

Abstract
To study double-strand break (DSB)-induced mutations in mammalian chromosomes, we stably transfected thymidine kinase (tk)-deficient mouse fibroblasts with a DNA substrate containing a recognition site for yeast endonuclease I-SceI embedded within a functional tk gene. Cells were then electroporated with a plasmid expressing endonuclease I-SceI to induce a DSB, and clones that had lost tk function were selected. In a previous study of DSB-induced tk-deficient clones, we found that ∼8% of recovered tk mutations involved the capture of one or more DNA fragments at the DSB site. Almost half of the DNA capture events involved the I-SceI expression plasmid, and several events involved retrotransposable elements. To learn whether only certain DNA sequences or motifs are efficiently captured, in the current work we electroporated an I-SceI expression plasmid along with HaeIII fragments of φX174 genomic DNA. We report that 18 out of 132 tk-deficient clones recovered had captured DNA fragments, and 14 DNA capture events involved one or more fragments of φX174 DNA. Microhomology existed at most junctions between φX174 DNA and genomic sequences. Our work suggests that virtually any extrachromosomal DNA molecule may be recruited for the patching of DSBs in a mammalian genome.
INTRODUCTION
In eukaryotic cells, DNA double-strand breaks (DSBs) may be repaired by homologous recombination or non-homologous end-joining (NHEJ) (1–12). During DSB repair via homologous recombination, the broken DNA sequence interacts with a homologous donor sequence and genetic information is exchanged between identical or nearly identical DNA sequences. Homologous recombination is considered to be an accurate mode of repair. NHEJ is accomplished by the joining of DNA ends with no interaction between the broken molecule and a donor sequence. In NHEJ there is no requirement for homology at the DNA termini being joined, although NHEJ may be facilitated by short terminal homologies (2). NHEJ may be accompanied by the deletion or gain of genetic material prior to healing of the DSB.
DSB repair mechanisms generally tend to produce little genetic change. For example, DSB-induced homologous recombination is often accomplished by gene conversion without associated crossovers (7,9,13,14) and NHEJ is typically accompanied by loss (or gain) of only a few nucleotides. Indeed, precise ligation of DSBs with no alteration of sequences may be a major pathway for repair in mammalian genomes (9). However, in contrast with the idea that repair generally minimizes genetic change, it has been reported that NHEJ is sometimes associated with the insertion of novel DNA fragments into DSBs in yeast (15–18), plant somatic cells (19,20) and mammalian cells (6,8,12,21). Such DNA capture events may play important and ongoing roles in the evolution of eukaryotic genomes. Previously, we observed that ∼8% of recovered genomic DSB repair events in mouse fibroblasts were accompanied by the insertion of DNA sequences (12). Strikingly, nearly half of the DNA capture events involved a specific fragment derived from the I-SceI expression plasmid that had been electroporated into the cells to induce a genomic DSB. Several other DNA capture events involved retrotransposable elements. From previous work it was not clear whether the capability of being captured at a DSB is a property displayed by a limited number of DNA motifs or if, instead, any sufficiently abundant extrachromosomal DNA may be captured efficiently at a DSB. In this work we addressed this question by transfecting φX174 DNA into mouse fibroblasts in which a genomic DSB was simultaneously induced with I-SceI. We report that a variety of fragments of the φX174 genome were captured at the DSB with high efficiency, suggesting that any extrachromosomal DNA molecule may be recruited for repair of a DSB in a mammalian genome.
MATERIALS AND METHODS
Cell culture
Mouse Ltk– cells and derivatives were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 0.1 mM minimal non-essential amino acids (Gibco) and 50 µg gentamicin sulfate/ml. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
DSB repair substrate and cell lines
The construction of plasmid pYL1 (Fig. 1A) was described previously (12). Plasmid pYL1 contains a herpes simplex virus type 1 tk gene on a 2.5 kb BamHI fragment. Inserted into the SstI site at position 963 of the tk gene, numbering according to Wagner et al. (22), is a 24 bp oligonucleotide containing the 18 bp recognition sequence for I-SceI (Fig. 1B). Insertion of the 24 bp sequence does not disrupt tk function. Mouse Ltk– cells were electroporated with pYL1 DNA and stable tk-positive transformants containing pYL1 and resistant to medium containing hypoxanthine/aminopterin/thymidine (HAT; Sigma) were selected as previously described (12). Based on Southern blotting analysis (data not shown), we determined that cell lines 2 and 4 each contain a single integrated copy of pYL1. We used these two cell lines in the current study as well as in our previous study (12).
