

ABSTRACT
Although the insulin receptor substrate (IRS) proteins IRS1 and IRS2 share considerable homology and activate common signaling pathways, their contributions to breast cancer are distinct. IRS1 has been implicated in the proliferation and survival of breast tumor cells. In contrast, IRS2 facilitates glycolysis, invasion, and metastasis. To determine the mechanistic basis for IRS2-dependent functions, we investigated unique structural features of IRS2 that are required for invasion. Our studies revealed that the ability of IRS2 to promote invasion is dependent upon upstream insulin-like growth factor 1 receptor (IGF-1R)/insulin receptor (IR) activation and the recruitment and activation of phosphatidylinositol 3-kinase (PI3K), functions shared with IRS1. In addition, a 174-amino-acid region in the IRS2 C-terminal tail, which is not conserved in IRS1, is also required for IRS2-mediated invasion. Importantly, this “invasion (INV) region” is sufficient to confer invasion-promoting ability when swapped into IRS1. However, the INV region is not required for the IRS2-dependent regulation of glucose uptake. Bone morphogenetic protein 2-inducible kinase (BMP2K) binds to the INV region and contributes to IRS2-dependent invasion. Taken together, our data advance the mechanistic understanding of how IRS2 regulates invasion and reveal that IRS2 functions important for cancer can be independently targeted without interfering with the metabolic activities of this adaptor protein.
KEYWORDS: breast cancer, IGF-1R, IRS2, invasion, PI3K
INTRODUCTION
Insulin receptor substrate 2 (IRS2) is a cytoplasmic adaptor protein that promotes breast cancer progression (1). Although IRS2 shares significant homology and the ability to activate common signaling pathways with its homologous family member IRS1, these adaptor proteins play distinct functional roles in breast cancer (2). The IRS1 gene is an estrogen receptor (ER)-regulated gene, and it is expressed at high levels in the ER+, luminal subtype of breast cancer (3–6). IRS1 interacts with the ER to positively regulate its transcriptional activity at estrogen response genes (6, 7). In this regard, tamoxifen response in breast cancer patients correlates with nuclear IRS1 expression (8). In vitro studies imply a role for IRS1 in the regulation of proliferation and survival in luminal breast carcinoma cells (9, 10). IRS1 expression decreases as ER expression or function is lost in more poorly differentiated, invasive breast tumors (11). In contrast, IRS2 is expressed at higher levels in ER− breast carcinoma cells of the basal-like/triple-negative breast cancer (TNBC) subtypes, and it regulates tumor cell migration, invasion, and glycolytic metabolism (1, 12–14). The different functions of IRS1 and IRS2 in breast cancer are further evidenced by the fact that mouse mammary tumors that lack IRS2 have significantly diminished ability to metastasize to the lungs, whereas tumors lacking IRS1 but expressing elevated IRS2 have enhanced metastatic potential (1, 15). IRS2 expression at the cell membrane in human breast tumors correlates with decreased overall survival, a finding that further supports a role for IRS2 in more aggressive tumor behavior (16).
The IRS proteins are recruited to cell surface receptors, where they are phosphorylated on tyrosine residues within their C-terminal tails, either directly by receptor tyrosine kinases or by associated nonreceptor kinases (i.e., the JAK family) (17, 18). These phosphorylation events generate SH2-binding sites for the recruitment and activation of signaling effectors that modify cell behavior. Common SH2-dependent binding partners that are recruited to IRS1 and IRS2 include phosphatidylinositol 3-kinase (PI3K), growth factor receptor-bound protein 2 (GRB-2), SHP2, and Src family kinases (SFKs) (19–23). The IRS proteins were first characterized as regulators of signaling downstream of the insulin receptor (IR) and the insulin-like growth factor 1 receptor (IGF-1R), but they can also serve as signaling intermediates of additional growth factor, cytokine, and integrin receptors (18, 24–28). Many of these receptors have been implicated in tumor development, growth, and metastasis, highlighting the importance of understanding the mechanism(s) by which the IRS proteins mediate their distinct downstream signaling outcomes.
The fact that IRS1 and IRS2 signal downstream of similar upstream receptors and activate common signaling pathways while the cellular responses to their signaling are unique implies that IRS function involves unique structural features of IRS1 and IRS2 that confer their distinct mechanisms of action. The IRS proteins have well-conserved, stable N-terminal PH and PTB domains that mediate their interactions with upstream receptors, followed by long, disordered tails that share less homology (29). They are considered to be intrinsically disordered proteins (IDPs) because of their overall absence of secondary or tertiary structure beyond the PH and PTB domains. This lack of stable structure is thought to allow dynamic intramolecular interactions to occur that rapidly integrate upstream signals to alter downstream function through the recruitment of signaling effectors (30). To date, interacting partners that bind uniquely to IRS1 or IRS2 that would explain their functional differences in cancer have not been reported. In the current study, we investigated the mechanism by which IRS2 selectively regulates one function, invasion. Our structure-function dissection of IRS2 identified a novel functional region within the C-terminal tail that is not conserved in IRS1, which we have termed the invasion (INV) region. This region is required for the ability of IRS2 to promote invasion but not glucose uptake by a mechanism that may involve the recruitment of novel effector molecules.
RESULTS
The IGF-1R/PI3K axis is involved in IRS2-mediated invasion.
In previous studies, we demonstrated that mouse mammary tumor cells and human breast carcinoma cells lacking IRS2 expression are deficient in their ability to invade (1, 31). In contrast, loss of IRS1 expression enhances invasion (1). To further our understanding of how IRS2 selectively regulates tumor cell invasion, double-Irs1/Irs2-null polyomavirus middle T mouse mammary tumor (PyMT:Irs1/2−/−) cells were used to assess Irs2 function in the absence of Irs1 expression. Restoration of IRS2 but not IRS1 expression in these double-Irs-null cells significantly increased invasion (Fig. 1A and 2C). IRS2 function is regulated by tyrosine phosphorylation in response to upstream receptor activation. To determine if IGF-1R or IR regulates IRS2-dependent invasion, assays were performed in the presence of the dual IGF-1R/IR small-molecule inhibitor BMS754807 (32). Cells were pretreated for 4 h and then incubated with inhibitor throughout the Matrigel Transwell invasion assay (Corning). Inhibition of IR/IGF-1R did not alter the invasion of PyMT:Irs1/2−/− cells expressing empty vector (EV). In contrast, IRS2-dependent tumor cell invasion was inhibited in a dose-dependent manner (Fig. 1A). Cells treated in parallel with BMS754807 were assayed for IGF-1R/IR phosphorylation and activation of AKT, as a measure of PI3K activity, to confirm inhibition of the pathway. Receptor inhibition was sustained throughout the time period of the assay at a concentration of 1 μM BMS754807 (Fig. 1B).
FIG 1.

