

Abstract
This study was aimed to detect a new mutation responsible for X-linked dilated cardiomyopathy with hyper-CKemia.
We studied a proband who presented with cardiac symptoms with hyper-CKemia, but no clinical skeletal involvement in physical examination, laboratory tests, electromyography, echocardiography, and magnetic resonance imaging (MRI) of cardiac muscles. Muscle biopsy for histopathology and immunohistochemistry for accessing sarcolemma changes. The next-generation sequencing and bioinformatics analysis were performed on the patient and Sanger sequencing was confirmed on the other 6 unaffected families.
The clinic investigations illustrated a dilated cardiomyopathy. Histopathology and immunohistochemistry showed dystrophic changes and an obvious reduction of dystrophin-N and δ-sarcoglycan, respectively. One hemizygous splicing pathogenic mutation c.31 + 1G > C of exon 1 in the DMD gene (chrX33229398, NM_00 4006) was finally identified in the patient and his nephew, but it was carried in his mother and sister.
A novel small mutation was identified at the first exon-intron boundary splicing site by next-generation sequencing and bioinformatics analysis.
Keywords: dilated cardiomyopathy, dystrophin gene, next-generation sequencing, novel splicing mutation
1. Introduction
Dystrophin (DMD) gene (MIM #300377) is initially located on chromosome Xp21.1; it occupies an extremely gigantic territory of 2.4-Mb gene and contains at least 79 exons,[1] which accounts for only 0.6% of the sequence.[2]DMD gene encodes a 427-kDa dystrophin protein; it is combined with the dystroglycan, sarcoglycan, and syntrophin/dystrobrevin to form a dystrophin glycoprotein complex (DGC). This DGC serves as a part of the cytoskeleton that connects the contractile apparatus and extracellular matrix to stabilize membrane.[3,4] There are 3 tissue-specific promoters produced by alternative splicing and transcription, since the 7 different promoters have been identified. Those tissues include the brain (in cortical neurons, heart, and cerebellar neurons), muscle (in skeletal muscle, smooth muscle, and T tubules in the cardiac myofibers), and Purkinje cell (in cerebellar).[5–8] Those promoters inevitably involve cardiomyocytes or central nervous system (CNS) impairments (e.g., intellectual disability) beside progressive weakness, dystrophy, or pseudohypertrophy of skeletal muscle. Mutations in the DMD gene include deletions (65%) around the 2 hot spots of exons 3 to 7 and 45 to 55,[9] duplications (6–10%), small mutations (10%), or other smaller rearrangements (nonsense mutations, splice mutations, frameshift small deletions or insertions, mid intronic variations). These mutations grossly disrupt the translational reading frame, which may lead to severe Duchenne muscular dystrophy (DMD) with a truncated and unfunctional protein lacks the cysteine-rich C-terminal region, also lead to the other traditionally allele form, such as the mild performance of Becker muscular dystrophy (BMD). It would change but maintain it with a semifunctional protein.[10,11] On the contrary, X-linked dilated cardiomyopathy (XLDCM) as a different type of dystrophinopathy mainly presents with preferential cardiac involvement with progressive ventricular dysfunction and chamber dilatation, but no clinical skeletal myopathy, only with increase of the level of serum creatine kinase (CK) in the first medical evaluation, which may be a variable in the severity and occurrence conditions of lethal myocardial failure.[12–14]
According to the previous reports, a defect in the universally conserved 5′end dystrophin gene may be linked to a selective transcriptional impairment, and then lead to a cardiac involvement, especially a subtle mutation, such as deletion, splicing, or insertion at the 5′portion of the muscle isoform promoter, the first muscle exon or intron. [15–17]
In this study, we found a novel small mutation in the first exon–intron boundary splicing site of the DMD gene, in a patient with higher serum CK level in his family. This small mutation is responsible for X-linked dilated cardiomyopathy.
2. Materials and methods
This study was approved by the ethics committee of the Second Hospital of Hebei Medical University, and informed consents were obtained from all individuals involved in the study.
