two selected cell clones. ALIC1 and ALIC2, ALIC-targeted cells that were transfected with Cre-Neo vector DNA and selected by G418 to remove the puromycin selection marker in the genome. CTL, control cells that carry the random insertion of donor vector were transfected with Cre-Neo vector DNA and selected by G418. ALIC1-2 clone cells carry the targeting allele. (C) Downregulation of IGF1R by cis competition in two selected cell clones (ALIC1 and ALIC2). Real-time PCR was performed to quantitate the expression level of IGF1R and IRAIN. All data shown are mean ± SD. **p < 0.01 as compared with the control cells (CTL). (D) Western blot of IGF1R in targeted cell clones. **p < 0.01 as compared with the control cells (CTL). All data shown are mean ± SD. (E) IGF1R rescue assay. The CMV promoter-IRAIN knockin (ALIC) cells were treated with IRAIN siRNA (siIRAIN) and control siRNA (siCT) using lipofectamine RNAiMax. Forty-eight hours after transfection, cells were collected for the measurement of IGF1R using qPCR. The PCR value was standardized by setting the CTL control as 1. Error bars represent the SE of the average of three independent PCR reactions. All data shown are mean ± SD. *p < 0.05 as compared with the siCT control. (F) IRAIN overexpression assay. MDA-MB-231 tumor cells were infected with lentiviruses carrying the 5K IRAIN cDNA insert or the copGFP vector control. After viral infection, cells were selected by puromycin and were collected for the quantitation of IRAIN and IGF1R using qPCR. The copGFP vector control was set as 1 for standardization. Error bars represent the SE of the average of three independent PCR reactions. All data shown are mean ± SD. **p < 0.01, *p < 0.05 as compared with the vector control.
The puromycin selection marker was removed from cell clone 7 by transfecting the cells with CMV-Cre-Neo vector DNA. Ultimately, two cell clones were used for subsequent studies (ALIC1, ALIC2). As a control, RIC cells were also treated with CMV promoter (pCMV)-Cre-Neo DNA (CTL1, CTL2). Two ALIC clones showed clear targeting to the IGF1R region as compared with the control cells (CTL) (Figure 2B). We amplified the whole targeting region, cloned it into a pJet vector, and performed DNA sequencing for the recombination sites (Figure S2). DNA sequencing confirmed the correct targeting of IGF1R/IRAIN by Cas9-mediated homologous recombination.
Using qPCR, we showed that IRAIN was increased by ALIC targeting, while IGF1R was significantly downregulated in ALIC1-2 cells (Figure 2C), presumably due to the antisense lncRNA cis competition. Using western blotting, we confirmed that the IGF1R protein was also downregulated in targeted cells (Figure 2D).
Rescue of IGF1R by IRAIN siRNAs in ALIC-Targeted Cells
To further delineate the role of ALIC targeting, we examined whether the cis inhibition of IGF1R could be rescued by IRAIN knockdown using small interfering RNAs (siRNAs). The CMV-IRAIN knockin ALIC cells were treated with three IRAIN siRNAs and were collected for quantitation of IGF1R and IRAIN. siRNA treatment decreased IRAIN lncRNA in ALIC-targeted cells (Figure S3). As expected, IGF1R was inhibited in cis by IRAIN in scramble control (siCT) control cells (ALIC + siCT). In siRNA-treated cells (ALIC + IRAIN-siRNA [siIRAIN]), however, the siRNA-induced IRAIN knockdown partially rescued the downregulated IGF1R (Figure 2E). These data suggest the siRNA-induced post-transcriptional knockdown of IRAIN lncRNA cannot fully release the cis inhibition of IGF1R-induced by ALIC targeting.
In a separate study, we also examined whether lentivirus-overexpressed IRAIN was able to affect the IGF1R. We used a lentivirus to overexpress the IRAIN 5K cDNA in MDA-MB-231 tumor cells. The copGFP lentiviral vector was used as the negative control (vector). After selection by puromycin, cells were collected for gene expression analysis. As seen in Figure 2F, tumor cells overexpressed IRAIN after lentiviral transfection. However, the trans overexpression of IRAIN did not reduce the expression of the endogenous IGF1R as originally expected. As a matter of fact, the IGF1R mRNA was slightly increased in these cells, although the specific mechanism is currently unknown.
Targeted Inhibition of IGF1R Alters the Phenotypes of MDA-MB-231 Tumor Cells
After the ALIC targeting of IGF1R, we examined the phenotypes of the MDA-MB-231 breast cancer cells. First, we measured cell proliferation in targeted cells. As shown in Figure 3A, the rebalance of the IGF1R/IRAIN ratio was associated with reduced proliferation in two MDA-MB-231 cell clones.
Figure 3.

Correction of the Abnormal IGF1R/IRAIN Production Alters the Phenotypes of Tumor Cells
(A) Cell proliferation. After ALIC targeting, two cell clones were collected for analysis of cell proliferation using the MTT assay. Cell growth was measured as the relative absorbance by setting 0 hour as 1. All experiments were performed in triplicate. The data shown are mean ± SD. (B) Tumor sphere colonies as measured by the soft agar assay. MDA-MB-231 cell colonies were stained on day 15. (C) Quantitation of tumor sphere colonies. Tumor spheres were stained by 0.005% crystal violet and quantitated as the average sphere colonies per microscope field. All data shown are mean ± SD. *p < 0.05, **p < 0.01 as compared with the CTL control. (D) Migration of MBA231 cells. Cell migration was measured by insert plate assay. (E) Quantitation of migrated cells. Cell growth was measured as absorbance at 590 nm. All data shown are mean ± SD. ***p < 0.001 as compared with the CTL control. Note the decreased cell migration in the two targeted cell clones. (F) Cell invasion. Cells that invaded through the collagen-coated membrane of the Transwell were stained with crystal violet 24 hr after cell seeding. (G) Quantitation of invaded cells. Cell growth was measured as absorbance at 590 nm. All data shown are mean ± SD. ***p < 0.001 as compared with the CTL control.
We also examined if ALIC targeting of the IGF1R pathway affected the formation of tumor spheres. After IGF1R knockdown, MDA-MB-231 cells exhibited a significant reduction in tumor spheres as compared with CTL control cells (Figures 3B and 3C).