IR/IGF-1R contributes to IRS2-mediated tumor cell invasion. PyMT:Irs1/2−/− cells expressing EV or IRS2 were treated with dimethyl sulfoxide (DMSO) or BMS754807 at the concentrations indicated for 4 h. (A) Matrigel Transwell invasion assays were performed for 5 h. The data shown represent the means ± standard errors of the mean (SEM) from three independent experiments. ns, no significant difference; **, P < 0.01 relative to EV-DMSO; ##, P < 0.01 relative to IRS2-DMSO. (B) Cells were stimulated with IGF-1 (50 ng/ml) for 5 h in the presence or absence of BMS754807. Cell extracts containing equivalent amounts of protein were immunoblotted with antibodies specific for IRS2, p-IGF-1R (Y1135/1136)/pIR (Y1150/1151), IGF-1R, pAKT (S473), AKT, or tubulin. (C) Schematic of Irs2 and Irs2-Y5F proteins. (D) Cell extracts from PyMT:Irs1/2−/− cells expressing EV, Irs2, or Irs2-Y5F were immunoblotted with antibodies specific for IRS2 and tubulin. (E and F) PyMT:Irs1/2−/− cells expressing EV, Irs2, or Irs2-Y5F were assayed for invasion. (E) Matrigel Transwell invasion assay. The data shown represent the means and SEM of the results of three independent experiments. (F) Matrigel-collagen I 3D invasion assay. The data shown represent the means and SEM of the results of a representative experiment performed three times independently. Representative images of colonies are shown on the right (magnification, ×10). **, P < 0.01 relative to EV; #, P < 0.05 relative to IRS2; ##, P < 0.01 relative to IRS2. Molecular weight markers (in kilodaltons) are indicated to the left of the immunoblots.
FIG 2.

The IRS2 C-terminal tail regulates tumor cell invasion. (A) Schematic of WT and mutant IRS proteins. (B) PyMT:Irs1/2−/− cells were stimulated with IGF-1 (50 ng/ml) for 10 min, and cell extracts were immunoprecipitated (IP) with an HA-specific antibody and immunoblotted with antibodies specific for HA or the p85 subunit of PI3K (p85). Whole-cell extracts (WCE) were also immunoblotted with antibodies specific for HA, pAkt (S473), pAkt (T308), Akt, and tubulin. (C and D) PyMT:Irs1/2−/− cells expressing IRS proteins were assayed for invasion. (C) Matrigel Transwell invasion assays. The data shown represent the means and SEM of the results of three independent experiments. (D) Matrigel-collagen I 3D invasion assays. The data shown represent the means and SEM of the results of a representative experiment performed three times independently. Representative images of colonies are shown below (magnification, ×10). (E) SUM-159:IRS1/2−/− cells expressing IRS proteins were assayed for invasion in a Matrigel Transwell invasion assay. The data shown represent the means and SEM of the results of four independent experiments. *, P < 0.05 relative to EV; **, P < 0.01 relative to EV; ##, P < 0.01 relative to IRS2. Molecular weight markers (in kilodaltons) are indicated to the left of the immunoblot.
Both IRS1 and IRS2 can recruit and activate PI3K, but only IRS2 promotes invasion (1). For this reason, we examined if the ability of IRS2 to regulate PI3K signaling is required for IRS2-mediated invasion. Previous work by our group identified four essential tyrosines in murine Irs2 (Y649, Y671, Y734, and Y814) that are conserved in human IRS2 (Y653, Y675, Y742, and Y823) and that are required for the recruitment of PI3K and activation of the downstream PI3K/AKT pathway in response to IGF-1 and insulin stimulation (33). PyMT:Irs1/2−/− cells expressing equivalent levels of Irs2 or Irs2-Y5F, an Irs2 mutant in which the essential tyrosines have been mutated to phenylalanine to prevent PI3K recruitment (Fig. 1C and D), were examined for their invasive potential. Cells expressing Irs2-Y5F showed a modest but significant increase in invasion over EV cells, but they were significantly less invasive than cells expressing wild-type (WT) Irs2 (Fig. 1E). To confirm the results of the two-dimensional (2D) Matrigel Transwell assay, cells were grown within a Matrigel-collagen I matrix to assess invasive potential in a three-dimensional (3D) environment that mimics the tumor matrix microenvironment in vivo (34). Poorly/less invasive cells grow as spherical colonies in this 3D matrix, whereas invasive cells invade the matrix to form branched colonies. Although Irs2- and Irs2-Y5F-expressing cells formed similar numbers of colonies, cells expressing Irs2-Y5F were significantly less invasive than cells expressing Irs2 (Fig. 1F). Our data support the notion that Irs2-dependent PI3K activation contributes to invasion. However, the partial reduction of invasion with the Irs2-Y5F mutant and the ability of IRS1 to activate PI3K imply that additional mechanisms also contribute to this regulation.
The IRS2 C-terminal tail participates in the regulation of invasion.
The IRS proteins lack stable tertiary-domain structure, with the exception of N-terminal PH and PTB domains that mediate recruitment to upstream receptors. To identify additional sequence requirements of IRS2 for promoting invasion, we sought to identify regions of the protein that are required for this functional outcome while preserving the ability of IRS2 to be recruited to upstream receptors and to interact with and activate PI3K. To do so, we generated an IRS2 truncation mutant lacking the C-terminal tail 3′ to the PI3K binding sites (IRS2Δ917) (Fig. 2A). A corresponding truncation mutant was also generated for IRS1 (IRS1Δ942) (Fig. 2A). Wild-type and mutant proteins were expressed in PyMT:Irs1/2−/− cells, and their abilities to interact with the PI3K regulatory subunit p85 in response to IGF-1 stimulation were examined. Both truncation mutants recruited PI3K in response to IGF-1 stimulation (Fig. 2B). To examine further the role of the C-terminal tail in the function of the IRS proteins, IRS1-IRS2 and IRS2-IRS1 chimeras were generated by swapping the corresponding C-terminal regions of each adapter protein (Fig. 2A). The chimeric proteins also maintained the ability to recruit PI3K. The IRS proteins are required for IGF-1R-dependent stimulation of PI3K activation, as evidenced by the lack of AKT activation in PyMT:Irs1/2−/− cells in response to IGF-1 stimulation (Fig. 2B, lane EV). Expression of either IRS1 or IRS2 restored the ability of IGF-1 to promote AKT phosphorylation at both the Thr308 and Ser473 sites, and Akt activation was maintained upon truncation or swapping of the C-terminal tails (Fig. 2B). Therefore, the C-terminal regions of IRS1 and IRS2 are not required for the IGF-1-dependent activation of the PI3K/AKT pathway.
Consistent with previous studies that have implicated IRS2 but not IRS1 in the promotion of tumor cell invasion, PyMT:Irs1/2−/− cells expressing IRS1 or the IRS1 truncation mutant (IRS1Δ942) invaded similarly to cells expressing EV (Fig. 2C). In contrast, truncation of the IRS2 C-terminal tail (IRS2Δ917) inhibited invasion relative to WT IRS2, indicating that sequences contained within the region are important for IRS2-dependent regulation of this function. To examine the sufficiency of the IRS2 C-terminal tail to enhance invasion, cells expressing the IRS1-IRS2 chimera, which contains the N-terminal portion of IRS1 and the C-terminal tail of IRS2, were assayed for their invasive potential. PyMT:Irs1/2−/− cells expressing the IRS1-IRS2 chimera exhibited an increase in invasion equivalent to what was observed for cells expressing WT IRS2. In contrast, cells expressing the IRS2-IRS1 chimera, which contains the N-terminal portion of IRS2 and the C-terminal tail of IRS1, failed to increase invasion above that of EV control cells (Fig. 2C). Cells expressing either WT IRS2 or the IRS1-IRS2 chimera also grew in a highly invasive manner within the Matrigel-collagen I matrix, whereas cells expressing IRS2Δ917 or the IRS2-IRS1 chimera exhibited little to no invasion, and the colonies formed were similar to those of cells expressing empty vector or IRS1 (Fig. 2D). Similar invasion results were obtained when the WT IRS proteins and their truncation mutants were expressed in an IRS1-IRS2 double-null SUM-159 human breast carcinoma cell line that was generated by clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 knockout (SUM-159:IRS1/2−/− cells) (Fig. 2E). Taken together, these data reveal that the C-terminal tail of IRS2 is both necessary and sufficient for IRS-dependent enhancement of invasion.