2.1. Subjects
The proband(III-3) was a 30-year-old man who suffered from a 10-year history of systemic fatigue and myalgia after exercise, especially when the weather was cold. Dilated cardiomyopathy was suspected on the basis of cardiac and respiratory progressive symptoms, such as palpitation and pant, high serum CK level, which was checked by electromyography, echocardiography, and magnetic resonance imaging (MRI) of cardiac muscles. However, he complained of worse chest tightness and dyspnea in daily activities (New York Heart Association functional classification, NYHA III)[18] within the preceding 3 months, which was caused by a severe cardiac insufficiency. A combination therapy of angiotensin-converting enzyme inhibitors (ACE-I) and β-blocker has been carried out for 3 years. His family history was pertinent; he has 2 uncles who died of dilated cardiomyopathy at the age of 20. His parents and sisters have no special performance. His little nephew was more than 2 years old when he was measuring genes, and he had no special performance. But with regret, there was no other information was available on the latter patients. Apart from his uncles, there was not any discomfort in the muscle or heart in his family members. The pedigree of this family is reported in Fig. 1A.
Figure 1.
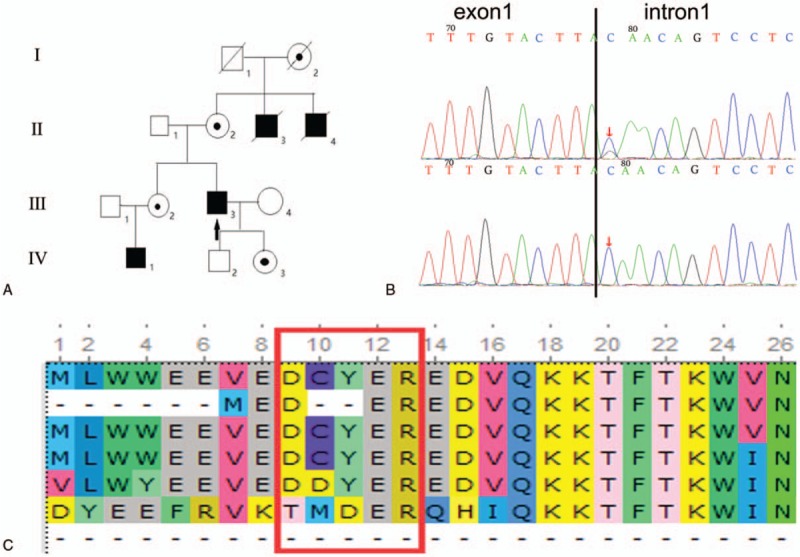
Pedigree of the family with X-linked dilated cardiomyopathy. The affection status is indicated by solid symbols (affected), clear symbols (unaffected), and circle with a dot (carrier) (A). The arrow represents the proband(III-3) with 1 hemizygous splicing mutation c.31 + 1G > C of exon 1(B, upper) as well as his nephew (IV-1) and 2 deceased uncles (II-3,4) but the mutation was carried in his mother (II-2), sister (III-2), and daughter (IV-3). The normal control had a homozygous base G (II-1, IV-2) (B, lower). The black borderline located at the end of exon 1 with the mutation in first base of intron 1 at its downstream (B). Multiple species alignment analysis showed the high evolutionary conservation of amino acid sequence at the splicing site (C). The meaning of the letters in figure C is as follows: H sapiens; P troglodytes; C lupus; M musculus; G gallus; D melanogaster; A gambiae.
2.2. Muscle biopsy
After informed consent, muscle biopsy specimens were obtained from the left biceps brachii of the proband, and the experimental procedures were in accordance with the standard protocol. Cryosections of 10 μm were cut from flash-frozen muscle biopsies in a cryostat operating at − 23°C and were processed for conventional histologic stains [hematoxylin and eosin (HE), modified Gomori trichrome (MGT), nicotinamide adenine dinucleotide phosphate-tetrazolium reductase (NADH-TR), adenosine triphosphatase (ATPase), periodic acid-Schiff (PAS) and oil red O (ORO)] and immunohistochemical reactions.[19] Immunohistochemistry was further confirmed by means of the monoclonal antibodies against DGC, which included dystrophy-N(amino-terminal), dystrophy-C(carboxyl-terminal), dystrophy-R(large central rod-domain), alpha-, beta-, gamma-, delta-sarcoglycan, and dysferlin (all from Vector Laboratories, San Francisco, America) for differential diagnosis.