Figure 1.

DNA repair substrate. (A) Plasmid pYL1, depicted in linear form, contains a tk gene (open rectangle) on a 2.5 kb BamHI fragment. Inserted into the tk gene is a 24 bp double-stranded oligonucleotide (stripped segment) containing the 18 bp recognition site for yeast endonuclease I-SceI. Also shown are two PCR primers positioned 1.5 kb apart flanking the I-SceI site. (B) Sequence of the 24 bp double-stranded oligonucleotide inserted into the tk gene. For simplicity, only the ‘top’ strand of the oligonucleotide sequence is shown. The recognition site for I-SceI is underlined, and the sites of staggered cleavage are indicated. Cleavage by I-SceI produces a 3′ overhang of the 4 nt sequence ATAA on the top strand.
DSB-induction and recovery of tk-deficient mutants
Plasmid pCMV-I-SceI, kindly provided by Maria Jasin (Sloan Kettering), contains a gene encoding I-SceI endonuclease under the control of the cytomegalovirus promoter and expressible in mouse cells (23). Prior to DSB-induction, cell lines containing pYL1 were grown in HAT medium for 4 days to kill cells that had spontaneously mutated to tk–. HAT selection was removed, and cells were allowed to continue to grow in DMEM for 5 additional days before DSB-induction. To induce a DSB, cells (5 × 106) were suspended in 800 µl phosphate buffered saline (PBS) containing 20 µg pCMV-I-SceI alone or in conjunction with either 5 µg of a HaeIII digest of φX174 DNA (New England Biolabs) or 5 µg of supercoiled φX174 RFI DNA (New England Biolabs). Cells were electroporated in a Bio-Rad Gene Pulser set to 1000 V and 25 µF. For mock experiments, cells were electroporated in PBS alone. After electroporation, cells were plated at a density of 105 cells per 150 cm2 flask. The cells were grown in DMEM for 6 days under no selection and then re-fed with medium supplemented with 5 µg trifluorothymidine (TFT) per ml to select for tk-deficient clones as described previously (12). Cells were re-fed with TFT-containing medium every 3 days for ∼1 week until TFT-resistant (TFTR) colonies were picked. Individual TFTR clones were propagated further and genomic DNA was prepared from clones as described (9).
Southern blotting analysis
DNA was transferred to nitrocellulose filters and hybridized with a 32P-labeled probe specific for the herpes simplex virus type 1 tk gene or for φX174 genomic DNA as previously described (24). The φX174-specific probe was made from a HaeIII digest of φX174 genomic DNA (New England Biolabs).
PCR and DNA sequencing analysis
A segment of the tk gene spanning the position of the I-SceI-induced DSB was amplified from genomic DNA isolated from TFTR clones using the primers AL1 (5′-CCAGCGTCTTGTCATTGGCG-3′) and AW-22 (5′-CGGTGGGGTATCGACAGAGT-3′). Primer AL1 is nucleotides 308–327 of the coding strand of the herpes simplex virus type 1 tk gene, while primer AW-22 is nucleotides 1786–1767 of the non-coding strand of the tk gene. PCR reactions were accomplished using Ready-To-Go PCR beads (Pharmacia) using a ‘touchdown’ PCR protocol. The annealing temperature initially was set to 72°C and was progressively decreased in steps of 2°C down to 62°C with two cycles at each temperature. An additional 20 cycles were carried out at an annealing temperature of 60°C. PCR products were routinely processed with a Presequencing Kit (Amersham) and then sequenced using a Sequenase Kit Version 2.0 (Amersham) using primer AW-28 (5′-TCTACACCACACAACACCGCC-3′), nucleotides 812–831 of the tk coding strand, and primer AW43 (5′-GGCAAGGTCGGCGGGATGAG-3′), nucleotides 1111–1092 of the non-coding strand of the tk gene. Some sequencing was accomplished by automated sequencing using a Licor 4000L at the DNA Sequencing and Synthesis Core Facility in the Department of Biological Sciences at the University of South Carolina.
