

TOR (target of rapamycin) has been previously implicated in transcriptional stimulation of the ribosomal protein (RP) genes via enhanced recruitment of NuA4 (nucleosome acetyltransferase of H4) to the promoters. However, it is not clearly understood how TOR enhances NuA4 recruitment to the promoters of the RP genes.
KEYWORDS: transcription, TBP, RNA polymerase II, NuA4, 19S proteasome subcomplex, TOR
ABSTRACT
TOR (target of rapamycin) has been previously implicated in transcriptional stimulation of the ribosomal protein (RP) genes via enhanced recruitment of NuA4 (nucleosome acetyltransferase of H4) to the promoters. However, it is not clearly understood how TOR enhances NuA4 recruitment to the promoters of the RP genes. Here we show that TOR facilitates the recruitment of the 19S proteasome subcomplex to the activator to enhance the targeting of NuA4 to the promoters of the RP genes. NuA4, in turn, promotes the recruitment of TFIID (transcription factor IID, composed of TATA box-binding protein [TBP] and a set of TBP-associated factors [TAFs]) and RNA polymerase II to the promoters of the RP genes to enhance transcriptional initiation. Therefore, our results demonstrate that TOR facilitates the recruitment of the 19S proteasome subcomplex to the promoters of the RP genes to promote the targeting of NuA4 for enhanced preinitiation complex (PIC) formation and consequently transcriptional initiation, hence illuminating TOR regulation of RP gene activation. Further, our results reveal that TOR differentially regulates PIC formation (and hence transcription) at the non-RP genes, thus demonstrating a complex regulation of gene activation by TOR.
INTRODUCTION
Transcription of the ribosomal protein (RP) genes is essential for ribosome biogenesis for translation and hence normal cellular growth and functions (1–3). In yeast, about 50% of RNA polymerase II is devoted to transcription of the RP genes (1–3). TOR (target of rapamycin; that is, inhibited by rapamycin [4]) has been shown to facilitate transcription of the RP genes by enhancing the association of NuA4 (nucleosome acetyltransferase of H4) with the promoters of the RP genes for histone H4 acetylation (1, 5). In the absence of TOR, Rpd3p-Sin3p mediates the repression of the RP genes (2, 5, 6). Further, TOR has been shown to control transcription of the RP genes via protein kinase A, which regulates YAK1 kinase to keep Crf1p corepressor in the cytoplasm (2, 7–10). When TOR is inactivated by rapamycin, Crf1p is phosphorylated by activated YAK1, shuttles into the nucleus, and competes with Ifh1p (interacts with Forkhead 1) coactivator of the activator Fhl1p (Forkhead-like 1), thereby inhibiting transcription of the RP genes (2, 7–10). Sfp1p, a regulator of cell size, has also been shown to control transcription of the RP genes via TOR (2, 7–10). Under optimal growth condition, Sfp1p is phosphorylated by TOR and localized into the nucleus and subsequently binds to the promoters of the RP genes to facilitate transcription. When TOR is inactivated by rapamycin, Sfp1p is dephosphorylated, resulting in transcriptional repression of the RP genes. Together, these studies demonstrate a complex transcriptional regulation of the RP genes by TOR.
Although previous studies (1, 2, 5, 6) demonstrated that TOR promotes transcription of the RP genes by enhancing the association of NuA4 with the promoters, it remains unknown how TOR facilitates the association of NuA4 lysine acetyltransferase (KAT) with the promoters of the RP genes (and hence histone H4 acetylation and transcription). To address this, we carried out experiments with several RP genes, such as RPS5, RPL2B, and RPS11B, with or without inhibition of TOR by rapamycin. Our results reveal that TOR inhibition reduces the targeting of the 19S base or subcomplex of the 26S proteasome (11–13) to the promoters of the RP genes and subsequently decreases the recruitment of NuA4 KAT, hence lowering histone H4 acetylation, preinitiation complex (PIC) formation, and transcription. Thus, our results decipher a new regulatory pathway of RP gene activation by TOR. Further, we show that TOR differentially regulates PIC formation (and hence transcription) at the non-RP genes, demonstrating a complex regulation of gene activation by TOR.
RESULTS
TOR facilitates the targeting of the 19S proteasome subcomplex to the UAS of an RP gene, RPS5, independently of activator Rap1p.