Identification of a region within the IRS2 C-terminal tail that regulates invasion but not glucose uptake.
The IRS2Δ917 truncation mutant lacks the final 421 amino acids (aa) of the IRS2 protein. To dissect further how this region contributes to the regulation of invasion, smaller truncation mutants that lacked either 324 amino acids (IRS2Δ1014) or 150 amino acids (IRS2Δ1188) were generated (Fig. 3A). These deletion mutants were expressed in PyMT:Irs1/2−/− cells and assayed for their responses to IGF-1 stimulation. As was observed for the larger truncation mutant (IRS2Δ917), deletion of smaller regions of the IRS2 C-terminal tail did not prevent PI3K recruitment or downstream AKT signaling (Fig. 3B).
FIG 3.

Identification of a region in the IRS2 C-terminal tail that regulates tumor cell invasion. (A) Schematic of WT and mutant IRS2 proteins. (B) Cell extracts from PyMT:Irs1/2−/− cells stimulated with IGF-1 (50 ng/ml) for 10 min were immunoprecipitated with an HA-specific antibody and immunoblotted with antibodies specific for HA or the p85 subunit of PI3K. Whole-cell extracts were also immunoblotted with antibodies specific for HA, pAkt (S473), pAkt (T308), Akt, and tubulin. (C) Matrigel Transwell invasion assays. The data shown represent the means and SEM of the results of five independent experiments. (D) Cell extracts from PyMT:Irs1/2−/− cells stimulated with IGF-1 (50 ng/ml) for 10 min were immunoprecipitated with an HA-specific antibody and immunoblotted with antibodies specific for HA or the p85 subunit of PI3K. Whole-cell extracts were also immunoblotted with antibodies specific for HA, pAkt (S473), Akt, and tubulin. The data shown in the graph below represent the means and SEM of p85 association with the IRS proteins from three independent experiments. No statistical significance was observed among the groups. (E) Matrigel Transwell invasion assays. The data shown represent the means and SEM of the results of three experiments. (F) Matrigel-collagen I 3D invasion assay. The data shown represent the means and SEM of the results of a representative experiment performed three times independently. *, P < 0.05 relative to EV; **, P < 0.01 relative to EV; #, P < 0.05 relative to IRS2; ##, P < 0.01 relative to IRS2. Molecular weight markers (in kilodaltons) are indicated to the left of the immunoblots.
PyMT:Irs1/2−/− cells expressing the IRS2Δ1014 and IRSΔ1188 truncation mutants were assayed for their invasive potential. Deletion of the C-terminal 150 amino acids (IRS2Δ1188) did not inhibit the ability of IRS2 to promote invasion compared with full-length IRS2 (Fig. 3C). In contrast, the IRS2Δ1014 mutant was deficient in promoting tumor cell invasion (Fig. 3C). These data indicate that sequences between amino acids 917 and 1188 are required for the ability of IRS2 to enhance tumor cell invasion. Internal deletion mutants missing amino acids 917 to 1014 (IRS2ΔCONT) or 1014 to 1188 (IRS2ΔINV) were generated (Fig. 3A), and these mutants maintained the ability to associate with PI3K and to activate the AKT pathway in response to IGF-1 stimulation (Fig. 3D). Deletion of the 174 amino acids between positions 1014 and 1188 rendered IRS2 incapable of promoting tumor cell invasion as measured by both Matrigel Transwell (Fig. 3E) and 3D Matrigel-collagen I (Fig. 3F) assays. However, IRS2 lacking the 95 amino acids between positions 917 and 1014 maintained full invasive potential (Fig. 3E and F).
We previously identified a role for IRS2 in the regulation of glucose uptake that is dependent upon the recruitment and activation of PI3K (13, 33). Knocking down the essential glucose transporter GLUT1 inhibited mammary tumor cell invasion, supporting a requirement for glucose uptake in invasion. To determine if the IRS2 C-terminal tail also participates in the regulation of glucose metabolism, PyMT:Irs1/2−/− cells expressing WT IRS1, WT IRS2, IRS2Δ917, and the two internal deletion mutants (IRS2ΔCONT and IRS2ΔINV) were assayed for glucose uptake. Cells lacking IRS protein expression (EV) or expressing only IRS1 did not increase glucose uptake significantly in response to IGF-1 stimulation (Fig. 4). In contrast, expression of WT IRS2 significantly enhanced the rate of IGF-1-stimulated glucose uptake (Fig. 4). Moreover, the full IRS2 C-terminal-tail deletion (IRS2Δ917) and the internal deletion mutants (IRS2ΔCONT and IRS2ΔINV) also increased glucose uptake in response to IGF-1 (Fig. 4). Therefore, the disruption of tumor cell invasion observed for the IRS2ΔINV mutant is not the result of deficient glucose uptake in these cells. Importantly, the ability of IRS2 to regulate invasion can be separated from its regulation of glucose metabolism.
FIG 4.
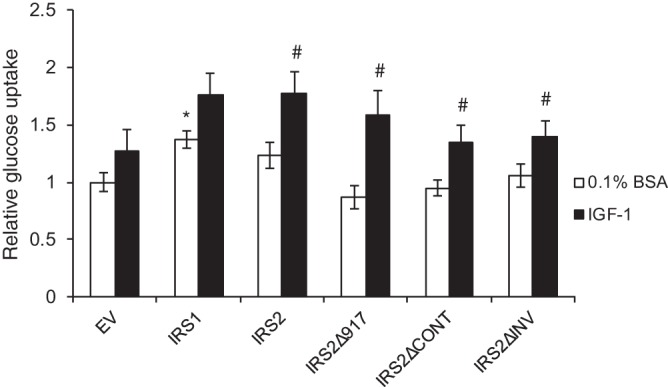
The INV region is not required for the IRS2-dependent regulation of glucose uptake. PyMT:Irs1/2−/− cells were grown in 0.1% BSA-DMEM with or without IGF-1 (25 ng/ml) for 16 to 24 h. Glucose uptake was measured and normalized to cell density. The data shown represent the means ± SEM of the results of five independent experiments. *, P < 0.05 relative to EV; #, P < 0.05 relative to unstimulated.
The INV region mediates essential interactions that promote invasion.
Given that IRS2 is an adaptor protein that lacks intrinsic kinase activity and mediates its functions through the recruitment and activation of signaling effectors, we hypothesized that intermolecular interactions that contribute to IRS2-dependent invasion occur within the INV region. To investigate further if essential interactions occur within this INV region, mVenus-tagged constructs containing the 174-aa INV region (aa 1014 to 1188), as well as two additional C-terminal regions (aa 917 to 1014 and 1188 to 1334) (Fig. 5A) that are not required for regulating invasion (Fig. 3), were generated. These mVenus-tagged regions were expressed by lentiviral infection and selection in SUM-159 breast carcinoma cells (Fig. 5B). Expression of the C-terminal regions did not alter the expression of endogenous IRS2 or interfere with IGF-1R-dependent PI3K/AKT activation (Fig. 5C). However, the INV region decreased invasion significantly (Fig. 5D). A decrease in invasion was also observed when the INV region was expressed in invasive human triple-negative MDA-MB-231 breast carcinoma cells (Fig. 5E).