We then tested the activity of ALIC targeting in altering cell migration and invasion. Using the Transwell assay, we showed that ALIC targeting significantly decreased cell migration in both cell clones (Figures 3D and 3E). For cell invasion, MDA-MB-231 cells were seeded on a Matrigel-coated Transwell chamber after the ALIC knockdown of IGF1R. The number of cells that passed through the Matrigel to the bottom of the insert was quantified. The CTL control cells exhibited the malignant phenotype of invading across the Matrigel membrane. However, ALIC targeting of IGF1R resulted in significantly decreased invasion activity (Figures 3F and 3G; p < 0.01).
Downregulation of the IGF1R Pathway Results in S Phase Cell Cycle Arrest
We used flow cytometry to analyze cell cycle in ALIC-targeting clone cells (Figure 4). After IGF1R knockdown by ALIC targeting, there was an increase in the number of cells in S phase (23.3% in CTL cells versus 32.20% in clone #1 and 40.1% in clone #2). In parallel, there was a decrease in the number of cells in G2 phase in the targeted breast cancer cells.
Figure 4.

Cell Cycle Analysis
(A–C) FACS analysis of MDA-MB-231 cells after the IGF1R cis competition targeting by upregulating IRAIN. ALIC1 (B), ALIC2 (C), targeting clones; CTL (A), control cells. (D) Quantitation of the cell cycle.
CpG Methylation in the IGF1R Promoter
We examined if the ALIC targeting altered the epigenotype in the overlapping IGF1R promoter. Sodium bisulfite sequencing was used to compare the status of DNA methylation in the IGF1R promoter (Figure 5A).
Figure 5.

The Status of DNA Methylation in the IGF1R Promoters
(A) Schematic diagram of the IGF1R/IRAIN gene locus and the location of PCR primers. (B) CpG DNA methylation of site 1. Genomic DNAs were extracted from breast cancer cells. After treated with sodium sulfite, the IRAIN promoter DNA was amplified and sequenced. Open circles, unmethylated CpGs; solid circles, methylated CpGs. All the CpG islands were unmethylated in both the CTL control and ALIC-targeted cells. (C) CpG DNA methylation of site 2. Note the equal DNA hypomethylation in both the CTL control and ALIC-targeted cells.
In CTL control cells that were transfected with the Cre-Neo control vector in parallel with the ALIC cells, there was minimal DNA methylation of CpG islands at region #1 near the proximal promoter region. In two targeted cell clones, we did not detect any changes in DNA methylation at the CpG sites (Figure 5B). Similarly, there were no changes in the level of CpG methylation in the distal promoter region (Figure 5C). Thus, the antisense tumor suppressor lncRNA-induced suppression of IGF1R is not associated with the epigenetic modification in the promoter region.20
Identification of Genome-wide Targets of IRAIN lncRNA
To examine if IRAIN might also target other genes, we used a “nuclear lncRNA reverse transcription-associated trap” (RAT) method20, 24 to identify a gene network associated with IRAIN lncRNA (Figure S4). After crosslinking the chromatin, IRAIN lncRNA was first reverse transcribed in situ in the nucleus using IRAIN-specific complementary primers (Table S1) in a reaction mixture containing biotin-deoxycytidine triphosphate (dCTP). The IRAIN-interacting chromatin complex was pulled down with streptavidin beads, and target gene DNAs were purified for library construction and DNA sequencing. By RAT-Seq, we found that in addition to IGF1R, IRAIN lncRNA also bound to many gene targets that are closely related to tumor progression (Figure 6A). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that IRAIN lncRNA might be involved in many signal pathways that are related to tumor growth (Figures 6B and S5). IRAIN binds to its gene targets with consensus motif sequences containing poly(A)s and poly(G)s (Figure 6C).
Figure 6.

IRAIN lncRNA Binding Targets
(A) The IRAIN lncRNA interactome. The IRAIN RNA interaction was drawn based on the enrichment of the top RAT-Seq pathway target genes. (B) The KEGG pathway of IRAIN lncRNA targets. (C) The consensus binding motifs of IRAIN lncRNA. (D) Quantitation of IRAIN target genes in ALIC1-targeted cells. Gene expression was measured by qPCR. The values were standardized by setting the CTL control as 1. Error bars represent the SE of the average of three independent PCR reactions. All data shown are mean ± SD. *p < 0.05, **p < 0.01 as compared with the CTL control.
We then used qPCR to measure the mRNA abundance of several IRAIN target genes (Figure 6D) in ALIC-targeted cells, including NM23, RHOG, PID1, IGF2R, and FKBP2. Although the ALIC targeting did not significantly affect all target genes examined, NM23 and FKBP3 were dramatically upregulated in ALIC cells. Similarly, the lentivirus-overexpressed IRAIN also affected these target genes in MDA-MB-231 breast cancer cells (Figure S6). NM23 is a well-established metastasis suppressor.25, 26 Thus, IRAIN may affect tumor cell growth by regulating its downstream gene targets.
Discussion
The increased activation of the IGF1R pathway is a well-established target for breast cancer drug therapy. We have recently identified IRAIN, an intragenic antisense lncRNA in the IGF1R locus. IRAIN is s tumor suppressor that may function as a “cis regulatory competitor” to compete with the overlapping promoter of IGF1R.20 In breast cancer, IRAIN lncRNA is downregulated.21 Lack of antisense cis competition releases the suppression of the IGF1R promoter. The activated IGF1R is associated with breast cancer metastasis. We have developed an ALIC strategy to correct the aberrant IGF1R/IRAIN pathway. In this approach, the weak promoter of IRAIN in tumors is activated by CRISPR Cas9 gene editing. The increased transcription of IRAIN competes in cis with the overlapping IGF1R promoter, leading to a rebalance of IGF1R/IRAIN transcription. Thus, this study may present a useful strategy to precisely target the aberrantly upregulated IGF1R pathway in breast cancer cells.
CRISPR-Cas9 technology has been employed as a simple, efficient, and relatively inexpensive technology to precisely edit genes for various applications, including transcriptional regulation, epigenetic control, and chromosome translocation.27, 28, 29 Recently, this editing system has been harnessed to correct disease genes in human embryo genomes.30, 31, 32 By tethering the C terminus of the catalytically inactive dCas9 to three epigenetic suppressor domains, we found that the synthetic dCas9 epi-suppressor gRNAs induced significant decreases in the transcription activity of the GRN target gene promoter in Hep3B hepatoma cells.33 In this study, we have successfully applied the Cas9 system to introduce a powerful promoter in front of the IRAIN lncRNA. The Cas9-mediated ALIC approach leads to an increase in IRAIN antisense lncRNA, which induces the downregulation of the sense IGF1R coding RNA through the cis transcription competition over the overlapping promoter.