We have recently demonstrated that the 19S base or subcomplex of the 26S proteasome is recruited to the upstream activation sequences (UASs) of the RP genes and promotes the targeting of NuA4 KAT to the activator Rap1p (1). NuA4 KAT, in turn, enhances the formation of the PIC at the core promoter and hence transcriptional stimulation (1). Further, previous studies (1, 2, 5, 6) have shown that the recruitment of NuA4 KAT to the promoters of the RP genes is impaired in the presence of rapamycin (which inhibits TOR), thus implying a role for TOR in facilitating the recruitment of NuA4 KAT to the promoters of the RP genes. However, it is not clear how TOR facilitates the recruitment of NuA4 KAT to the promoters of the RP genes. TOR may promote recruitment of NuA4 KAT directly or via the 19S proteasome subcomplex (as the 19S proteasome subcomplex has been previously shown to promote the targeting of NuA4 KAT to the promoters of the RP genes [1]). To test this, we analyzed the recruitment of the 19S proteasome subcomplex to the promoter of an RP gene, RPS5, in the presence and absence of rapamycin. For this purpose, we tagged the Rpt2p and Rpt6p components of the 19S proteasome subcomplex by Myc epitope at their chromosomal loci (such tagging did not alter cellular growth [14]) and then performed a chromatin immunoprecipitation (ChIP) assay at the RPS5 UAS (as our previous studies demonstrated the association of the 19S proteasome subcomplex with the UAS of the RPS5 gene [1]). We found that the recruitment of the Rpt2p and Rpt6p components of the 19S proteasome subcomplex to the UAS of RPS5 was decreased in the presence of rapamycin (Fig. 1A). Such reduced levels of Rpt2p and Rpt6p at the RPS5 UAS following rapamycin treatment could be due to decreased stabilities of Rpt2p and Rpt6p following rapamycin treatment. To test this possibility, we analyzed the stabilities of Rpt2p and Rpt6p with or without rapamycin treatment. Our Western blot analysis revealed that the levels of Rpt2p and Rpt6p were not impaired following rapamycin treatment (Fig. 1B). Actin was monitored as a loading control (since actin is not regulated by rapamycin [15–17]), and its level was not changed following rapamycin treatment (Fig. 1B). Thus, TOR inhibition by rapamycin does not alter the stabilities of Rpt2p and Rpt6p but rather reduces their recruitment to the RPS5 UAS. Therefore, our results support a role for TOR in promoting the recruitment of the 19S proteasome subcomplex to the RPS5 UAS. Such function of TOR in enhancing the recruitment of the 19S proteasome subcomplex to the RPS5 UAS could be mediated via the activator Rap1p or independently of the activator. If TOR facilitates the recruitment of the 19S proteasome subcomplex independently of Rap1p, the recruitment of Rap1p but not the 19S proteasome subcomplex to the RPS5 UAS would not be impaired following rapamycin inhibition of TOR. To test this, we analyzed the association of Rap1p with the RPS5 UAS in the presence and absence of rapamycin. Our ChIP analysis revealed that the inhibition of TOR by rapamycin did not impair the recruitment of Rap1p to the RPS5 UAS (Fig. 1C) but rather decreased the targeting of the 19S proteasome subcomplex (Fig. 1A). Together, these results support the idea that TOR facilitates the targeting of the 19S proteasome subcomplex to the RPS5 UAS independently of the activator Rap1p. Consistently, the interaction between 19S proteasome subcomplex and Rap1p was decreased following rapamycin inhibition of TOR (Fig. 1D and E).
FIG 1.