RESULTS
Experimental system
To study DSB repair-associated mutations in a mammalian genome, mouse Ltk– fibroblast cell lines were isolated that contain a stably integrated copy of the DNA substrate pYL1 (Fig. 1A). Plasmid pYL1 contains a functional herpes simplex virus tk gene with an inserted 24 bp oligonucleotide containing the 18 bp recognition site for yeast endonuclease I-SceI (Fig. 1B). To induce a genomic DSB in the integrated tk gene, cells were electroporated with plasmid pCMV-I-SceI, which expresses endonuclease I-SceI in mammalian cells. Due to the large size of the I-SceI recognition site (18 bp), it is likely that only a single genomic DSB is induced by expression of I-SceI. Linearized or supercoiled forms of φX174 DNA were electroporated along with pCMV-I-SceI in experiments designed to assess whether transfected DNA sequences may be efficiently captured at a genomic DSB. Mutagenic DSB repair events were recovered by selecting for cells that lost tk function and thus gained resistance to the thymidine analog TFT. Selection for loss-of-tk-function should in principle allow recovery of virtually any type of non-lethal DSB-induced mutation, including capture of DNA sequences.
Recovery of DSB-induced TFTR clones
Two cell lines (2 and 4) were isolated that contain one copy of construct pYL1. Electroporation of cells with pCMV-I-SceI alone or in conjunction with a HaeIII digest of φX174 DNA brought about a significant increase in the recovery of TFTR clones compared with mock electroporations (Table 1).
Table 1. TFTR colony frequencies (×105) following DSB-inductiona.
| Cell line | Type of DNA electroporated | |||
| |
pCMV-I-SceI |
pCMV-I-SceI + φX174/HaeIII |
pCMV-I-SceI + φX174 RF I |
Mock |
| 2 | 138 | 116 | 76 | 1.3 |
| 4 | 51 | 60 | 58 | 1.9 |
aCells were electroporated with either pCMV-I-SceI alone, pCMV-I-SceI plus a HaeIII digest of φX174 DNA, pCMV-I-SceI plus supercoiled φX174 RF I DNA, or PBS alone (mock) and then selected in TFT as described in Materials and Methods. The number of TFTR colonies recovered was divided by the number of cells plated following electroporation; this quotient was multiplied by 105 and is presented in the table.
tk sequences were PCR-amplified from genomic DNA isolated from a total of 132 TFTR clones (64 TFTR clones derived from cell line 2, 68 TFTR clones derived from cell line 4) following electroporation with pCMV-I-SceI plus the HaeIII digest of φX174 DNA. Primers were used that were positioned ∼1.5 kb apart from one another and flanked the I-SceI recognition site originally present in the integrated tk gene (see Fig. 1A). As expected, a PCR product of 1.5 kb was generated when parental genomic DNA was used as template (Fig. 2, lane 12). Among TFTR clones, 109 clones (82%) produced PCR products of ∼1.5 kb, suggestive of small insertions or deletions at the DSB site as seen previously (12). These clones were not analyzed further. Eighteen TFTR clones (13.6%) produced PCR products that were visibly larger than 1.5 kb (for example, Fig. 2, lanes 2 and 10). To be scored as ‘large’ in our assay, a PCR product had to be ∼1.6 kb or larger. The large PCR products were suggestive of capture of DNA sequences at the I-SceI-induced DSB. In our earlier work (12), a similar percentage of DSB repair events recovered from the very same two cell lines was associated with DNA capture. The large PCR products were sequenced and are described in further detail below. Five of the clones (3.8%) did not give PCR products. These clones, which either lost the entire integrated substrate or suffered a gross chromosomal rearrangement, were not studied further.
Figure 2.

Representative PCR analysis of DSB-induced mutations. PCR was performed on samples of genomic DNA isolated from TFTR clones recovered from cells after electroporation with pCMV-I-SceI and a HaeIII digest of φX174 DNA. The pair of primers used were positioned 1.5 kb apart and flanked the I-SceI site (Fig. 1A). Shown here is a representative analysis of parental cell line 2 (lane 12) and 10 TFTR clones derived from cell line 2 (lanes 2–11). Parental cell line 2 generated a 1.5 kb PCR product. Among the 10 TFTR clones, seven clones (lanes 3–7, 9 and 11) displayed PCR products indistinguishable in size from that of the parent. Two clones (lanes 2 and 10) displayed PCR products clearly larger than that of the parent whereas one failed to produce a PCR product (lane 8). Lane 1 displays a negative control PCR reaction to which no template DNA was added. A total of 132 TFTR clones isolated from cell lines 2 and 4 were analyzed by PCR in a similar manner. See text for further details.