It is known that several lncRNAs regulate their target genes in cis by directly binding to their promoters and enhancers.34, 35, 36 Airn is expressed on mouse chromosome 17 from a paternal promoter located in intron 2 of the Igf2r gene, which is expressed exclusively from the maternal allele. Airn transcriptionally overlaps the Igf2r promoter. Airn expression is necessary to initiate the cis paternal-specific silencing of the Igf2r gene.37, 38 In developing ventral forebrain, Evf2 lncRNA controls expression of Dlx5, Dlx6, and Gad1 in both trans and cis by recruiting DLX and MECP2 transcription factors to important DNA regulatory elements in the intergenic region.39 Expression of lncRNAs is required for initiating X chromosome inactivation.40, 41, 42, 43 For example, the spreading of Xist lncRNA along one X chromosome in cis initiates chromosome-wide silencing. The X (inactive)-specific transcript, antisense (Tsix) overlaps with the entire Xist gene in an antisense orientation and silences Xist on the active X chromosome. Previously, we demonstrated that Kcnq1ot1 lncRNA uses its 5′ region RNA as a scaffold to orchestrate a long-range intrachromosomal loop between the imprinting control region KvDMR1 and the Kcnq1 promoter. With this spatial proximity, the imprinting signal in the KvDMR1 can be directly delivered to the Kcnq1 promoter in cis.44 Additionally, many enhancer DNA elements can encode unpolyadenylated enhancer RNAs (eRNAs) that tightly regulate neighboring coding genes.45, 46 However, these eRNAs function primarily as activators rather than repressors when targeting promoters.47, 48
In the current study, using the CRISPRCas9 gene-editing system, we also show that the actively transcribed IRAIN antisense lncRNA is able to downregulate in cis the expression of the overlapping IGF1R. These data suggest a useful approach to modulate gene activity efficiently using a cis competition mechanism. This is in contrast to siRNA and locked nucleic acids (LNA) oligo techniques, which function at the posttranscriptional level. siRNAs bind to the target RNA and induce RNA degradation by the Dicer-Ago2 pathway, inducing the inhibition of gene expression or translation. The data from our rescue assay also demonstrate that the IRAIN siRNA rescue experiment only shows a minor restoration in IGF1R mRNA levels, while IRAIN levels are almost back to normal levels. The siRNA-mediated post-transcriptional knockdown of IRAIN cannot fully counteract the cis inhibition of IGF1R induced by ALIC targeting. This suggests that it is not the sequence of IRAIN but the cis transcription competition that is most important of the effect on IGF1R. Future studies are needed to address whether the siRNA-mediated knockdown in the rescue assay or the trans overexpressed IRAIN is able to alter the phenotypes in breast cancer cells, including xenograft tumor studies in vivo.
Using nuclear RNA RAT sequencing (RAT-Seq), we have identified a gene network associated with IRAIN lncRNA. We found that IRAIN bound to many target genes that are closely related to tumor progression. We used qPCR to quantitate target genes in ALIC-targeted cells, including NM23, RHOG, PID1, IGF2R, and FKBP3. Among them, NM23 and FKBP3 were significantly upregulated in ALIC cells. NM23 has been extensively studied for its role as a metastasis suppressor.25, 26 In humans, at least eight NM23 family genes that encode for nucleoside diphosphate kinases or for homologous isoforms have been identified. NM23 participates in the regulation of a broad spectrum of cellular pathways involved in development, differentiation, proliferation, endocytosis, and apoptosis.49, 50 Ectopic expression of NM23 has been shown to reduce metastasis in several tumor models.51 The molecular mechanisms for the role of NM23 as a metastasis suppressor have so far remained unclear. There is a suggestion that NM23 and its interacting partner STRAP may physically interact with p53 and positively regulate p53-induced apoptosis and cell cycle arrest.52 In addition, NM23 may regulate many genes with relevance to breast cancer, including MET, EDG2, L1CAM, SMO, and PTN.53 FKBP3 (FK506-binding protein 3), a member of the immunophilin protein family, plays a role in immunoregulation and basic cellular processes involving protein folding and trafficking by binding to the immunosuppressants FK506 and rapamycin.54 We found that ectopic expression of the IRAIN 5K cDNA also significantly upregulated FKBP3 (Figure S6). Thus, IRAIN may also directly affect tumor metastasis by affecting its downstream gene targets, like NM23 metastasis suppressor.
In summary, the IGF1R-IRAIN locus is dysregulated in breast cancers due to the upregulated IGF1R growth signal and the downregulated IRAIN lncRNA tumor suppressor. We show that targeted activation of IRAIN by ALIC targeting suppressed IGF1R using a cis competition mechanism. Rebalance of the IGF1R pathway provides a molecular basis for the development of precision therapy against breast cancer. Thus, ALIC targeting may provide a useful strategy to target IGF1R in precision tumor therapy. Future studies are needed to learn if ALIC can be used to develop novel therapies against other cancer target genes that may not have an antisense lncRNA.
Materials and Methods
Construction of the IGF1R Targeting and Donor Vectors
In breast cancer cells, IGF1R is upregulated in parallel with the downregulated IRAIN tumor suppressor lncRNA. To precisely target this unbalanced IGF1R/IRAIN pathway, we used a CRISPRCas9 gene-editing system to activate the weak IRAIN promoter in breast cancer cells (Figures 1A and 1B). We constructed the Cas9-IRAIN-gRNA-targeting vector by cloning two IRAIN promoter gRNAs into the lenti-CRISPR-EGFP-gRNA vector (Addgene Plasmid #51761). A U6-gRNA1-T5-H1-gRNA2 cassette was synthesized by joining the H1 promoter with gRNA oligonucleotides from the IRAIN promoter: IRAIN-gRNA1, 5′-GCCCCAGTCCGCGTCCCACT-3′; IRAIN-gRNA2, 5′-CATTTCTTACCAGACGTTTA-3′; IRAIN-gRNA3, 5′-CCACCTAAACGACTTAGTAA-3′; IRAIN-gRNA4, 5′-GCCCAGATCTGTGGAAGGTC-3′; respectively (Figures S1A and S1B). The expression cassette was inserted downstream of the U6 promoter in the vector using Pme I and Not I.