FIG 5.

The IRS2 INV region acts in a dominant-negative manner. (A) Schematic depicting the C-terminal regions of IRS2 tagged with mVenus (green ovals). (B) (Top) Fluorescence and phase contrast images of SUM-159 cells expressing the individual regions (magnification, ×10). (Bottom) Whole-cell extracts were immunoblotted with antibodies specific for GFP and tubulin. (C) Cell extracts from SUM-159 cells expressing the IRS2 C-terminal regions and stimulated with IGF-1 (50 ng/ml) for 10 min were immunoblotted with antibodies specific for IRS2, pIGF-1R (Y1135/1136), IGF-1R, pAKT (T308), pAKT (S473), AKT, and tubulin. (D and E) SUM-159 cells (D) and MDA-MB-231 cells (E) expressing the IRS2 C-terminal regions were evaluated for invasion by Matrigel Transwell invasion assay. The data shown represent the means and SEM of the results of three independent experiments. *, P < 0.05 relative to EV; **, P < 0.01 relative to EV. Molecular weight markers (in kilodaltons) are indicated to the left of the immunoblots.
The ability of the INV region to act in a dominant-negative manner and inhibit invasion suggested that there are binding partners of this region of IRS2 that are important for the regulation of invasion. To identify novel interacting proteins, we used tandem affinity purification, followed by mass spectrometry (TAP-MS). For this purpose, the IRS2-INV region (aa 1014 to 1188) was tagged with a flexible 3×Flag-6×His tag and transiently transfected into SUM-159 cells. TAP-MS analysis identified several interacting proteins of the INV region, including desmoplackin, desmoglein, and desmocollin-1, which are members of the desmosome cell adhesion complex (35). In addition, the INV region pulled down bone morphogenetic protein 2-inducible kinase (BMP2K), a serine threonine kinase that has been previously reported to play a role in osteoblast differentiation (36, 37). Given the potential of BMP2K to function as a signaling kinase downstream of IRS2, we validated its interaction with full-length IRS2. BMP2K-specific antibodies coimmunoprecipitated IRS2, and this interaction was enhanced in IGF-1-stimulated cells (Fig. 6A). Although the BMP2K antibody recognizes all three human BMP2K isoforms (BMP2K-1, -2, and -3) by immunoblotting, only BMP2K-2 and BMP2K-3 were present in the immunoprecipitation. BMP2K isoforms 2 and 3 lack the C-terminal half of BMP2K-1, which supports the idea that IRS2 interacts with the N-terminal portion of BMP2K.
FIG 6.

BMP2K interacts with IRS2 and contributes to IRS2-dependent invasion. (A) MDA-MB-231 cells expressing IRS2-3×Flag-6×His were stimulated with IGF-1 (50 ng/ml) for 10 min. Cell extracts were immunoprecipitated with BMP2K-specific antibodies and immunoblotted with antibodies specific for either IRS2 or BMP2K. The data shown in the graph represent the means and SEM of the results of three independent experiments. Light, short exposure; dark, long exposure. (B) shRNAs targeting Bmp2k (shBmp2k#1 and shBmp2k#2) were expressed in PyMT:Irs1/2−/− cells expressing IRS2. Whole-cell extracts were immunoblotted with antibodies specific for Bmp2k, IRS2, and tubulin. (C) Matrigel Transwell invasion assay. The data shown represent the means and SEM of the results of three independent experiments. (D) Cells were stimulated with IGF-1 (50 ng/ml) for 10 min, and whole-cell extracts were immunoblotted with antibodies specific for IRS2, Bmp2k, pAkt (S473), Akt, and tubulin. The graph below represents the means and SEM of the results of three independent experiments. **, P < 0.01 relative to EV; #, P < 0.05 relative to IRS2-shGFP; ##, P < 0.01 relative to IRS2-shGFP. Molecular weight markers (in kilodaltons) are indicated to the left of the immunoblots.
To assess the contribution of BMP2K to IRS2-dependent invasion, short hairpin RNAs (shRNAs) targeting BMP2K were expressed in PyMT:Irs1/2−/− cells expressing WT-IRS2. There are only two murine isoforms of Bmp2k, and the expression of both Bmp2k-1 and Bmp2k-2 was decreased by ≥95% (Fig. 6B). Suppression of Bmp2k expression significantly decreased the IRS2-dependent enhancement of invasion (Fig. 6C). Akt activation was not inhibited by the loss of Bmp2k expression, indicating that Bmp2k is not required for IRS2-dependent PI3K activation (Fig. 6D).
DISCUSSION
Our study identifies a novel invasion-promoting region within IRS2 and advances the mechanistic understanding of how IRS1 and IRS2 regulate distinct functional outcomes. We established that the ability of IRS2 to promote invasion is dependent upon upstream IGF-1R/IR activation and recruitment and activation of PI3K, functions that are shared by IRS1. However, additional sequences within the C-terminal tail of IRS2 are also required for IRS2-mediated invasion, and these sequences were sufficient to confer the ability to promote invasion when swapped into IRS1. A 174-amino-acid region within the IRS2 C-terminal tail, which we term the INV region, is essential for enhancing invasion. Importantly, the INV region is not required for the IRS2-dependent regulation of glucose uptake, which provides evidence that IRS2 functions important for cancer progression can be disrupted while preserving the metabolic functions of the adaptor protein. We demonstrated that the INV region acts in a dominant-negative manner to inhibit invasion when expressed exogenously and identified BMP2K as one binding partner that interacts with this region and contributes to IRS2-dependent invasion. Taken together, our data highlight the important role of the IRS C-terminal tails in determining the unique functions of these adaptor proteins and identify a novel mechanism by which IRS2 regulates invasion.
Our structure/function analysis of IRS2 revealed important mechanistic information regarding how the IRS proteins regulate their unique functions. The IRS proteins are most similar in their N-terminal PH and PTB domains, and they are recruited to common cell surface receptors through these structured regions (38). C-terminal to these domains, the IRS proteins are less homologous across family members, although tyrosine residues located within canonical SH2 domain binding motifs are conserved. PI3K, GRB2, SHP2, and SFKs are recruited to these phosphotyrosine sites in both IRS1 and IRS2, and therefore, these interactions are not sufficient to explain their divergent functions (19–23). However, our data establish that the C-terminal tails do play an important role in their unique functional properties, as swapping these regions between IRS1 and IRS2 confers on IRS1 the ability to regulate invasion. The intrinsically disordered nature of the IRS C-terminal tails is proposed to facilitate dynamic structural changes in response to upstream stimuli to allow rapid recruitment and activation of signaling effectors. This model is based upon another adaptor protein, Gab1, which has a structured PH domain in the N terminus and a long, disordered C-terminal tail, similar to the IRS proteins (30). Phosphorylation of a serine residue in the Gab1 tail alters intramolecular interactions with the PH domain to generate “loop structures” that serve as docking sites for signaling effectors (39). We predict that sequences within the INV region in the IRS2 C-terminal tail participate in intramolecular interactions to form binding domains for unique effectors, such as BMP2K, that cooperate with PI3K to regulate invasion (Fig. 7).