Capture of transfected φX174 DNA at a genomic DSB
Sequence analyses of the 18 large PCR products revealed that DNA fragments ranging from 180 to 1622 bp had indeed been captured at the I-SceI-induced DSB (Table 2). In most of these clones, DNA capture was accompanied by deletions of 1–40 bp from one or both DNA termini at the DSB site. In several clones, capture of DNA was accompanied with the addition of a few nucleotides that may be derived from the single-strand ATAA overhang produced by I-SceI cleavage (Fig. 1B). No clone had captured a fragment from pCMV-I-SceI. This latter observation was notable because in our previous work involving electroporation of pCMV-I-SceI alone, 10 out of 21 DNA capture events involved a fragment of pCMV-I-SceI (12). Fourteen clones had captured one or more fragments of φX174 DNA (Table 2). Twelve clones captured one continuous φX174 DNA sequence. In 11 of these cases, internal segments of the HaeIII fragments were captured with the sites of joining between φX174 and genomic DNA positioned 6–867 bp away from the original ends of HaeIII fragments of φX174. Only one clone (2-D1) captured an original φX174 HaeIII fragment in its entirety. Clones 4-B11 and 4-F7 each contained portions of three distinct HaeIII fragments of φX174 DNA. The captured φX174 fragments are illustrated schematically in Figure 3. The inserts were derived from various regions of the φX174 genome and there was no evidence for preferential capture of any particular sequence from the φX174 genome.
Table 2. DNA sequences captured at a chromosomal DSB.
| Clonea |
Upstream change (bp)b |
Downstream change (bp)c |
Length of captured DNA sequence (bp)d |
Source of captured DNA |
| 2-A1 | –5 | +1(A) | 778 | φX174 |
| 2-A4 | +1(A) | –34 | 197 | φX174 |
| 2-A7 | –1 | –2 | 180 | φX174 |
| 2-A9 | –1 | 0 | 515 | E.coli |
| 2-B1 | –7 | –40 | 1490 | IAP-UIe |
| 2-B4 | –2 | +2(AA) | 585 | φX174 |
| 2-B9 | –9 | +3(TAA) | 1234 | φX174 |
| 2-C7 | –8 | +2(AA) | 350 | IAP |
| 2-D1 | –6 | –22 | 310 | φX174 |
| 2-D11 | –2 | –21 | 240 | φX174 |
| 4-A12 | 0 | +3(TAA) | 561 | φX174 |
| 4-B5 | –4 | –25 | 180 | UI |
| 4-B9 | 0 | 0 | 223 | φX174 |
| 4-B11 | –2 | –22 | 1360 | φX174 |
| 4-D9 | –1 | –2 | 200 | φX174 |
| 4-E4 | +3(ATA) | 0 | 196 | φX174 |
| 4-E8 | –1 | 0 | 362 | φX174 |
| 4-F7 | –8 | –5 | 1622 | φX174 |
aThe first character of each clone name refers to the cell line (2 or 4) from which the TFTR clone was recovered.
bNumber of bases deleted (–) or gained (+) upstream from the I-SceI cut site on the top strand of the integrated pYL1 substrate (see Fig. 1B). These changes are in addition to the captured DNA sequence. In clones involving the gain of several bases, the actual sequences added are indicated in parentheses.
cNumber of bases deleted (–) or gained (+) downstream from the I-SceI cut site on the top strand of the integrated pYL1 substrate. The changes are in addition to the captured DNA sequence. In clones involving the gain of several bases, the actual sequences added are indicated in parentheses.
dThe lengths of inserts are defined so as to minimize the length of deletions in genomic sequences in cases in which there exist microhomologies at new junctions.
eUI, sequences of unidentified origin. These sequences are not present in GenBank or EMBL.
Figure 3.

Captured DNA sequences from φX174. The φX174 genome is represented by the thick horizontal line. The 11 HaeIII fragments of the φX174 genome are indicated, numbered from left to right (diagram is not to scale). HaeIII sites in the φX174 genome are depicted by the downward arrows. Indicated by horizontal lines below the map of φX174 are the DNA sequences captured in various clones. Note that clones 4-B11 and 4-F7 each contain three discrete fragments of φX174 DNA. With the exception of clone 2-D1, none of the clones captured an original HaeIII fragment in its entirety. Further information regarding captured fragments is provided in Figure 5.
Capture of transfected DNA at a DSB is not accompanied by random integration
It was conceivable that the cells that captured DNA at the DSB may have been particularly proficient at incorporating exogenous DNA at all genomic loci. We were thus curious to learn whether capture of φX174 DNA at the I-SceI-induced DSB was associated with additional random integration of φX174 DNA at one or more additional sites in the genome. DNA from clones that had captured φX174 DNA were digested with BamHI and analyzed on a Southern blot using a φX174-specfic probe (Fig. 4). As there are no BamHI sites in φX174 DNA or the tk gene, we expected each clone to display a single hybridizing band if there were no sites of integration of φX174 other than the I-SceI-induced DSB. We expected the size of the hybridizing fragment containing captured φX174 DNA to be roughly equal to the size of the BamHI fragment from pYL1 (2.5 kb) plus the length of the captured φX174 DNA. As shown in Figure 4, the φX174 probe hybridized to a single band in all clones analyzed, indicating that in cells in which φX174 DNA was captured at the DSB there were no additional sites of random integration of φX174 DNA. Our observations indicate that transfected DNA specifically integrated at the genomic DSB site.