The donor vector was constructed by PCR amplification of two arm fragments from the IRAIN locus and cloned into a targeting vector containing both the pCMV-loxP-puromycin-loxP positive selection cassette and the pPGK-thymidine kinase (TK) negative selection marker (Figure 1C).55 Two arm fragments were amplified from the human genome DNA using the following primers: SJ016F, 5′-TCAAAATTTTATCGATATTATCTGGCTATCACTCAGAACCT-3′, and SJ017, 5′-GCGTATATCTGGCCCGTACATCTTCGAAGATAAGTACGGTTTAGAAGACACG-3′ for the Arm 1 fragment; and SJ022, 5′-CTGAGTCGACTCTAGACAGCCTTTCTGAATTGCCCCGGT-3′, and SJ023, 5′-TTAAATCGACGCTAGCCCTCGGCTGTGACCTTCAGCGAGC-3′ for the Arm 2 fragment. They were cloned into the Cas9 donor vector using Cla1/BsrG1 and Xba1/Nhe1 sites, respectively. The pCMV-controlled puromycin in the vector was used as the positive selection marker and the pPGK-TK cassette as the negative selection marker.
Targeting the IGF1R/IRAIN Pathway in Breast Cancer Cells
IGF1R targeting was accomplished by introducing a strong CMV promoter immediately upstream of IRAIN using the CRISPRCas9 gene-editing system. First, the IGF1R locus was targeted by Cas9 using four IGF1R gRNAs in two targeting vectors. Second, a donor vector that contains a strong CMV promoter was used to target the IGF1R locus through homologous recommendation. After targeting, the CMV promoter drove the overexpression of the antisense IRAIN lncRNA, which in turn competes with the transcription of the sense IGF1R coding mRNA (Figure 1C).
Breast cancer cell line MDA-MB-231 was purchased from American Type Culture Collection (ATCC). Cells were grown in RP1640 media, supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Both the targeting vector and the donor vectors were co-transfected into MDA-MB-231 cells using Lipofectamine 2000 (Invitrogen, CA) following the protocol provided by the kit. Two days after co-transfection, cells were positively selected using 1 mg/mL puromycin for 3 days and then were negatively selected using 250 μM ganciclovir55 (Figure S1C). Treatment of cells with ganciclovir should reduce the random insertion of the donor vector in the genome. A total of twelve stable cell clones were collected, and genomic DNA was purified to screen for homologous knockin recombination, using primers located in the genomic DNA and vectors (Table S1). As the study control, MDA-MB-231 cells were transfected with only the donor vector DNA and were selected with puromycin. These control cells contained randomly inserted donor vector DNA in the genome (RIC cells).
Removal of Puromycin Selection Marker in Targeting Cells
Among targeted cells, clone 7 cells that contained the minimum randomly integrated vector DNA were selected for the study. The cells were transfected with a pCMV-Cre-Neo vector DNA using Lipofectamine 2000 (Invitrogen, CA) and were treated with G418 (1 mg/mL, Invitrogen, CA). The expression of the Cre recombinase removed the puromycin marker gene at the targeted IGF1R and IRAIN loci and generated the pCMV-IRAIN knockin cells (ALIC targeting). As a control, RIC cells that carry the random insertion of the donor vector DNA were also transfected with pCMV-Cre-Neo DNA and were treated with G418 in parallel with the ALIC cells (CTL cells).
Validation of ALIC Targeting by DNA Sequencing
ALIC targeting produced cell clones that contain some randomly inserted donor vector DNA, which may interfere with the signal in Southern blotting. Thus, we used PCR to amplify the entire targeted region and confirmed ALIC targeting by DNA sequencing. The targeted region was amplified by primers SJ131 and JH583 (Table S1) and was cloned into a pJet vector for sequencing. The regions covering the donor arms and the inserted promoter were validated by DNA sequencing.
Quantitation of IGF1R/IRAIN Expression
Total RNA was extracted from tissues by TRI-REAGENT (Sigma, CA), and cDNA was synthesized with RNA reverse transcriptase as previously described.56, 57 The resulting cDNA was used to quantitate the expression of IGF1R coding RNA and IRAIN lncRNA using primers listed in Table S1. In brief, PCR was performed under liquid wax in a 6 μL reaction containing 2 μL of 3 × Klen-TaqI Mix, 2 μL cDNA, and 1 μL of each 2.5 μM primer. After incubation at 95°C for 2 min, IGF1R and IRAIN cDNAs were amplified by 32 cycles of 95°C for 30 s, 65°C for 30 s of annealing, and 72°C for 35 s of extension, and finally with extension at 72°C for 5 min. Amplified PCR products of the expected size were quantified by densitometric measurements and normalized to β-actin values. For qPCR analysis, cDNA samples were amplified using CFX96 real-time system (Bio-Rad) by SYBR PrimeScript RT-PCR Kit (Takara). mRNA levels were quantitated by normalizing Ct value over β-actin (housekeeping gene) as previously described.58
The IGF1R-Targeting Rescue Assay
To further test the role of ALIC targeting, we determined whether the phenotypes of the IGF1R-targeted cells could be rescued by knockdown of IRAIN using siRNAs. Three IRAIN-specific RNAi oligonucleotides were chemically synthesized by Wiewsolid Biotech (Beijing, China). Cells were plated in 6-well plates at 5 × 105 per well. Twenty-four hours after plating, 40 pmol of siIRAIN and scramble controls siCTs (Table S1) were transfected into cells using Lipofectamine RNAiMax (Invitrogen, CA) following the manufacturer’s protocol. Forty-eight hours after transfection, cells were collected and total RNA was isolated using Trizol (Sigma, MO). The expression of IGF1R and IRAIN was measured by qPCR.
IRAIN Overexpression Assay
To examine whether a virally expressed IRAIN lncRNA would affect IGF1R in trans, we overexpressed IRAIN in MDA-MB-231 tumor cells. As previously reported,20 a 5.4-kb full-length IRAIN cDNA was cloned into pCMV-copGFP/Puro vector to construct the IRAIN 5K cDNA vector (IRAIN5K). Lentiviruses were packaged in 293 cells and were used to infect MDA-MB-231 cells. As a control, MDA-MB-231 cells were infected with the CMV-copGFP/Puro empty vector (Vector). After puromycin selection, cells that stably expressed IRAIN or the control vector were collected for gene analysis. qPCR was used to measure the expression of IGF1R and target genes.