TOR facilitates the targeting of the 19S proteasome subcomplex to the UAS of RPS5 for enhanced recruitment of NuA4 KAT for histone H4 acetylation. (A) TOR facilitates the targeting of the 19S proteasome subcomplex to the UAS of RPS5. Yeast strains expressing Myc-tagged Rpt2p and Rpt6p were grown in YPD (yeast extract, peptone, 2% dextrose) up to an OD600 of 0.9 at 30°C and then treated with 100 nM rapamycin (Sigma) for next 30 min prior to formaldehyde-based in vivo cross-linking. The ChIP assay was performed as described in Materials and Methods. Immunoprecipitation was performed using a mouse monoclonal antibody against the c-Myc epitope tag (9E10; Santa Cruz Biotechnology, Inc.). A primer pair located at the UAS of the RPS5 gene (see Materials and Methods) was used for PCR analysis of the immunoprecipitated DNA samples. The ratio of immunoprecipitate over the input in the autoradiogram was measured and is represented as the ChIP signal. The ChIP signal without rapamycin treatment (−R) was set to 100. The ChIP signal following rapamycin treatment (+R) was normalized with respect to 100. The normalized ChIP signal (represented as normalized occupancy) is plotted in the form of a histogram. (B) Western blot analysis of Rpt2p-Myc, Rpt6p-Myc and actin with or without rapamycin treatment. (C) Inhibition of TOR by rapamycin does not alter the recruitment of activator Rap1p to the RPS5 UAS but rather impairs the targeting of NuA4 KAT. (D and E) Coimmunoprecipitation (Co-IP) assay for analysis of interaction of the 19S proteasome with Rap1p in the presence and absence of rapamycin treatment. Results from panel D are plotted in the form of a histogram in panel E. (F and G) Growth analysis of the yeast strain bearing Myc-tagged Esa1p or Esa1p in solid and liquid YPD growth media. (H) Western blot analysis of Esa1p-Myc and actin with or without rapamycin treatment. (I) Histone H4 acetylation at the RPS5 promoter is impaired following rapamycin inhibition of TOR.
TOR-mediated recruitment of the 19S proteasome subcomplex facilitates the targeting of NuA4 KAT to the RPS5 promoter for histone H4 acetylation and PIC formation.
Since the 19S proteasome subcomplex has been shown to promote the targeting of NuA4 KAT to the activator Rap1p at the RPS5 UAS (1), the recruitment of NuA4 KAT would thus likely be similarly impaired following the inhibition of TOR by rapamycin. To test this, we analyzed the recruitment of NuA4 KAT to the RPS5 UAS with or without rapamycin treatment. For this purpose, we tagged the Esa1p (KAT) component of NuA4 (such tagging did not alter cellular growth [Fig. 1F and G]) and then performed the ChIP assay at the RPS5 UAS (as previous studies demonstrated the association of NuA4 KAT with the UASs of the RP genes [1]) with or without rapamycin inhibition of TOR. We found that recruitment of NuA4 KAT (Esa1p-Myc) to the RPS5 UAS was impaired following rapamycin treatment (Fig. 1C). However, the stability of Esa1p was not altered in response to rapamycin treatment (Fig. 1H). Thus, the inhibition of TOR by rapamycin impairs the recruitment of NuA4 KAT to the RPS5 UAS. Therefore, TOR promotes the recruitment of the 19S proteasome subcomplex, which subsequently enhances the targeting of NuA4 KAT to the RPS5 UAS.
Since NuA4 KAT is involved in acetylation of histone H4, the inhibition of TOR by rapamycin would impair histone H4 acetylation at the promoter of RPS5. To test this, we analyzed the level of histone H4 acetylation at the RPS5 promoter using the ChIP assay. We found that histone H4 acetylation at the RPS5 promoter was decreased following rapamycin inhibition of TOR (Fig. 1I). However, such a decreased level of histone H4 acetylation at the RPS5 promoter following TOR inhibition by rapamycin could be due to eviction (or loss) of histone H3-H4 tetramer from the RPS5 promoter. To test this, we analyzed the level of histone H3 (as a representative component of histone H3-H4 tetramer) at the RPS5 promoter following rapamycin treatment. Our ChIP analysis revealed that the level of histone H3 at the RPS5 promoter was not impaired following rapamycin treatment (Fig. 1I). Thus, TOR inhibition by rapamycin impairs histone H4 acetylation. Taken together, our results reveal that TOR promotes the targeting of NuA4 KAT via the 19S proteasome subcomplex to the RPS5 UAS for histone H4 acetylation.
In addition to facilitating the recruitment of NuA4 KAT to the RPS5 UAS via the 19S proteasome subcomplex, TOR may also have an additional role in enhancing directly the recruitment of NuA4 KAT to the RPS5 UAS. If so, the decrease of NuA4 KAT recruitment to the RPS5 UAS would be much more than that of the 19S proteasome subcomplex following inhibition of TOR by rapamycin. Intriguingly, we found that the extent of decrease in the recruitment of the 19S proteasome subcomplex to the RPS5 UAS was the same as that of the NuA4 KAT following rapamycin inhibition of TOR (Fig. 1A and C). Further, we have recently demonstrated that the 19S proteasome subcomplex facilitates the targeting of the NuA4 KAT to the activator Rap1p (1). These results support the idea that TOR facilitates the targeting of the 19S proteasome subcomplex to the RPS5 UAS to enhance the recruitment of NuA4 KAT. Hence, the level of histone H4 acetylation at the RPS5 promoter was decreased following inhibition of TOR by rapamycin (Fig. 1I). Thus, our results provide a novel regulatory network for TOR-19S proteasome subcomplex-NuA4 KAT at the UAS of RPS5 for histone H4 acetylation.