2.3. Targeted sequence capture and next-generation sequencing
DNA was extracted from peripheral blood by QIAamp DNA Mini Kit (Tingen, Beijing, China). Target sequences, including the DMD genes, were enriched by using the GenCap custom enrichment kit (MyGenostics Inc., Beijing, China). Briefly, 3 μg DNA were fragmented into 350 to 450 bp length using Covaris Acousitic System. The DNA fragments were then processed by end-repairing, a-tailing and adaptor ligation, a 4-cycle pre-capture PCR amplification, targeted sequences capture. Captured DNA fragments were eluted and amplified by 15-cycle postcapture PCR. The final products were sequenced with 150-bp paired-end reads on an Illumina HiSeq 2000 (Illumina, San Diego, CA) platform, according to the standard manual.
2.4. Data analysis
Image analysis, error estimation, base calling, and generating the primary sequence data on the raw data were processed by the Illumina pipeline (version 1.3.4). The low-quality reads and the 3’ and 5’ adapters were filtered by Trim Galore. The clean short-reads were mapped to human genome (hg19) using BWA software (Burrows Wheeler Aligner http://sourceforge.net/projects/bio-bwa/). SOAPsnp software (http: //soap.genomics.org.cn/) and SAM tools Pileup software (http://sourceforge.net/projects/samtools/) were used to detect single nucleotide variants (SNPs) and small insertion and deletions (small INDEL). Variants were annotated by ANNOVAR software (http://www.openbioinformatics.org/annovar/), and allele frequency in 1000 Genomes (www.1000genomes.org), ESP6500 (evs.gs.washington.edu/EVS/), and ExAc_ALL (exac.broadinstitute.org/). Mutations were described in accordance with the current mutation nomenclature guidelines (http://varnomen.hgvs.org/) and analyzed in silico by the Mutation Taster (http://www.mutationtaster.org/).
2.5. Validation by sanger sequencing
To validate the pathogenic variant from next-generation sequencing (NGS) experiment, genomic DNA from all available family members were obtained for Sanger sequencing. The captured DNA was amplified as per the following procedure: 98°C 30 seconds, 15 × (98°C 25 seconds, 65°C 30 seconds, 72°C 30 seconds), 72°C 5 minutes. The PCR product was purified and sequenced with SPRI beads (Beckman Coulter, Brea, CA) and ABI Prism 3100 genetic analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc., California, America) according to the manufacturer's protocol, respectively. Sites of variation were identified through a comparison of DNA sequences with the corresponding GenBank (www.ncbi.nlm.nih.gov) reference sequences. Two pair primers primer were designed for the sequence centers on splicing mutation as follows: forward primer: 5’-TGCTTCTTTGCAAACTACTGTGAT-3’ and reverse primer: 5’-GAGGCAATTACCTTCGGAGAA-3’.
2.6. Bioinformatics analysis
The human Splicing Finder (http://www.umd.be/HSF3/technicaltips.html) was used as a splicing prediction tool to perform in silico analysis, in order to predict the effect of the novel intronic variant on the splicing of the DMD gene.
3. Results
3.1. Clinical findings
Apart from systemic heart discomfort, he never had any clinical signs of skeletal myopathy. His laboratory tests including thyroid function examination was normal, but a higher CK was 1297 IU/L. In addition, cardiac physical examination yielded that there was a systolic murmur of apical portion during auscultation, and the murmur sounded like a wind, and a shifted left of heart lower bound also appeared. He was neurologically intact clinically. The electrocardiogram (ECG) showed bidirectional high T waves in the right precordial leads (V2–6) and deep Q waves in leads I, aVL. Echocardiography results demonstrated that the proband's myocardium was thickened, the left ventricle was enlarged, and systolic dysfunction had ejection fraction of 13%. MRI of cardiac muscles reveled wall motion abnormalities, partial fibrosis with a scar in the left ventricular myocardium, and a secondary enlargement and lower diastolic dysfunction.