Western Blot Analysis of IGF1R in ALIC-Targeted Cells
Total cell protein was extracted with radioimmunoprecipitation assay (RIPA) buffer supplemented with cocktail protease inhibitor (Roche), and the protein concentration was determined by a Pierce BCA protein assay kit as previously described.59 Twenty micrograms of total cell protein were separated on 12% SDS-PAGE and electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes (0.45 μm, Millipore, Billerica, MA, USA). After blocking with 5% skim milk, the membranes were incubated with monoclonal antibodies against IGF1R (Santa Cruz Biotechnology, CA) and were detected with the enhanced chemiluminescence system (ECL, Thermo). Protein expression levels were determined semiquantitatively by densitometric analysis with the Quantity One software (Bio-Rad, CA).60, 61
RAT Assay
A nuclear lncRNA RAT assay20, 62 was used in this study to determine the genome-wide targets that interact with IRAIN lncRNA (Figure S4). In brief, stable MDA-MB-231 clone cells were cross-linked with 2% formaldehyde and lysed with cell lysis buffer (10 mM Tris [pH 8.0], 10 mM NaCl, 0.2% NP-40, 1× protease inhibitors). The nuclei were centrifuged and suspended in 1× reverse transcription buffer, and gene strand-specific reverse transcription was performed in situ in the nucleus with biotin-dCTP. The cells were incubated for 50 min with Maxima Reverse Transcriptase (Thermo Fisher Scientific, CA) at 60°C, and the reaction was stopped by heating at 85°C for 5 min. The nuclei were lysed by adding 0.3% SDS at 37°C for 1 hr. Triton X-100 was then added to a final concentration of 1.8% to sequester the SDS. The biotinylated-IRAIN cDNA/chromatin DNA complex was purified by streptavidin magic beads (Invitrogen, CA). After washing, the pulled-down sample was treated with 10 mg/mL proteinase K at 65°C overnight to reverse the cross-links. Following incubation with 0.4 μg/mL RNase A for 30 min at 37°C, the IRAIN cDNA/DNA was extracted and sonicated on ice using a Branson sonicator. The sonicated DNAs were further digested by Mbo1 into small fragments and ligated with the NEBNext adaptors (NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina) to construct the library. The library DNAs were subjected to Illumina sequencing by Shanghai Biotechnology (Shanghai, China).
RAT-Seq Data Analysis
The RAT-Seq reads were mapped to the human genome (hg38) using Bowtie2.63 MACs2 was used to identify the DNA regions that interacted with the IRAIN lncRNA. Target genes were obtained from GENCODE64 and the upstream 2 kb and downstream 1 kb of the transcription start sites were defined as the gene promoter regions. The peaks were visualized using the Integrative Genomics Viewer (IGV) tools.65, 66
Functional Enrichment Analysis and GSEA
The GseaPreranked tool in GSEAv2.067 was used to perform gene set enrichment analysis (GSEA) on gene promoters that were bound by IRAIN. We obtained the gene sets from the Gene Set Knowledgebase (GSKB) (https://bioconductor.org/packages/release/data/experiment/html/gskb.html) for pathways and functional categories. The genes that interacted with IRAIN were sorted by the qValue (−log10) of the peak signal. Statistical significance was assessed by comparing the enrichment score to enrichment results generated from 1,000 random permutations of the gene sets to obtain p values.
Tumor Sphere-Forming Assay
The tumor clonogenic assay was performed using the method as previously described.56, 68 Cells were digested, centrifuged, and re-suspended in DMEM to form single-cell suspensions. Cells were diluted to 2 × 103 cells/mL in 0.25 mL DMEM-agarose. The plates were incubated at 37°C, 5% CO2. After ∼2 weeks, colonies were visualized by staining with 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma, MO) for 3 hr. Cloning forming efficiency (CFE) was defined as the number of colonies/number of inoculated cells × 100%.
Cell Proliferation Assay
Cell survival was measured using the MTT assay as previously described.58, 69 In brief, cells (1 × 104/well) were plated onto 96-well plates and were incubated with 20 μL 5 mg/mL MTT (Sigma, MO) per well at 37°C for 4 hr. The absorbance was measured at 490 nm. Cell viability (%) was calculated based on the following equation: Cell viability (%)1/4(Asample/Acontrol) × 100%, where Asample and Acontrol represent the absorbance of the sample and control wells, respectively.
Cell Migration and Invasion Assay
Cell migration and invasion assays were carried out using Transwell membrane filters inserted in 24-well tissue culture plates (6.5-mm diameter, 8-μm pore size) (Corning, MA) following the method as previously described.70, 71 For the migration assay, 4 × 104 cells suspended in serum-free medium were seeded on the top chamber of Transwell filters. Serum-containing medium was added to the bottom chamber and incubated for 16 hr at 37°C. The nonmigrating cells were removed by wiping the upper side of the filter, and the migrating cells on the bottom side of the filter were fixed with 4% formaldehyde and stained with crystal violet.
Cell invasion was measured using a similar protocol except that cells were seeded in Transwell chambers coated with Matrigel (Invitrogen, CA, USA). Each assay represents the average of three independent experiment.56
Cell Cycle Analysis by Flow Cytometry
Cells were washed twice with PBS, pelleted, and fixed with cold 70% ethyl alcohol for at least 30 min. After being washed twice with cold PBS, cells were incubated with 200 μg/mL RNase A for 30 min at 37°C. The cells were stained with 100 μg/mL propidium iodide for 30 min at room temperature. The samples were immediately analyzed by flow cytometry. Cell cycle phase distribution was determined using Cell Quest Pro software.56, 59
Statistical Analysis
The experimental data are expressed as mean ± SD. Data were analyzed using SPSS software (version 16.0; SPSS, IL). Student’s t test or one-way ANOVA (Bonferroni test) was used to compare statistical differences for variables among treatment groups. Results were considered statistically significant at p < 0.05.
Author Contributions
J.-F.H., J.C., J.S., W.L., and S.Z. conceived and designed the study; L.P., X.W., L.K., Z.L., Y.N., Z.D., D.Y., L.Z., L.J., N.C., and D.L. performed the experiments; J.-F.H. wrote the paper; A.R.H. reviewed and edited the manuscript. All authors read and approved the manuscript.
Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grants 31430021 and 81272294), the National Basic Research Program of China (973 Program) (2015CB943303), the Natural Science Foundation of Jilin Science and Technique (grant 20180101117JC), and the California Institute of Regenerative Medicine (CIRM) (grant RT2-01942) to J.-F.H.; the National Natural Science Foundation of China (grants 81372835 and 81670143) and the Jilin Science and Technique International Collaboration (grant 20130413010GH) to W.L.; the Key Project of Chinese Ministry of Education (grant 311015), the National Natural Science Foundation of China (grant 81672275), the National Key Research and Development Program of China (grant 2016YFC13038000), the Natural Science Foundation of Jilin Province (grant 20150101176JC), the Research on Chronic Noncommunicable Diseases Prevention and Control of National Ministry of Science and Technology (grant 2016YFC1303804), and the National Health Development Planning Commission Major Disease Prevention and Control of Science and Technology Plan of Action, Cancer Prevention and Control (grant ZX-07-C2016004) to J.C.; the Junior National Natural Science Foundation of China (grant #81502277) to L.K.; the Junior National Natural Science Foundation of China (grant #81600150) and the Bethune B project grant of Jilin University (#2015211) to J.S.; the Jilin Science and Technique International Collaboration grant (20180414065GH) to D.Y.; and the Department of Veterans Affairs.
Footnotes
Supplemental Information includes six figures and one table and can be found with this article online at https://doi.org/10.1016/j.omtn.2018.04.013.
Contributor Information
Jiuwei Cui, Email: cuijiuwei@jlu.edu.cn.
Ji-Fan Hu, Email: jifan@stanford.edu.
Supplemental Information
References
- 1.Devin J.L., Bolam K.A., Jenkins D.G., Skinner T.L. The influence of exercise on the insulin-like growth factor axis in oncology: physiological basis, current, and future perspectives. Cancer Epidemiol. Biomarkers Prev. 2016;25:239–249. doi: 10.1158/1055-9965.EPI-15-0406. [DOI] [PubMed] [Google Scholar]
- 2.Singh P., Alex J.M., Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med. Oncol. 2014;31:805. doi: 10.1007/s12032-013-0805-3. [DOI] [PubMed] [Google Scholar]
- 3.Kasprzak A., Kwasniewski W., Adamek A., Gozdzicka-Jozefiak A. Insulin-like growth factor (IGF) axis in cancerogenesis. Mutat. Res. Rev. Mutat. Res. 2017;772:78–104. doi: 10.1016/j.mrrev.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Solarek W., Czarnecka A.M., Escudier B., Bielecka Z.F., Lian F., Szczylik C. Insulin and IGFs in renal cancer risk and progression. Endocr. Relat. Cancer. 2015;22:R253–R264. doi: 10.1530/ERC-15-0135. [DOI] [PubMed] [Google Scholar]
- 5.Denduluri S.K., Idowu O., Wang Z., Liao Z., Yan Z., Mohammed M.K., Ye J., Wei Q., Wang J., Zhao L., Luu H.H. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015;2:13–25. doi: 10.1016/j.gendis.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Motallebnezhad M., Aghebati-Maleki L., Jadidi-Niaragh F., Nickho H., Samadi-Kafil H., Shamsasenjan K., Yousefi M. The insulin-like growth factor-I receptor (IGF-IR) in breast cancer: biology and treatment strategies. Tumour Biol. 2016;37:11711–11721. doi: 10.1007/s13277-016-5176-x. [DOI] [PubMed] [Google Scholar]
- 7.Bergman D., Halje M., Nordin M., Engström W. Insulin-like growth factor 2 in development and disease: a mini-review. Gerontology. 2013;59:240–249. doi: 10.1159/000343995. [DOI] [PubMed] [Google Scholar]
- 8.Voudouri K., Berdiaki A., Tzardi M., Tzanakakis G.N., Nikitovic D. Insulin-like growth factor and epidermal growth factor signaling in breast cancer cell growth: focus on endocrine resistant disease. Anal. Cell. Pathol. (Amst.) 2015;2015:975495. doi: 10.1155/2015/975495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapuis N., Tamburini J., Cornillet-Lefebvre P., Gillot L., Bardet V., Willems L., Park S., Green A.S., Ifrah N., Dreyfus F. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica. 2010;95:415–423. doi: 10.3324/haematol.2009.010785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallardo A., Lerma E., Escuin D., Tibau A., Muñoz J., Ojeda B., Barnadas A., Adrover E., Sánchez-Tejada L., Giner D. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br. J. Cancer. 2012;106:1367–1373. doi: 10.1038/bjc.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat. Rev. Cancer. 2012;12:159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 12.Pierre-Eugene C., Pagesy P., Nguyen T.T., Neuillé M., Tschank G., Tennagels N., Hampe C., Issad T. Effect of insulin analogues on insulin/IGF1 hybrid receptors: increased activation by glargine but not by its metabolites M1 and M2. PLoS ONE. 2012;7:e41992. doi: 10.1371/journal.pone.0041992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danielsen S.A., Eide P.W., Nesbakken A., Guren T., Leithe E., Lothe R.A. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim. Biophys. Acta. 2015;1855:104–121. doi: 10.1016/j.bbcan.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Farabaugh S.M., Boone D.N., Lee A.V. Role of IGF1R in breast cancer subtypes, stemness, and lineage differentiation. Front. Endocrinol. (Lausanne) 2015;6:59. doi: 10.3389/fendo.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rieder S., Michalski C.W., Friess H., Kleeff J. Insulin-like growth factor signaling as a therapeutic target in pancreatic cancer. Anticancer. Agents Med. Chem. 2011;11:427–433. doi: 10.2174/187152011795677454. [DOI] [PubMed] [Google Scholar]
- 16.Marzec K.A., Baxter R.C., Martin J.L. Targeting insulin-like growth factor binding protein-3 signaling in triple-negative breast cancer. BioMed Res. Int. 2015;2015:638526. doi: 10.1155/2015/638526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochnik A.M., Baxter R.C. Combination therapy approaches to target insulin-like growth factor receptor signaling in breast cancer. Endocr. Relat. Cancer. 2016;23:R513–R536. doi: 10.1530/ERC-16-0218. [DOI] [PubMed] [Google Scholar]