FIG 7.

Model of IRS2 function. Upstream receptor activation stimulates dynamic intramolecular interactions within IRS2 that facilitate recruitment of PI3K and BMP2K to enhance invasion. SS, disulfide bond; P, tyrosine phosphorylation site.
We identified BMP2K as a novel IRS2-interacting protein that contributes to the ability of IRS2 to promote invasion. BMP2K, also known as BIKE (36), is a member of the human Numb-associated protein kinase (NAK) family, which also includes adaptor-associated kinase 1 (AAK1), cyclin G-associated kinase (GAK), and myristoylated and palmitoylated serine/threonine kinase 1 (MPSK1) (37). The limited information about BMP2K includes a role in attenuating osteoblast differentiation and genetic association with myopia and hip dysplasia (36, 40, 41). BMP2K has also been identified as a clathrin-coated-vesicle-associated protein, likely through its interaction with NUMB, a tumor suppressor that regulates endocytic receptor trafficking (42, 43). Trafficking of growth factor and integrin receptors has been implicated in cancer cell migration and invasion (44, 45), and this could be one mechanism by which BMP2K contributes to IRS2-dependent invasion. BMP2K is a constitutive kinase based upon its crystal structure, but its substrates are unknown (46). Future identification of these substrates will be important for elucidation of the mechanism by which BMP2K contributes to IRS2-dependent invasion.
The ability of IRS2 to enhance invasion is dependent upon the activation of PI3K. IRS1 also activates PI3K, but the functional outcome of this signaling is proliferation, not invasion (47). How each of these adaptor proteins can activate PI3K in response to common upstream receptor stimuli but regulate diverse functional outcomes has remained an open question. Although the INV region is not required for the recruitment and activation of PI3K, binding partners that interact with the region may cooperate with PI3K to enhance invasion. Alternatively, interactions with the region could modify the downstream outcomes of PI3K signaling to elicit divergent outcomes. In this regard, the differential requirement for an intact microtubule cytoskeleton in the IRS-dependent activation of AKT suggests that one mechanism by which interactions with the INV region could alter PI3K signaling outcomes is through the regulation of IRS2 trafficking that localizes PI3K signaling to intracellular locations necessary for regulating invasion (48). This could impact access to distinct pools of PI3K downstream effectors that promote invasion. Although BMP2K is not required for PI3K activation, the interaction of BMP2K with NUMB could impact endocytic trafficking and contribute to this mechanism of differential signaling. Interactions that regulate the strength and duration of the PI3K signal could also determine differential downstream functional outcomes. For example, interaction of the HECT-type ubiquitin ligase NEDD4 with IRS2 promotes monoubiquitination of C-terminal lysine residues (49). This ubiquitin modification promotes IGF-1-dependent tyrosine phosphorylation of IRS2 and enhances downstream signaling. However, NEDD4 binds to IRS2 through its conserved PH and PTB domains, not the INV region, and the specificity of this mechanism of regulation for IRS2, and not IRS1, has not been demonstrated (49).
A key outcome of our study is the demonstration that the mechanism by which IRS2 regulates invasion is distinct from the mechanism by which it regulates glucose uptake. Although the two IRS2 functions share a requirement for PI3K activation, as demonstrated by the deficiency of cells expressing the Irs2-Y5F mutant that is defective in PI3K recruitment in invading or enhancing glucose uptake (33), the INV region is uniquely required for regulating invasion. These mechanistic differences could be exploited to selectively target IRS2-mediated invasion, which contributes to metastasis, without affecting IRS2-mediated metabolic processes in normal tissues. Drugs that target IGF-1R have been investigated in clinical trials for several types of cancer, and the results overall have been disappointing, as multiple specific inhibitors have failed to show efficacy in preventing disease progression (50, 51). One reason for the failure of IGF-1R inhibitors is the upregulation of the insulin receptor to compensate for the loss of IGF-1R signaling (52, 53), but targeting both receptors disrupts normal metabolic homeostasis. Our results provide a rationale for considering IRS2 and IRS2-interacting proteins viable alternative targets for the inhibition of the IGF-1R/IR pathway in cancer. The identification of the INV region and BMP2K opens the door to developing novel approaches that would allow the disruption of tumor functions while maintaining normal metabolic function.
MATERIALS AND METHODS
Cells and transfection.
MDA-MB-231 cells were obtained from the ATCC Cell Biology Collection and grown in RPMI medium (Gibco) containing 10% fetal bovine serum (FBS) (Sigma). SUM-159 cells were a kind gift from Stephen Ethier (University of Michigan) and were grown in F-12 medium (Gibco) containing 5% FBS (Sigma), 5 μg/ml insulin (Sigma), and 1 μg/ml hydrocortisone (Sigma). PyMT cells were grown in low-glucose (1-g/liter) Dulbecco's modified Eagle's medium (DMEM) (Corning) containing 10% FBS. All the cells tested negative for mycoplasma using the Morwell MD Biosciences EZ PCR mycoplasma test kit (no. 409010).
PyMT mammary tumor cells were isolated from female FVB MMTV-PyMT:Irs1fl/flIrs2fl/fl mice, and PyMT:Irs1−/− Irs2−/− cells were generated by infection with adenoviral Cre recombinase as described previously (33). IRS1/IRS2 double-null SUM-159 cells were generated by CRISPR/Cas9-mediated gene editing using guide RNAs (gRNAs) that targeted an early 5′ exon region for either IRS1 (gRNA sequence: GCATGCTCTTGGGTTTGCGCAGG) or IRS2 (gRNA sequence: AACCACAGCGTGCGCAAGTGCGG). The gRNAs were subcloned into the pSpCas9(BB)-2A-GFP plasmid (Addgene; 48138). Cells were transfected with the CRISPR plasmid containing the IRS1 gRNA using Lipofectamine 2000 (Invitrogen) and sorted by flow cytometry for the green fluorescent protein (GFP)-high population to obtain IRS1−/− cells. The IRS1−/− cells were transfected with the CRISPR plasmid containing the IRS2-specific gRNA and sorted for GFP-high cells to generate SUM-159:IRS1−/− IRS2−/− cells. The cells were transfected with human IRS1, IRS2, and IRS mutants in pcDNA3.1 and selected in 500 μg/ml G418 (Gibco).
Mutagenesis and cloning.
Hemagglutinin (HA)-tagged human IRS1 and IRS2 were kindly provided by Adrian Lee (University of Pittsburgh, Pittsburgh, PA). Murine Irs2 was a kind gift from Morris White (Children's Hospital, Boston, MA), and the Irs2-Y5F mutant was generated as described previously (33). Human IRS deletions and chimeras were generated by PCR amplification using Q5 Hot Start High-Fidelity DNA polymerase (M0493S; New England BioLabs), and the PCR products were ligated into vectors using a Quick Ligation kit (M2200S; New England BioLabs). IRS chimeras were generated by PCR amplification of IRS1 and IRS2 N-terminal and vector sequences and C-terminal sequences. An AgeI site was introduced into both sections without altering the amino acid sequence (underlined in primer sequences in Table 1). IRS1-IRS2 and IRS2-IRS1 chimeras were generated by digestion of the PCR products with AgeI and HindIII and ligation of the swapped C-terminal regions with the IRS1 and IRS2 N termini. mVenus-tagged IRS2 regions were generated by PCR amplification from pcDNA-IRS2-HA and digestion with NheI and NotI overnight. The digested PCR products were ligated into the pCDH-mVenus vector that was generated by ligation of mVenus from the mVenus N1 plasmid (Addgene; 27793). The PCR primers (Table 1) were purchased from Integrated DNA Technologies.