Figure 4.
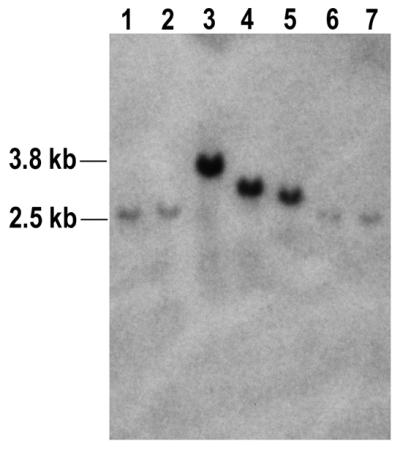
Representative Southern blotting analyses of clones that captured DNA. Samples of genomic DNA (15 µg each) isolated from the 14 clones that had captured φX174 DNA were digested with BamHI and analyzed on a Southern blot using a φX174-specific probe. Analysis of seven clones is shown here. Lanes 1–7 display DNA from clones 2-D11, 2-D1, 2-B9, 2-A1, 2-B4, 2-A7 and 2-A4, respectively. Each clone displayed a single hybridizing band, indicating an absence of additional sites of random integration of φX174 DNA. As the probe used was constructed from the entire φX174 genome, hybridization intensity for each clone is roughly proportional to the length of φX174 DNA captured.
DNA capture involves joining of DNA molecules preferentially at sites of microhomology
Although we transfected cells with a HaeIII digest of φX174 DNA, internal segments of the HaeIII fragments rather than the original fragments themselves were predominantly captured at the DSB. As indicated in Figure 5, in all clones (with the exception of clone 2-D1) genomic sequences were joined to φX174 DNA at sites located 6–867 bp from the original termini of the HaeIII fragments. Sequence analysis revealed microhomologies of 1–4 bp at most sites of DNA joining (Fig. 5). Considering all the clones that had captured φX174 DNA, there are a total of 32 novel genomic/φX174 or φX174/φX174 DNA junctions. Illustrated in Figure 6 are the numbers of junctions displaying various lengths of microhomology compared with the distribution we would have expected to recover had DNA joining occurred at random sites. From this analysis, we conclude that DNA capture involves the joining of DNA molecules preferentially at sites of microhomology.
Figure 5.

Microhomologies at DNA junctions. Shown are the nucleotide sequences at the sites of joining between captured φX174 fragments and genomic DNA. For each junction, the upper sequence presented is from the genomic sequence near the DSB site and the lower sequence is from the captured φX174 DNA. The underlined nucleotides are those actually found at the junctions in each clone. Nucleotides in boldface represent overlapping microhomologies at sites of DNA joining. Information about the captured φX174 sequences is presented between the junction sequences. Numbers not enclosed within parentheses are the first and last nucleotides of the captured φX174 sequences; numbers in parentheses indicate the number of nucleotides deleted from the original termini of the respective HaeIII fragment of the φX174 genome. Clones 4-B11 and 4-F7, at the bottom of the figure, each captured three fragments of φX174 DNA and thus display four sites of DNA joining. For these latter clones, two genomic DNA/φX174 junctions and two φX174/φX174 junctions are shown.
Figure 6.

Distribution of microhomologies at junctions between genomic DNA and captured sequences. Among all the clones which had captured φX174 DNA, there are a total of 32 genomic/φX174 or φX174/φX174 DNA junctions. Shown by the open bars are the number of junctions recovered displaying various lengths of microhomology. Black bars show the expected distribution of microhomologies at junctions had DNA joining occurred at random sites. The expected distribution was determined by the equation P(x) = (x + 1)(1/4)x(3/4)2 where P(x) is the probability of finding x nucleotides of homology at a junction (28). The value of P is then multiplied by the total number of junctions analyzed (which equals 32) to calculate the distribution. This analysis is modeled after a similar analysis developed by Merrihew et al. (26).