3.2. Histopathology and immunohistochemistry abnormalities
Histopathology typically exhibited dystrophic changes, such as moderate variation in myofiber size, accompanied by circular atrophy fibers. There were occasional degeneration, necrosis, and regeneration, but no increase in interstitial connective tissue. Immunohistochemistry illustrated an obvious reduction of dystrophin-N and δ-sarcoglycan (Fig. 2).
Figure 2.
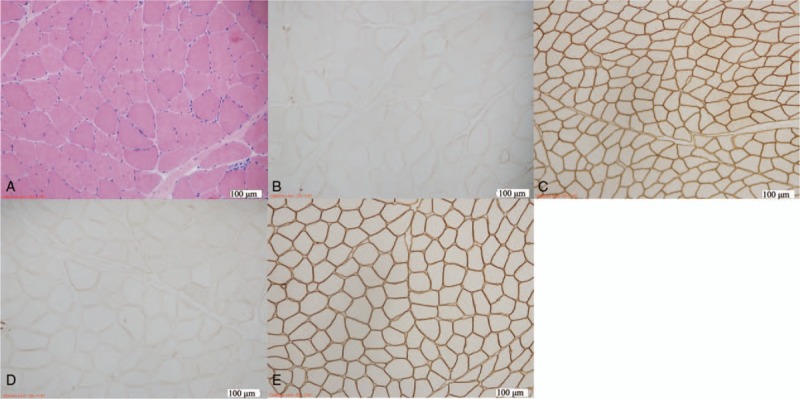
Muscle biopsy from left biceps brachii of the proband. Hematoxylin and eosin (H&E) staining (A) showed moderate variation in myofiber size. Immunohistochemical staining of DGC illustrated an obvious reduction of the number of dystrophin-N(B) and δ-sarcoglycan(D) compared with control, respectively (C,E). Magnification (A,B,C,D,E) × 100.
3.3. Identification of pathogenic changes
The average sequencing depth was more than 288 reads. Generally, more than 99.92% of the targeted disease gene regions were sequenced. Coverage of the targeted bases greater than 10 reads was 99.64%, and more than 20 reads was 98.97%. This mutation was neither described in 1000 genome project nor in ESP6500, and it was never observed in the ExAC Browser database before. Combining “disease-causing” prediction analysis with conservation of altered the amino acid, only 1 hemizygous splicing mutation c.31 + 1G > C of exon 1 in the DMD gene (chrX33229398, NM_00 4006) was finally confirmed. Meanwhile, the hemizygotic mutation was found in his nephew, but it was carried in her mother and sister, and was found by Sanger sequencing. Combined with the mode of inheritance in his family, our data support that it was a pathogenic mutation (Fig. 1B, C).
3.4. Bioinformatics prediction
The human Splicing Finder predicted that the 5′donor splice site of intron 1 would disappear due to the mutation. This might lead to 3 types of aberrant splicing, including 2 different intron retentions, and a donor site break to create a new splice site, but there was no exon skipping (Fig. 3).
Figure 3.
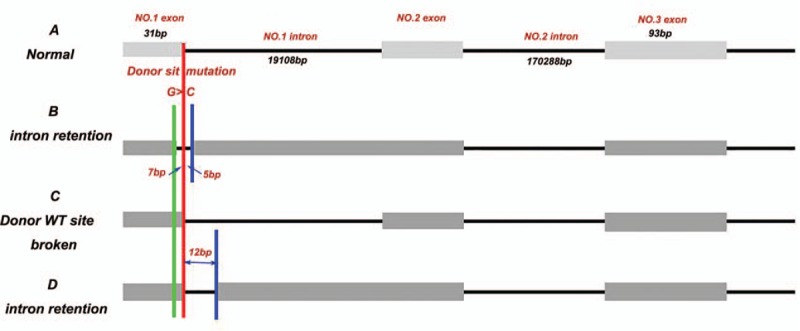
Bioinformatics prediction of the effect of the mutation on the splicing of the DMD gene. The red borderline indicated the position of the splice donor site mutation. (A) Normal splicing. (B) Aberrant splicing with 7 bp upstream and 5 bp downstream intron retention around the splice donor site. (C) Aberrant splicing with donor WT site broken. (D) Aberrant splicing with 12 bp intron retention downstream the splice donor site.