TABLE 1.
Primers used for PCR

aAgeI sites are underlined.
Invasion assays.
Matrigel Transwell invasion assays were performed as described previously (1, 54). Cells were preincubated with BMS754807 (S1124; Selleckchem) for 4 h prior to the assays, and the inhibitor was also present in the upper and lower wells of the Transwell chamber during the assays. For 3D invasion assays, cells were suspended in a mixture of Matrigel (2 mg/ml) and collagen I (1 mg/ml) and plated over a base gel layer of the same matrix composition in 8-well chamber glass slides (Falcon). The gel was overlaid with complete-serum-containing medium, which was changed every 2 days for 8 days. All colonies were imaged and scored for the extent of cell invasion/branching (Diaphot 300 microscope; Nikon, Melville, NY) after 8 days. Matrigel (number 354230) and collagen I (number 354236) were obtained from Corning Discovery Labware, Inc. (Bedford, MA).
Immunoblotting and immunoprecipitation.
Cells were serum starved overnight (human cells) or for 4 h (PyMT cells) in serum-free medium. The cells were stimulated with human recombinant IGF-1 (R&D Systems, Minneapolis, MN) for the time periods indicated in the figure legends prior to extraction. For whole-cell extract immunoblots, cells were solubilized at 4°C in RIPA lysis buffer (25 mM Tris, pH 8.0, 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, 1% Nonidet P-40, 150 mM sodium chloride, 10 mM sodium fluoride, 1 mM sodium orthovanadate) containing protease inhibitors (Roche, Basel, Switzerland). Cell extracts containing equivalent amounts of protein were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked for 1 h with a 50 mM Tris buffer, pH 7.5, containing 0.15 M NaCl, 0.05% Tween 20, and 5% (wt/vol) dry milk or 5% bovine serum albumin (BSA); incubated overnight at 4°C in the same buffer containing primary antibodies; and then incubated for 1 h in 5% blocking buffer with milk containing peroxidase-conjugated secondary antibodies. Proteins were detected by enhanced chemiluminescence (Bio-Rad, Hercules, CA).
For immunoprecipitations, cells were extracted in either a 20 mM Tris buffer, pH 7.4, containing 1% Nonidet P-40, 0.137 M NaCl, 10% glycerol, 10 mM sodium fluoride, 1 mM sodium orthovanadate, and protease inhibitors (Roche) or a 40 mM HEPES buffer, pH 7.5, containing 120 mM NaCl, 1% Triton X-100, 10 mM sodium fluoride, 1 mM sodium orthovanadate, and protease inhibitors (Roche). Aliquots of cell extracts containing equivalent amounts of protein were precleared for 30 min with IgG and protein G-Sepharose beads and then incubated for 3 h or overnight at 4°C with specific antibodies and protein G-Sepharose beads (GE Healthcare) with constant agitation. The beads were washed three times in extraction buffer. Laemmli sample buffer was added to the samples, and immune complexes were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted as described above.
The following antibodies were used for immunoblotting and immunoprecipitation: IRS2 (4502; Cell Signaling, Danvers, MA), p85 (05-212; Millipore, Billerica, MA), HA (11867423001; Roche), phospho-IGF-1Rβ (Y1135/1136)/IRβ (Y1150/1151) (3024; Cell Signaling), IGF-1Rβ (3025; Cell Signaling), phospho-AKT S473 (9271 and 4060; Cell Signaling), phospho-AKT T308 (2965; Cell Signaling), AKT (sc-8312, Santa Cruz; 9272, Cell Signaling), BMP2K (sc-134284; Santa Cruz), α-tubulin (T5168; Sigma-Aldrich), GFP (ab6556; Abcam), HA (11867423001; Roche), peroxidase-conjugated goat anti-rabbit IgG (111-035-144; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), and peroxidase-conjugated goat anti-mouse IgG (711-035-151; Jackson ImmunoResearch Laboratories, Inc.).
Tandem affinity purification and mass spectrometry.
SUM-159 cells transiently expressing INV-3×Flag-6×His were extracted in a 50 mM Tris, pH 7.4, buffer containing 250 mM NaCl, 0.1% Triton X-100, 10 mM sodium fluoride, 1 mM sodium orthovanadate, and protease inhibitors (Roche). Aliquots of cell extracts were incubated for 3 h with FLAG-M2 agarose beads (Sigma) that had been equilibrated in lysis buffer. The beads were washed and eluted with 3× FLAG peptide (Sigma) diluted in lysis buffer. The FLAG elution was incubated with Talon (Millipore) agarose beads for 2 h before washing and elution with lysis buffer containing 250 mM imidazole. The Talon bead elution was trichloroacetic acid (TCA) precipitated and reconstituted in 1× Laemmli sample buffer. Samples were resolved by SDS-PAGE and analyzed by liquid chromatography-tandem mass spectrometry (LC–MS-MS) at the University of Massachusetts Medical School Mass Spectrometry Facility.
Glucose uptake assays.
Cells were grown to near confluence, washed with phosphate-buffered saline (PBS), and then incubated in 0.1% BSA-DMEM (1 g/liter glucose) for 16 to 24 h in the presence or absence of IGF-1 (25 ng/ml). Glucose levels were measured using a glucose assay kit (Sigma) according to the instructions of the manufacturer. Cell density per well was determined by crystal violet staining, and glucose uptake was expressed as a rate measurement (millimolar per milligram per hour) normalized to the cell density.
Statistical analysis.
Statistical analysis between two groups was performed using the two-tailed unpaired Student t test. A P value of <0.05 was considered to indicate statistical significance.
ACKNOWLEDGMENTS
We thank Art Mercurio for helpful comments on the manuscript.
This work was supported by National Institutes of Health (NIH) grant CA142782 (L.M.S.) and NIH F31 predoctoral fellowship CA180706 (J.M.-M.).
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
We declare that no conflict of interest exists.
J.M.-M. and L.M.S. were involved in the conception and design of the project and wrote the manuscript; J.J., S.Z., and S.S.C. were involved with the acquisition and analysis of data. We all reviewed and approved the final version of the manuscript.