Transfected linear DNA molecules are captured more efficiently than are supercoiled molecules
From the results described above, it was clear that HaeIII-digested φX174 DNA was captured at the I-SceI-induced genomic DSB more frequently than the cotransfected pCMV-I-SceI DNA. One possible explanation for this observation was that linear DNA is more efficiently captured than circular, supercoiled DNA (the disposition of the electroporated pCMV-I-SceI DNA). To address this question, we cotransfected cell lines 2 and 4 with 20 µg pCMV-I-SceI DNA (supercoiled) plus 5 µg supercoiled φX174 RF I DNA. Out of a total of 69 TFTR colonies analyzed by PCR as above, six clones appeared to have captured a DNA sequence at the DSB based on the size of PCR products (data not shown). The PCR products of these six clones were further analyzed by Southern blotting using either pCMV-I-SceI or φX174 DNA as a probe. Two out of six PCR products hybridized to pCMV-I-SceI and none hybridized to φX174 DNA (data not shown), indicating that two clones had captured pCMV-I-SceI DNA and no clone had captured φX174 DNA. This contrasted sharply with the observation that 14 out of 18 capture events involved φX174 DNA when the φX174 DNA was electroporated as a set of HaeIII fragments. This result suggests that linear DNA fragments are more readily captured at a genomic DSB than are circular, supercoiled DNA molecules.
Capture of other sequences at a genomic DSB
In experiments in which cell lines 2 and 4 were transfected with pCMV-I-SceI plus a HaeIII digest of φX174 DNA, four of 18 TFTR clones had captured DNA sequences from sources other than φX174 DNA (Table 2). Clone 2-A9 had an insert of 515 bp of continuous DNA sequence from the Escherichia coli genome, which presumably was electroporated as a contaminant along with the φX174 or pCMV-I-SceI DNA. Clone 4-B5 captured a DNA fragment of 180 bp from an unidentified source. Clones 2-C7 and 2-B1 each captured a long terminal repeat (LTR) of the intracisternal A particle (an endogenous, moderately repetitive retrovirus-like sequence). The latter two clones are notable in that we previously reported several other instances of capture of similar LTR sequences (12). In addition to the LTR sequence, clone 2-B1 captured a sequence of unidentified origin.
DISCUSSION
We previously reported (12) that capture of DNA sequences at a DSB accompanies nearly 10% of recovered DSB repair events in mouse fibroblasts. Although our previous work was consistent with the notion that any abundant extrachromosomal sequence may be captured [as had been suggested for DNA capture in yeast (18)], our work was equally consistent with the possibility that only certain specific DNA sequences or motifs are readily captured.
To gain further insight into this latter issue, in the current work we cotransfected φX174 DNA along with pCMV-I-SceI to learn if φX174 sequences may be readily captured at the DSB. We used a HaeIII digest of φX174 DNA as a model for non-specific, low-complexity DNA having no homology to the DSB locus. Similar to previous findings for cell lines 2 and 4 (12), 13.6% of DSB-induced TFTR clones recovered from cell lines 2 and 4 in the current work captured DNA at the DSB. However, in the current work 14 out of 18 DNA capture events involved capture of one or more segments of φX174 and none involved pCMV-I-SceI (Table 2). Further, there was no preference for capture of any particular region of the φX174 genome. These results suggest that virtually any abundant extrachromosomal DNA sequence may be captured at a genomic DSB.
It is likely that the high frequency of capture of pCMV-I-SceI in our previous work (12) was due to the high relative abundance of pCMV-I-SceI sequences. In the current work, the absolute intracellular abundance of pCMV-I-SceI was presumably the same as in previous work as the same amount of pCMV-I-SceI DNA was electroporated. Indeed, pCMV-I-SceI was actually transfected in a 4-fold excess over φX174 fragments in terms of mass (20 versus 5 µg). The reduction in pCMV-I-SceI capture concomitant with the high frequency of φX174 DNA capture suggests that the φX174 DNA fragments ‘out-competed’ pCMV-I-SceI for interactions with the genomic DSB. However, in transfections with supercoiled φX174 RFI DNA, two out of six DNA capture events involved pCMV-I- SceI and none involved φX174. These results indicated that linearized φX174 DNA is more readily captured than supercoiled φX174 DNA. From our experiments we cannot determine whether linear DNA molecules are intrinsically more prone to being captured or if linear molecules are more efficiently transfected into the nucleus during electroporation.