4. Discussion
In summary, clinical investigations were indicative of a severe myocardial dilatation and dysfunction. Combined with dystrophic changes on histopathology, decrease of dystrophin-N and δ-sarcoglycan on immunohistochemistry, variants analysis and bioinformatics prediction, it was reasonable to draw a conclusion regarding the splicing mutation c.31 + 1G > C of exon 1 in the DMD gene of the proband and his nephew, which was carried in his mother, sister, and daughter, as well as 2 of his dead uncles. This mutation was the pathogenic mutation and it was in support of the X-linked inheritance.
This intron mutation was G to C transversion at the position + 1 of the first muscle exon-intron boundary with a consensus sequence G100 T100 A/G95 A70 G80 T45 in eukaryotes (where the numbers indicated the percentage of nucleotide conservation), which commonly affects the splice sites, branch sites, or create cryptic splice sites.[20] In general, classical splicing mutations always affect normal critical constitutive or alternative sites around exons, accounting for 15% of the human mutation databases,[21] but up to 34% of DMD,[22] which approximately 70% of the carriers are from the carrier's mother.[23]
In our patient, this novel splice site mutation is in the conserved base at the 5′ donor splice site of intron 1, including putative regulatory sequences and numerous pseudo-exons,[5] which might be activated by this deep intronic single mutations. It was a pity that the muscle for RNA samples of the proband was degraded and was unfeasible to design a hybrid minigene to transfection of HeLa cells for splicing assay because of the huge intron 1.[24] So, we could not confirm the splicing mutation, but only assume these 2 different intron retentions and a new splice site from the RNA transcript, through computational prediction. It has deleterious effect on the precursor (pre) mRNAs splicing. It generated an aberrant dystrophin protein that contributed to DMD, which was different from previous 2 mutations with replacing a G with a A or T at the same base.[25,26] According to the previous report, loss of dystrophin was due to the deletion of the muscle dystrophin isoform(M) at the promoter region and exon 1, which could be compensated by upregulation of the other full-length brain(B) and cerebella Purkinje(P) isoform and several muscle-specific regulatory elements. These muscle-specific regulatory elements included dyftrophin enhancer 1 (DME1), myocyte-specific enhancer factor 2 (MEF2), myocyte-specific enhancer factor 1 (MEF2), which bind the myogenic MyoD1 factor, and “E” box preserved at enormous first intron (>200 kb) but absent in the heart.[27,28] In other words, it was able to prevent the myopathy and was compatible with the selective heart involvement from transcriptional initiation at alternative promoters in the patients. Furthermore, this theory may promote a new therapy for dilated cardiomyopathy patients especially those with no muscle weakness, but has a mutation in the first exon–intron boundary of the muscle dystrophin isoform gene, through activation of brain or Purkinje promoters and regulatory elements in the heart, to some certain degree. On the contrary, reduction or loss of N-terminal dystrophin binding to F-acting is associated with all forms of DCM.[29,30] The changed DGC structure of our patient with a decrease of dystrophin-N and δ-sarcoglycan interrupted interactions with intracellular signaling molecules,[31] especially increased susceptibility to mechanical stress with exercise overload on the myocardium.[32]As a consequence, implantation of a left ventricular assist device to reduce mechanical stress or design novel therapies to determine N-terminal dystrophin abnormalities may provide a new way for treatment and delaying progression of DCM and sudden death.
However, his nephew was at the age of 2 years, he had the same mutation but no myocardial abnormalities, or delayed milestones were found. Even if the child is too young to get sick before age 18 years,[33] some cardiac function evaluations, especially the test of serum CK level, are still necessary for prevention.
In conclusion, we reported a novel splice site mutation (c.31 + 1G > C) of exon 1 in the DMD gene to broaden the mutation spectrum of DMD with X-linked dilated cardiomyopathy. Furthermore, this high sensitivity, high throughput, and low time cost amplicon-targeted NGS technology provided a new perspective in huge DMD gene sequencing, especially in undetectable variations deep in intronic mutations and genetic counselling of obligate female carriers as well.