REFERENCES
- 1.Nagle JA, Ma Z, Byrne MA, White MF, Shaw LM. 2004. Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol Cell Biol 24:9726–9735. doi: 10.1128/MCB.24.22.9726-9735.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mardilovich K, Pankratz SL, Shaw LM. 2009. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun Signal 7:14. doi: 10.1186/1478-811X-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee AV, Jackson JG, Gooch JL, Hilsenbeck SG, Coronado-Heinsohn E, Osborne CK, Yee D. 1999. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol 13:787–796. doi: 10.1210/mend.13.5.0274. [DOI] [PubMed] [Google Scholar]
- 4.Molloy CA, May FE, Westley BR. 2000. Insulin receptor substrate-1 expression is regulated by estrogen in the MCF-7 human breast cancer cell line. J Biol Chem 275:12565–12571. doi: 10.1074/jbc.275.17.12565. [DOI] [PubMed] [Google Scholar]
- 5.Koda M, Sulkowska M, Kanczuga-Koda L, Sulkowski S. 2005. Expression of insulin receptor substrate 1 in primary breast cancer and lymph node metastases. J Clin Pathol 58:645–649. doi: 10.1136/jcp.2004.022590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sisci D, Morelli C, Garofalo C, Romeo F, Morabito L, Casaburi F, Middea E, Cascio S, Brunelli E, Ando S, Surmacz E. 2007. Expression of nuclear insulin receptor substrate 1 in breast cancer. J Clin Pathol 60:633–641. doi: 10.1136/jcp.2006.039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morelli C, Garofalo C, Sisci D, del Rincon S, Cascio S, Tu X, Vecchione A, Sauter ER, Miller WH Jr, Surmacz E. 2004. Nuclear insulin receptor substrate 1 interacts with estrogen receptor alpha at ERE promoters. Oncogene 23:7517–7526. doi: 10.1038/sj.onc.1208014. [DOI] [PubMed] [Google Scholar]
- 8.Migliaccio I, Wu MF, Gutierrez C, Malorni L, Mohsin SK, Allred DC, Hilsenbeck SG, Osborne CK, Weiss H, Lee AV. 2010. Nuclear IRS-1 predicts tamoxifen response in patients with early breast cancer. Breast Cancer Res Treat 123:651–660. doi: 10.1007/s10549-009-0632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nolan MK, Jankowska L, Prisco M, Xu S, Guvakova MA, Surmacz E. 1997. Differential roles of IRS-1 and SHC signaling pathways in breast cancer cells. Int J Cancer 72:828–834. doi: 10.1002/(SICI)1097-0215(19970904)72:5<828::AID-IJC20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 10.Byron SA, Horwitz KB, Richer JK, Lange CA, Zhang X, Yee D. 2006. Insulin receptor substrates mediate distinct biological responses to insulin-like growth factor receptor activation in breast cancer cells. Br J Cancer 95:1220–1228. doi: 10.1038/sj.bjc.6603354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schnarr B, Strunz K, Ohsam J, Benner A, Wacker J, Mayer D. 2000. Down-regulation of insulin-like growth factor-I receptor and insulin receptor substrate-1 expression in advanced human breast cancer. Int J Cancer 89:506–513. doi: 10.1002/1097-0215(20001120)89:6<506::AID-IJC7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 12.Jackson JG, Zhang X, Yoneda T, Yee D. 2001. Regulation of breast cancer cell motility by insulin receptor substrate-2 (IRS-2) in metastatic variants of human breast cancer cell lines. Oncogene 20:7318–7325. doi: 10.1038/sj.onc.1204920. [DOI] [PubMed] [Google Scholar]
- 13.Pankratz SL, Tan EY, Fine Y, Mercurio AM, Shaw LM. 2009. Insulin receptor substrate-2 regulates aerobic glycolysis in mouse mammary tumor cells via glucose transporter 1. J Biol Chem 284:2031–2037. doi: 10.1074/jbc.M804776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porter HA, Perry A, Kingsley C, Tran NL, Keegan AD. 2013. IRS1 is highly expressed in localized breast tumors and regulates the sensitivity of breast cancer cells to chemotherapy, while IRS2 is highly expressed in invasive breast tumors. Cancer Lett 338:239–248. doi: 10.1016/j.canlet.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Z, Gibson SL, Byrne MA, Zhang J, White MF, Shaw LM. 2006. Suppression of insulin receptor substrate 1 (IRS-1) promotes mammary tumor metastasis. Mol Cell Biol 26:9338–9351. doi: 10.1128/MCB.01032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark JL, Dresser K, Hsieh CC, Sabel M, Kleer CG, Khan A, Shaw LM. 2011. Membrane localization of insulin receptor substrate-2 (IRS-2) is associated with decreased overall survival in breast cancer. Breast Cancer Res Treat 130:759–772. doi: 10.1007/s10549-011-1353-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myers MG Jr, Sun XJ, White MF. 1994. The IRS-1 signaling system. Trends Biochem Sci 19:289–293. doi: 10.1016/0968-0004(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 18.Johnston JA, Wang LM, Hanson EP, Sun XJ, White MF, Oakes SA, Pierce JH, O'Shea JJ. 1995. Interleukins 2, 4, 7, and 15 stimulate tyrosine phosphorylation of insulin receptor substrates 1 and 2 in T cells. Potential role of JAK kinases. J Biol Chem 270:28527–28530. [DOI] [PubMed] [Google Scholar]
- 19.Myers MG Jr, Backer JM, Sun XJ, Shoelson S, Hu P, Schlessinger J, Yoakim M, Schaffhausen B, White MF. 1992. IRS-1 activates phosphatidylinositol 3′-kinase by associating with src homology 2 domains of p85. Proc Natl Acad Sci U S A 89:10350–10354. doi: 10.1073/pnas.89.21.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myers MG Jr, Wang LM, Sun XJ, Zhang Y, Yenush L, Schlessinger J, Pierce JH, White MF. 1994. Role of IRS-1-GRB-2 complexes in insulin signaling. Mol Cell Biol 14:3577–3587. doi: 10.1128/MCB.14.6.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tobe K, Tamemoto H, Yamauchi T, Aizawa S, Yazaki Y, Kadowaki T. 1995. Identification of a 190-kDa protein as a novel substrate for the insulin receptor kinase functionally similar to insulin receptor substrate-1. J Biol Chem 270:5698–5701. doi: 10.1074/jbc.270.11.5698. [DOI] [PubMed] [Google Scholar]
- 22.Sun XJ, Pons S, Asano T, Myers MG Jr, Glasheen E, White MF. 1996. The Fyn tyrosine kinase binds Irs-1 and forms a distinct signaling complex during insulin stimulation. J Biol Chem 271:10583–10587. doi: 10.1074/jbc.271.18.10583. [DOI] [PubMed] [Google Scholar]
- 23.Myers MG Jr, Mendez R, Shi P, Pierce JH, Rhoads R, White MF. 1998. The COOH-terminal tyrosine phosphorylation sites on IRS-1 bind SHP-2 and negatively regulate insulin signaling. J Biol Chem 273:26908–26914. doi: 10.1074/jbc.273.41.26908. [DOI] [PubMed] [Google Scholar]
- 24.Sun XJ, Miralpeix M, Myers MG Jr, Glasheen EM, Backer JM, Kahn CR, White MF. 1992. Expression and function of IRS-1 in insulin signal transmission. J Biol Chem 267:22662–22672. [PubMed] [Google Scholar]
- 25.Myers MG Jr, Sun XJ, Cheatham B, Jachna BR, Glasheen EM, Backer JM, White MF. 1993. IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3′-kinase. Endocrinology 132:1421–1430. doi: 10.1210/endo.132.4.8384986. [DOI] [PubMed] [Google Scholar]
- 26.Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MG Jr, Glasheen E, Lane WS, Pierce JH, White MF. 1995. Role of IRS-2 in insulin and cytokine signalling. Nature 377:173–177. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- 27.Jackson JG, White MF, Yee D. 1998. Insulin receptor substrate-1 is the predominant signaling molecule activated by insulin-like growth factor-I, insulin, and interleukin-4 in estrogen receptor-positive human breast cancer cells. J Biol Chem 273:9994–10003. doi: 10.1074/jbc.273.16.9994. [DOI] [PubMed] [Google Scholar]