Although we infer from our results reported here that any abundant extrachromosomal sequence may be captured at a genomic DSB, our previous work (12) did indicate that a specific fragment of pCMV-I-SceI spanning the SV40 and ColE1 origins was particularly prone to capture. Our collective observations suggest that the origin sequences on pCMV-I-SceI (non-functional in mouse Ltk– cells) are either hotspots for DNA breakage or hotspots for invasion by DNA termini (see below). We rule out the possibility that the origins on pCMV-I-SceI serve as special sequences needed to deliver extrachromosomal DNA to the genomic DSB as the efficient capture of φX174 fragments reported here demonstrates that, in general, transfected DNA is efficiently delivered to a DSB.
We can imagine two mechanisms for the capture of transfected DNA into a genomic DSB. DNA may be joined to both DSB termini by NHEJ. Alternatively, DNA may be ‘copied’ into the genomic DSB by a break-induced replication (BIR) process (reviewed in 25) in which one or both 3′ DNA ends from the genomic DSB invades a transfected molecule (likely at a site of microhomology) and primes DNA synthesis using the transfected molecule as a template. In the case of a one-ended invasion, the other end of the genomic DSB would join with the newly copied DNA via NHEJ. The finding of microhomologies at sites of joining between captured DNA and genomic sequences (Figs 5 and 6) is consistent with either NHEJ or BIR. The fact that most of the φX174 inserts are internal fragments rather than the original HaeIII fragments argues that if NHEJ is often involved in capture then substantial degradation of fragment ends typically occurs prior to most capture events. If BIR is an operative mechanism for capture, invasion of genomic DNA termini at internal sites of microhomology may occur with little or no degradation of the HaeIII fragment ends. We cannot distinguish between invasion at internal sites versus exonucleolytic digestion of fragment ends, although earlier work by others (2) suggests that random integration into a mammalian genome frequently occurs with the loss of very few nucleotides from the termini of transfected DNA fragments. We also note that Merrihew et al. (26) have presented evidence for a ‘copy-join’ mechanism for random integration into a mammalian genome. In this scenario, integration of transfected DNA is preceded by one or more rounds of invasion of a terminus of the transfected DNA molecule into genomic sites of microhomology. During these events, the invading terminus serves as a primer for DNA synthesis resulting in the addition of genomic sequences to the end of the transfected molecule. The transfected molecule may thus ‘pick up’ segments of genomic sequence from several loci prior to integrating. In a somewhat analogous fashion, we propose that DNA termini from a broken chromosome may invade transfected DNA molecules at sites of microhomology, allowing the genome to ‘pick up’ segments of transfected sequences.
Our work makes it evident that DNA transfected into a mammalian cell will preferentially integrate at a genomic DSB. In our hands, stable integration of a selectable marker typically occurs at a frequency of less than one in 104 electroporated mouse Ltk– cells (data not shown). φX174 DNA was captured in about one in 103 electroporated cells (Table 1). Considering the small ‘target’ size of the I-SceI-induced DSB in the context of the entire genome, it is unequivocal that the transfected φX174 DNA preferentially integrated at the DSB. It would appear that there exists a mechanism that results in the efficient delivery of transfected DNA to the site of a genomic DSB. We note previous reports of preferential integration of transfected DNA at unstable mammalian loci (25) and at an induced fragile site (27), consistent with the proposition that DNA efficiently integrates at sites of genomic breakage. Selective integration of transfected DNA at a genomic DSB may have future application in methodologies for targeted genome alterations.
Our work suggests that mammalian cells exhibit one or more robust DSB repair pathways that can apparently lead to patching of genomic DSBs by virtually any DNA molecule that happens to be available. To maintain genomic integrity, cells undoubtedly have regulatory mechanisms for selecting the DNA sequences with which the ends of a broken chromosome will interact. How cells may control DSB repair to help avoid promiscuous exchanges is a topic requiring further investigation.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Barbara Criscuolo Waldman for helpful comments on the manuscript. This work was supported by grants from the American Cancer Society (NP-949) and the National Institute of General Medical Sciences (GM47110) to A.S.W.
References
- 1.Roth D. and Wilson,J. (1986) Nonhomologous recombination in mammalian cells: role for short sequence homologies in the joining reaction. Mol. Cell. Biol., 6, 4295–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roth D. and Wilson,J. (1988) Illegitimate recombination in mammalian cells. In Kucherlapati,R. and Smith,G.R. (eds), Genetic Recombination. American Society for Microbiology, Washington, DC, pp. 621–653.