Author contributions
Conceptualization: Jin Tang, Xueqin Song, Huiqing zhang.
Data curation: Jin Tang.
Formal analysis: Jin Tang, Xueqin Song, Guang Ji.
Funding acquisition: Xueqin Song, Guang Ji.
Investigation: Jin Tang.
Methodology: Hongran Wu, Shuyan Sun, Chi Zhang.
Project administration: Hongran Wu, Shuyan Sun, Shan Lu, Chi Zhang.
Resources: Shan Lu, Yuan Li, Chi Zhang.
Software: Hongran Wu, Shan Lu.
Supervision: Jin Tang, Yuan Li.
Validation: Jin Tang, Yuan Li, Huiqing zhang.
Visualization: Huiqing zhang.
Writing – original draft: Jin Tang, Huiqing zhang.
Writing – review & editing: Huiqing zhang.
Footnotes
Abbreviations: BMD = Becker muscular dystrophy, CK = creatine kinase, CNS = central nervous system, DGC = dystrophin glycoprotein complex, DMD = Duchenne muscular dystrophy, DMD = Dystrophin, DME1 = dyftrophin enhancer 1, MEF2 = myocyte-specific enhancer factor 1, MEF2 = myocyte-specific enhancer factor 2, MRI = magnetic resonance imaging, SNP = single nucleotide variants, XLDCM = X-linked dilated cardiomyopathy.
The authors have no conflicts of interest.
References
- [1].Anand A, Vinish M, Prabhakar S. A case of manifesting carrier with DMD phenotype. Acta Medica (Hradec Kralove) 2009;52:167–70. [DOI] [PubMed] [Google Scholar]
- [2].Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet 1993;3:283–91. [DOI] [PubMed] [Google Scholar]
- [3].Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature 1989;338:259–62. [DOI] [PubMed] [Google Scholar]
- [4].Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol 2006;7:762–73. [DOI] [PubMed] [Google Scholar]
- [5].Muntoni F, Wilson L, Marrosu G, et al. A mutation in the dystrophin gene selectively affecting dystrophin expression in the heart. J Clin Invest 1995;96:693–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Boyce FM, Beggs AH, Feener C, et al. Dystrophin is transcribed in brain from a distant upstream promoter. Proc Natl Acad Sci U S A 1991;88:1276–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Górecki DC, Monaco AP, Derry JM, et al. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum Mol Genet 1992;1:505–10. [DOI] [PubMed] [Google Scholar]
- [8].Holder E, Maeda M, Bies RD. Expression and regulation of the dystrophin Purkinje promoter in human skeletal muscle, heart, and brain. Hum Genet 1996;97:232–9. [DOI] [PubMed] [Google Scholar]
- [9].Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- [10].Beggs AH, Hoffman EP, Snyder JR, et al. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am J Hum Genet 1991;49:54–67. [PMC free article] [PubMed] [Google Scholar]
- [11].Monaco AP, Bertelson CJ, Liechti-Gallati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988;2:90–5. [DOI] [PubMed] [Google Scholar]
- [12].Towbin JA, Hejtmancik JF, Brink P, et al. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993;87:1854–65. [DOI] [PubMed] [Google Scholar]
- [13].Cohen N, Muntoni F. Multiple pathogenetic mechanisms in X linked dilated cardiomyopathy. Heart 2004;90:835–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Diegoli M, Grasso M, Favalli V, et al. Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J Am Coll Cardiol 2011;58:925–34. [DOI] [PubMed] [Google Scholar]
- [15].Yoshida K, Ikeda S, Nakamura A, et al. Molecular analysis of the Duchenne muscular dystrophy gene in patients with Becker muscular dystrophy presenting with dilated cardiomyopathy. Muscle Nerve 1993;16:1161–6. [DOI] [PubMed] [Google Scholar]
- [16].Muntoni F, Cau M, Ganau A, et al. Brief report: deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N Engl J Med 1993;329:921–5. [DOI] [PubMed] [Google Scholar]