- 18.Chen H.X., Sharon E. IGF-1R as an anti-cancer target—trials and tribulations. Chin. J. Cancer. 2013;32:242–252. doi: 10.5732/cjc.012.10263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beckwith H., Yee D. Minireview: were the IGF signaling inhibitors all bad? Mol. Endocrinol. 2015;29:1549–1557. doi: 10.1210/me.2015-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun J., Li W., Sun Y., Yu D., Wen X., Wang H., Cui J., Wang G., Hoffman A.R., Hu J.F. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic Acids Res. 2014;42:9588–9601. doi: 10.1093/nar/gku549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang L., Sun J., Wen X., Cui J., Wang G., Hoffman A.R., Hu J.F., Li W. Aberrant allele-switch imprinting of a novel IGF1R intragenic antisense non-coding RNA in breast cancers. Eur. J. Cancer. 2015;51:260–270. doi: 10.1016/j.ejca.2014.10.031. [DOI] [PubMed] [Google Scholar]
- 22.Feng J., Sun Y., Zhang E.B., Lu X.Y., Jin S.D., Guo R.H. A novel long noncoding RNA IRAIN regulates cell proliferation in non small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015;8:12268–12275. [PMC free article] [PubMed] [Google Scholar]
- 23.Lian Y., Wang J., Feng J., Ding J., Ma Z., Li J., Peng P., De W., Wang K. Long non-coding RNA IRAIN suppresses apoptosis and promotes proliferation by binding to LSD1 and EZH2 in pancreatic cancer. Tumour Biol. 2016;37:14929–14937. doi: 10.1007/s13277-016-5380-8. [DOI] [PubMed] [Google Scholar]
- 24.Wang H., Ge S., Qian G., Li W., Cui J., Wang G., Hoffman A.R., Hu J.F. Restoration of IGF2 imprinting by polycomb repressive complex 2 docking factor SUZ12 in colon cancer cells. Exp. Cell Res. 2015;338:214–221. doi: 10.1016/j.yexcr.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 25.Takács-Vellai K. The metastasis suppressor Nm23 as a modulator of Ras/ERK signaling. J. Mol. Signal. 2014;9:4. doi: 10.1186/1750-2187-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall J.C., Collins J., Marino N., Steeg P. The Nm23-H1 metastasis suppressor as a translational target. Eur. J. Cancer. 2010;46:1278–1282. doi: 10.1016/j.ejca.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donohoue P.D., Barrangou R., May A.P. Advances in industrial biotechnology using CRISPR-Cas systems. Trends Biotechnol. 2018;36:134–146. doi: 10.1016/j.tibtech.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Birling M.C., Herault Y., Pavlovic G. Modeling human disease in rodents by CRISPR/Cas9 genome editing. Mamm. Genome. 2017;28:291–301. doi: 10.1007/s00335-017-9703-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higashijima Y., Hirano S., Nangaku M., Nureki O. Applications of the CRISPR-Cas9 system in kidney research. Kidney Int. 2017;92:324–335. doi: 10.1016/j.kint.2017.01.037. [DOI] [PubMed] [Google Scholar]
- 30.Liang P., Xu Y., Zhang X., Ding C., Huang R., Zhang Z., Lv J., Xie X., Chen Y., Li Y. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 2015;6:363–372. doi: 10.1007/s13238-015-0153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang X., He W., Huang Y., Yu Q., Chen Y., Gao X., Sun X., Fan Y. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. J. Assist. Reprod. Genet. 2016;33:581–588. doi: 10.1007/s10815-016-0710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang L., Zeng Y., Du H., Gong M., Peng J., Zhang B., Lei M., Zhao F., Wang W., Li X., Liu J. CRISPR/Cas9-mediated gene editing in human zygotes using Cas9 protein. Mol. Genet. Genomics. 2017;292:525–533. doi: 10.1007/s00438-017-1299-z. [DOI] [PubMed] [Google Scholar]
- 33.Wang H., Guo R., Du Z., Bai L., Li L., Cui J., Li W., Hoffman A.R., Hu J.-F. Epigenetic targeting of Granulin in hepatoma cells by synthetic CRISPR dCas9 epi-suppressors. Mol. Ther. Nucleic Acids. 2018;11:23–33. doi: 10.1016/j.omtn.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engreitz J.M., Ollikainen N., Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat. Rev. Mol. Cell Biol. 2016;17:756–770. doi: 10.1038/nrm.2016.126. [DOI] [PubMed] [Google Scholar]
- 35.Krishnan J., Mishra R.K. Emerging trends of long non-coding RNAs in gene activation. FEBS J. 2014;281:34–45. doi: 10.1111/febs.12578. [DOI] [PubMed] [Google Scholar]
- 36.Kornienko A.E., Guenzl P.M., Barlow D.P., Pauler F.M. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59. doi: 10.1186/1741-7007-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santoro F., Mayer D., Klement R.M., Warczok K.E., Stukalov A., Barlow D.P., Pauler F.M. Imprinted Igf2r silencing depends on continuous Airn lncRNA expression and is not restricted to a developmental window. Development. 2013;140:1184–1195. doi: 10.1242/dev.088849. [DOI] [PubMed] [Google Scholar]
- 38.Latos P.A., Pauler F.M., Koerner M.V., Şenergin H.B., Hudson Q.J., Stocsits R.R., Allhoff W., Stricker S.H., Klement R.M., Warczok K.E. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science. 2012;338:1469–1472. doi: 10.1126/science.1228110. [DOI] [PubMed] [Google Scholar]
- 39.Bond A.M., Vangompel M.J., Sametsky E.A., Clark M.F., Savage J.C., Disterhoft J.F., Kohtz J.D. Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci. 2009;12:1020–1027. doi: 10.1038/nn.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Froberg J.E., Yang L., Lee J.T. Guided by RNAs: X-inactivation as a model for lncRNA function. J. Mol. Biol. 2013;425:3698–3706. doi: 10.1016/j.jmb.2013.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J.T., Bartolomei M.S. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–1323. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 42.Jégu T., Aeby E., Lee J.T. The X chromosome in space. Nat. Rev. Genet. 2017;18:377–389. doi: 10.1038/nrg.2017.17. [DOI] [PubMed] [Google Scholar]
- 43.Furlan G., Rougeulle C. Function and evolution of the long noncoding RNA circuitry orchestrating X-chromosome inactivation in mammals. Wiley Interdiscip. Rev. RNA. 2016;7:702–722. doi: 10.1002/wrna.1359. [DOI] [PubMed] [Google Scholar]