- 28.Shaw LM. 2001. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the alpha6beta4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol Cell Biol 21:5082–5093. doi: 10.1128/MCB.21.15.5082-5093.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhe-Paganon S, Ottinger EA, Nolte RT, Eck MJ, Shoelson SE. 1999. Crystal structure of the pleckstrin homology-phosphotyrosine binding (PH-PTB) targeting region of insulin receptor substrate 1. Proc Natl Acad Sci U S A 96:8378–8383. doi: 10.1073/pnas.96.15.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simister PC, Schaper F, O'Reilly N, McGowan S, Feller SM. 2011. Self-organization and regulation of intrinsically disordered proteins with folded N-termini. PLoS Biol 9:e1000591. doi: 10.1371/journal.pbio.1000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mardilovich K, Shaw LM. 2009. Hypoxia regulates insulin receptor substrate-2 expression to promote breast carcinoma cell survival and invasion. Cancer Res 69:8894–8901. doi: 10.1158/0008-5472.CAN-09-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, Chen C, Discenza L, Menard K, Li A, Trainor G, Vyas D, Kramer R, Attar RM, Gottardis MM. 2009. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther 8:3341–3349. doi: 10.1158/1535-7163.MCT-09-0499. [DOI] [PubMed] [Google Scholar]
- 33.Landis J, Shaw LM. 2014. Insulin receptor substrate 2-mediated phosphatidylinositol 3-kinase signaling selectively inhibits glycogen synthase kinase 3beta to regulate aerobic glycolysis. J Biol Chem 289:18603–18613. doi: 10.1074/jbc.M114.564070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen-Ngoc KV, Shamir ER, Huebner RJ, Beck JN, Cheung KJ, Ewald AJ. 2015. 3D culture assays of murine mammary branching morphogenesis and epithelial invasion. Methods Mol Biol 1189:135–162. doi: 10.1007/978-1-4939-1164-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huber O, Petersen I. 2015. 150th anniversary series: desmosomes and the hallmarks of cancer. Cell Commun Adhes 22:15–28. doi: 10.3109/15419061.2015.1039642. [DOI] [PubMed] [Google Scholar]
- 36.Kearns AE, Donohue MM, Sanyal B, Demay MB. 2001. Cloning and characterization of a novel protein kinase that impairs osteoblast differentiation in vitro. J Biol Chem 276:42213–42218. doi: 10.1074/jbc.M106163200. [DOI] [PubMed] [Google Scholar]
- 37.Smythe E, Ayscough KR. 2003. The Ark1/Prk1 family of protein kinases. Regulators of endocytosis and the actin skeleton. EMBO Rep 4:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White MF. 2002. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab 283:E413–E422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 39.Wolf A, Eulenfeld R, Bongartz H, Hessenkemper W, Simister PC, Lievens S, Tavernier J, Feller SM, Schaper F. 2015. MAPK-induced Gab1 translocation to the plasma membrane depends on a regulated intramolecular switch. Cell Signal 27:340–352. doi: 10.1016/j.cellsig.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 40.Liu HP, Lin YJ, Lin WY, Wan L, Sheu JJ, Lin HJ, Tsai Y, Tsai CH, Tsai FJ. 2009. A novel genetic variant of BMP2K contributes to high myopia. J Clin Lab Anal 23:362–367. doi: 10.1002/jcla.20344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao L, Zhou Z, Wang S, Jiao Q, Wu J, Ma F, Fan L, Chen M, Ying H. 2017. A recurrent mutation in bone morphogenetic proteins-2-inducible kinase gene is associated with developmental dysplasia of the hip. Exp Ther Med 13:1773–1778. doi: 10.3892/etm.2017.4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santolini E, Puri C, Salcini AE, Gagliani MC, Pelicci PG, Tacchetti C, Di Fiore PP. 2000. Numb is an endocytic protein. J Cell Biol 151:1345–1352. doi: 10.1083/jcb.151.6.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krieger JR, Taylor P, Gajadhar AS, Guha A, Moran MF, McGlade CJ. 2013. Identification and selected reaction monitoring (SRM) quantification of endocytosis factors associated with Numb. Mol Cell Proteomics 12:499–514. doi: 10.1074/mcp.M112.020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caswell P, Norman J. 2008. Endocytic transport of integrins during cell migration and invasion. Trends Cell Biol 18:257–263. doi: 10.1016/j.tcb.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 45.Porther N, Barbieri MA. 2015. The role of endocytic Rab GTPases in regulation of growth factor signaling and the migration and invasion of tumor cells. Small GTPases 6:135–144. doi: 10.1080/21541248.2015.1050152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sorrell FJ, Szklarz M, Abdul Azeez KR, Elkins JM, Knapp S. 2016. Family-wide structural analysis of human Numb-associated protein kinases. Structure 24:401–411. doi: 10.1016/j.str.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Myers MG Jr, Zhang Y, Aldaz GA, Grammer T, Glasheen EM, Yenush L, Wang LM, Sun XJ, Blenis J, Pierce JH, White MF. 1996. YMXM motifs and signaling by an insulin receptor substrate 1 molecule without tyrosine phosphorylation sites. Mol Cell Biol 16:4147–4155. doi: 10.1128/MCB.16.8.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mercado-Matos J, Clark JL, Piper AJ, Janusis J, Shaw LM. 2017. Differential involvement of the microtubule cytoskeleton in insulin receptor substrate 1 (IRS-1) and IRS-2 signaling to AKT determines the response to microtubule disruption in breast carcinoma cells. J Biol Chem 292:7806–7816. doi: 10.1074/jbc.M117.785832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fukushima T, Yoshihara H, Furuta H, Kamei H, Hakuno F, Luan J, Duan C, Saeki Y, Tanaka K, Iemura S, Natsume T, Chida K, Nakatsu Y, Kamata H, Asano T, Takahashi S. 2015. Nedd4-induced monoubiquitination of IRS-2 enhances IGF signalling and mitogenic activity. Nat Commun 6:6780. doi: 10.1038/ncomms7780. [DOI] [PubMed] [Google Scholar]
- 50.Yee D. 2012. Insulin-like growth factor receptor inhibitors: baby or the bathwater? J Natl Cancer Inst 104:975–981. doi: 10.1093/jnci/djs258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.King H, Aleksic T, Haluska P, Macaulay VM. 2014. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat Rev 40:1096–1105. doi: 10.1016/j.ctrv.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belfiore A, Malaguarnera R. 2011. Insulin receptor and cancer. Endocr Relat Cancer 18:R125–R147. doi: 10.1530/ERC-11-0074. [DOI] [PubMed] [Google Scholar]
- 53.Forest A, Amatulli M, Ludwig DL, Damoci CB, Wang Y, Burns CA, Donoho GP, Zanella N, Fiebig HH, Prewett MC, Surguladze D, DeLigio JT, Houghton PJ, Smith MA, Novosiadly R. 2015. Intrinsic resistance to cixutumumab is conferred by distinct isoforms of the insulin receptor. Mol Cancer Res 13:1615–1626. doi: 10.1158/1541-7786.MCR-15-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rohatgi RA, Janusis J, Leonard D, Bellve KD, Fogarty KE, Baehrecke EH, Corvera S, Shaw LM. 2015. Beclin 1 regulates growth factor receptor signaling in breast cancer. Oncogene 34:5352–5362. doi: 10.1038/onc.2014.454. [DOI] [PMC free article] [PubMed] [Google Scholar]