- 3.Derbyshire M.K., Epstein,L.H., Young,C.S.H., Munz,P.L. and Fishel,R. (1994) Nonhomologous recombination in human cells. Mol. Cell. Biol., 14, 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choulika A., Perrin,A., Dujon,B. and Nicolas,J.-F. (1995) Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson S.P. and Jeggo,P.A. (1995) DNA double-strand break repair and V(D)J recombination: involvement of DNA-PK. Trends Biochem. Sci., 20, 412–415. [DOI] [PubMed] [Google Scholar]
- 6.Sargent R.G., Brenneman,M.A. and Wilson,J.H. (1997) Repair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol. Cell. Biol., 17, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taghian D.G. and Nickoloff,J.A. (1997) Chromosomal double-strand breaks induce gene conversion at high frequency in mammalian cells. Mol. Cell. Biol., 17, 6386–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang F., Han,M., Romanienko,P.J. and Jasin,M. (1998) Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl Acad. Sci. USA, 95, 5172–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Y., Lukacsovich,T. and Waldman,A.S. (1999) Multiple pathways for repair of DNA double-strand breaks in mammalian chromosomes. Mol. Cell. Biol., 19, 8353–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haber J.E. (2000) Partners and pathways: repairing a double-strand break. Trends Genet., 16, 259–264. [DOI] [PubMed] [Google Scholar]
- 11.Van Gent D.C., Hoeijmakers,J.H.J. and Kanaar,R. (2001) Chromosomal stability and the DNA double-strand break connection. Nature Rev. Genet., 2, 196–206. [DOI] [PubMed] [Google Scholar]
- 12.Lin Y. and Waldman,A.S. (2001) Capture of DNA sequences at double-strand breaks in mammalian chromosomes. Genetics, 158, 1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richardson C., Moynahan,M.E. and Jasin,M. (1998) Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocation. Genes Dev., 12, 3831–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson R.D. and Jasin,M. (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J., 19, 3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore J.K. and Haber,J.E. (1996) Capture of retrotransposon DNA at the sites of chromosomal double-strand breaks. Nature, 383, 644–646. [DOI] [PubMed] [Google Scholar]
- 16.Teng S.C., Kim,B. and Gabriel,A. (1996) Retrotransposon reverse-transcriptase-mediated repair of chromosomal breaks. Nature, 383, 641–644. [DOI] [PubMed] [Google Scholar]
- 17.Ricchetti M., Fairhead,C. and Dujon,B. (1999) Mitochondrial DNA repairs double-strand breaks in yeast chromosomes. Nature, 402, 96–100. [DOI] [PubMed] [Google Scholar]
- 18.Yu. X. and Gabriel,A. (1999) Patching broken chromosomes with extranuclear cellular DNA. Mol. Cell, 4, 873–881. [DOI] [PubMed] [Google Scholar]
- 19.Salomon S. and Puchta,H. (1998) Capture of genomic and T-DNA sequences during double-strand break repair in somatic plant cells. EMBO J., 17, 6086–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirik A., Salomon,S. and Puchta,H. (2000) Species-specific double strand break repair and genome evolution in plants. EMBO J., 19, 5562–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van deWater N., Williams,R., Ockelford,P. and Browe,P. (1998) A 20.7 kb deletion within the factor VIII gene associated with LINE element insertion. Thromb. Haemost., 79, 938–942. [PubMed] [Google Scholar]
- 22.Wagner M.J., Sharp,J.A. and Summers,W.C. (1981) Nucleotide sequence of the thymidine kinase gene of herpes simplex virus type 1. Proc. Natl Acad. Sci. USA, 78, 1441–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rouet P., Smith,F. and Jasin,M. (1994) Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl Acad. Sci. USA, 91, 6064–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lukacsovich T., Yang,D. and Waldman,A.S. (1994) Repair of a specific double-strand break generated within a mammalian chromosome by yeast endonuclease I-SceI. Nucleic Acids Res., 22, 5649–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haber J.E. (1999) DNA recombination: the replication connection. Trends Biochem. Sci., 24, 271–275. [DOI] [PubMed] [Google Scholar]
- 26.Merrihew R.V., Marburger,K., Pennington,S., Roth,D.B. and Wilson,J.H. (1996) High-frequency illegitimate integration of transfected DNA at preintegrated target sites in a mammalian genome. Mol. Cell. Biol., 16, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rassool F.V., McKeithan,T.W., Neilly,M.E., van Melle,E., Espinosa,R. and Le Beau,M.M. (1991) Preferential integration of marker DNA into the chromosomal fragile site at 3p14: an approach to cloning fragile sites. Proc. Natl Acad. Sci. USA, 88, 6657–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roth D.B., Porter,T.N. and Wilson,J.H. (1985) Mechanisms of nonhomologous recombination in mammalian cells. Mol. Cell. Biol., 5, 4295–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]