- [17].Yoshida K, Nakamura A, Yazaki M, et al. Insertional mutation by transposable element, L1, in the DMD gene results in X-linked dilated cardiomyopathy. Hum Mol Genet 1998;7:1129–32. [DOI] [PubMed] [Google Scholar]
- [18].Hurst JW. The value of using the entire New York Heart Association's classification of heart and vascular disease. Clin Cardiol 2006;29:415–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ljunggren A, Duggan D, McNally E, et al. Primary adhalin deficiency as a cause of muscular dystrophy in patients with normal dystrophin. Ann Neurol 1995;38:367–72. [DOI] [PubMed] [Google Scholar]
- [20].Horowitz DS, Kreiner AR. Mechanisms for selecting 5′ splice site in mammalian pre-mRNA splicing. Trends Genet 1994;10:100–6. [DOI] [PubMed] [Google Scholar]
- [21].Cooper TA, Mattox W. The regulation of splice-site selection, and its role in human disease. Am J Hum Genet 1997;61:259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tuffery-Giraud S, Chambert S, Demaille J, Claustres M. Point mutations in the dystrophin gene: evidence for frequent use of cryptic splice sites as a result of splicing defects. Hum Mutat 1999;14:359–68. [DOI] [PubMed] [Google Scholar]
- [23].Tuffery-Giraud S, Chambert S, Demaille J, et al. Point mutations in the dystrophin gene: evidence for frequent use of cryptic splice sites as a result of splicing defects. Hum Mutat 1999;14:359–68. [DOI] [PubMed] [Google Scholar]
- [24].Wang Z, Lin Y, Qiu L, et al. Hybrid minigene splicing assay verified the pathogenicity of a novel splice site variant in the dystrophin gene of a Chinese patient with typical Duchenne muscular dystrophy phenotype. Clin Chem Lab Med 2016;54:1435–40. [DOI] [PubMed] [Google Scholar]
- [25].Milasin J, Muntoni F, Severini GM, et al. A point mutation in the 5’ splice site of the dystrophin gene first intron responsible for X-linked dilated cardiomyopathy. Hum Mol Genet 1996;5:73–9. [DOI] [PubMed] [Google Scholar]
- [26].Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 2015;36:1123–35. [DOI] [PubMed] [Google Scholar]
- [27].Bastianutto C, Bestard JA, Lahnakoski K, et al. Dystrophin muscle enhancer 1 is implicated in the activation of non-muscle isoforms in the skeletal muscle of patients with X-linked dilated cardiomyopathy. Hum Mol Genet 2001;10:2627–35. [DOI] [PubMed] [Google Scholar]
- [28].Muntoni F, Melis MA, Ganau A, et al. Transcription of the dystrophin gene in normal tissues and in skeletal muscle of a family with X-linked dilated cardiomyopathy. Am J Hum Genet 1995;56:151–7. [PMC free article] [PubMed] [Google Scholar]
- [29].Feng J, Yan JY, Buzin CH, et al. Comprehensive mutation scanning of the dystrophin gene in patients with nonsyndromic X-linked dilated cardiomyopathy. J Am Coll Cardiol 2002;40:1120–4. [DOI] [PubMed] [Google Scholar]
- [30].Vatta M, Stetson SJ, Perez-Verdia A, et al. Molecular remodeling of dystrophin in patients with end-stage cardiomyopathies and reversal in patients treated with assist device therapy. Lancet 2002;359:936–41. [DOI] [PubMed] [Google Scholar]
- [31].Nakamura A. X-linked dilated cardiomyopathy: a cardiospecific phenotype of dystrophinopathy. Pharmaceuticals (Basel) 2015;8:303–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Melacini P, Fanin M, Danieli GA, et al. Myocardial involvement is very frequent among patients affected with subclinical Becker's muscular dystrophy. Circulation 1996;94:3168–75. [DOI] [PubMed] [Google Scholar]
- [33].Darras BT, Miller DT, Urion DK. Dystrophinopathies. 1993;Seattle, WA: University of Washington, Seattle, 1993-2018. [PubMed] [Google Scholar]