- 44.Zhang H., Zeitz M.J., Wang H., Niu B., Ge S., Li W., Cui J., Wang G., Qian G., Higgins M.J. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J. Cell Biol. 2014;204:61–75. doi: 10.1083/jcb.201304152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim T.K., Hemberg M., Gray J.M., Costa A.M., Bear D.M., Wu J., Harmin D.A., Laptewicz M., Barbara-Haley K., Kuersten S. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lai F., Orom U.A., Cesaroni M., Beringer M., Taatjes D.J., Blobel G.A., Shiekhattar R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501. doi: 10.1038/nature11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng J.H., Pan D.Z., Tsai Z.T., Tsai H.K. Genome-wide analysis of enhancer RNA in gene regulation across 12 mouse tissues. Sci. Rep. 2015;5:12648. doi: 10.1038/srep12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prabhu V.V., Siddikuzzaman, Grace V.M., Guruvayoorappan C. Targeting tumor metastasis by regulating Nm23 gene expression. Asian Pac. J. Cancer Prev. 2012;13:3539–3548. doi: 10.7314/apjcp.2012.13.8.3539. [DOI] [PubMed] [Google Scholar]
- 50.Kim H.D., Youn B., Kim T.S., Kim S.H., Shin H.S., Kim J. Regulators affecting the metastasis suppressor activity of Nm23-H1. Mol. Cell. Biochem. 2009;329:167–173. doi: 10.1007/s11010-009-0109-2. [DOI] [PubMed] [Google Scholar]
- 51.Marino N., Nakayama J., Collins J.W., Steeg P.S. Insights into the biology and prevention of tumor metastasis provided by the Nm23 metastasis suppressor gene. Cancer Metastasis Rev. 2012;31:593–603. doi: 10.1007/s10555-012-9374-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung H., Seong H.A., Ha H. NM23-H1 tumor suppressor and its interacting partner STRAP activate p53 function. J. Biol. Chem. 2007;282:35293–35307. doi: 10.1074/jbc.M705181200. [DOI] [PubMed] [Google Scholar]
- 53.Horak C.E., Lee J.H., Elkahloun A.G., Boissan M., Dumont S., Maga T.K., Arnaud-Dabernat S., Palmieri D., Stetler-Stevenson W.G., Lacombe M.L. Nm23-H1 suppresses tumor cell motility by down-regulating the lysophosphatidic acid receptor EDG2. Cancer Res. 2007;67:7238–7246. doi: 10.1158/0008-5472.CAN-07-0962. [DOI] [PubMed] [Google Scholar]
- 54.Kim K.H., Yeo S.G., Yoo B.C., Myung J.K. Identification of calgranulin B interacting proteins and network analysis in gastrointestinal cancer cells. PLoS ONE. 2017;12:e0171232. doi: 10.1371/journal.pone.0171232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu J.F., Vu T.H., Hoffman A.R. Genomic deletion of an imprint maintenance element abolishes imprinting of both insulin-like growth factor II and H19. J. Biol. Chem. 1997;272:20715–20720. doi: 10.1074/jbc.272.33.20715. [DOI] [PubMed] [Google Scholar]
- 56.Zhao X., Liu X., Wang G., Wen X., Zhang X., Hoffman A.R., Li W., Hu J.F., Cui J. Loss of insulin-like growth factor II imprinting is a hallmark associated with enhanced chemo/radiotherapy resistance in cancer stem cells. Oncotarget. 2016;7:51349–51364. doi: 10.18632/oncotarget.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han W., Li W., Zhang X., Du Z., Liu X., Zhao X., Wen X., Wang G., Hu J.F., Cui J. Targeted breast cancer therapy by harnessing the inherent blood group antigen immune system. Oncotarget. 2017;8:15034–15046. doi: 10.18632/oncotarget.14746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yin H., Chen N., Guo R., Wang H., Li W., Wang G., Cui J., Jin H., Hu J.F. Antitumor potential of a synthetic interferon-alpha/PLGF-2 positive charge peptide hybrid molecule in pancreatic cancer cells. Sci. Rep. 2015;5:16975. doi: 10.1038/srep16975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu D., Du Z., Li W., Chen H., Ye S., Hoffman A.R., Cui J., Hu J.F. Targeting Jurkat T lymphocyte leukemia cells by an engineered interferon-alpha hybrid molecule. Cell. Physiol. Biochem. 2017;42:519–529. doi: 10.1159/000477601. [DOI] [PubMed] [Google Scholar]
- 60.Song W., Li W., Li L., Zhang S., Yan X., Wen X., Zhang X., Tian H., Li A., Hu J.F., Cui J. Friend leukemia virus integration 1 activates the Rho GTPase pathway and is associated with metastasis in breast cancer. Oncotarget. 2015;6:23764–23775. doi: 10.18632/oncotarget.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang H., Chao K., Ng S.C., Bai A.H., Yu Q., Yu J., Li M., Cui Y., Chen M., Hu J.F., Zhang S. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol. 2016;17:58. doi: 10.1186/s13059-016-0901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang H., Li W., Guo R., Sun J., Cui J., Wang G., Hoffman A.R., Hu J.F. An intragenic long noncoding RNA interacts epigenetically with the RUNX1 promoter and enhancer chromatin DNA in hematopoietic malignancies. Int. J. Cancer. 2014;135:2783–2794. doi: 10.1002/ijc.28922. [DOI] [PubMed] [Google Scholar]
- 63.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang S., Zhong B., Chen M., Yang L., Yang G., Li Y., Wang H., Wang G., Li W., Cui J. Epigenetic reprogramming reverses the malignant epigenotype of the MMP/TIMP axis genes in tumor cells. Int. J. Cancer. 2014;134:1583–1594. doi: 10.1002/ijc.28487. [DOI] [PubMed] [Google Scholar]
- 69.Zhai Y., Chen X., Yu D., Li T., Cui J., Wang G., Hu J.F., Li W. Histone deacetylase inhibitor valproic acid promotes the induction of pluripotency in mouse fibroblasts by suppressing reprogramming-induced senescence stress. Exp. Cell Res. 2015;337:61–67. doi: 10.1016/j.yexcr.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 70.Li L., Song W., Yan X., Li A., Zhang X., Li W., Wen X., Zhou L., Yu D., Hu J.F., Cui J. Friend leukemia virus integration 1 promotes tumorigenesis of small cell lung cancer cells by activating the miR-17-92 pathway. Oncotarget. 2017;8:41975–41987. doi: 10.18632/oncotarget.16715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang L., Guo H., Lin C., Yang L., Wang X. Enrichment and characterization of cancer stem-like cells from a cervical cancer cell line. Mol. Med. Rep. 2014;9:2117–2123. doi: 10.3892/mmr.2014.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.