We have recently demonstrated that NuA4 KAT promotes formation of the PIC at the core promoter of RPS5 (1). Such stimulatory function of NuA4 KAT has been implicated to be mediated via histone H4 acetylation (1, 18–24). Thus, an impaired recruitment of NuA4 KAT to the RPS5 UAS (and hence histone H4 acetylation) following rapamycin inhibition of TOR would decrease formation of the PIC at the RPS5 core promoter. To test this, we analyzed the recruitment of TATA box binding protein (TBP) (which nucleates the assembly of general transcription factors for the PIC formation [11, 12]) in the presence and absence of rapamycin. We found that the recruitment of TBP to the RPS5 core promoter was impaired following rapamycin treatment (Fig. 2A). Likewise, the recruitment of TBP-associated factor 1 (TAF1p), a specific component of transcription factor IID (TFIID), and RNA polymerase II (Rpb1p, the largest subunit of RNA polymerase II) was also impaired following rapamycin treatment (Fig. 2A). We have previously demonstrated that TAF1p and/or other TAFs are required for recruitment of TBP to the RPS5 promoter, while TBP is dispensable for recruitment of TAF1p or other TAFs (25), and TAFs are targeted to the RPS5 promoter by activator and NuA4 (26–30). Further, TFIID is required for initiation of PIC formation and hence recruitment of RNA polymerase II to the core promoter (25). General transcription factors such as TFIIB and Mediator are not required for recruitment of TFIID or TAFs at the RPS5 promoter (26) but rather are essential for initiation of RPS5 transcription (31). Thus, impaired recruitment of TBP, TAF1p, and RNA polymerase II to the RPS5 core promoter in response to rapamycin treatment indicates that TOR facilitates PIC formation at the RPS5 core promoter. Consistently, transcription of RPS5 was decreased following rapamycin inhibition of TOR (Fig. 2B). As a control, we showed that the recruitment of TBP and RNA polymerase II to the core promoter of a rapamycin-independent gene, ACT1, was not altered following rapamycin treatment (Fig. 2C), and consistently, transcription of ACT1 was not changed when TOR is inhibited by rapamycin (Fig. 2B). Thus, an impaired targeting of NuA4 KAT to Rap1p via the 19S proteasome subcomplex following rapamycin inhibition of TOR lowers PIC formation (and hence transcriptional initiation) at the promoter of an essential RPS5 gene. Consistently, cellular growth was impaired following temperature-sensitive (ts) inactivation of the 19S proteasome subcomplex at 37°C (Fig. 2D). Like in Saccharomyces cerevisiae, human RPS5 was also facilitated by TOR (Fig. 2E). TSR2 and MGP were used as controls (Fig. 2E and F), as these genes are known to be stimulated and repressed, respectively, by TOR (32–35).
FIG 2.

TOR facilitates formation of the preinitiation complex (PIC) at the RPS5 promoter to enhance transcription. (A) Inhibition of TOR by rapamycin decreases the recruitment of TBP, TAF1p, and Rpb1p to the core promoter of RPS5. Yeast cells were grown as for Fig. 1A. (B) Reverse transcription-PCR (RT-PCR) analysis. Rapamycin inhibition of TOR impairs transcription of the RPS5 gene. (C) Inhibition of TOR by rapamycin does not impair the recruitment of TBP and Rpb1p to the ACT1 promoter. (D) Growth analysis of rpt4-ts (or sug2-ts) and wild-type (WT) strains in liquid YPD growth medium at the nonpermissive temperature (37°C). Yeast cells were initially grown at 23°C up to an OD600 of about 0.1 prior to switching temperature to 37°C, and then OD600 was measured at different time points for growth analysis. (E and F) RT-PCR analysis of RPS5, TSR2, and MGP in HEK293 cells following rapamycin inhibition of TOR.
TOR facilitates the targeting of the 19S proteasome subcomplex to enhance transcription complex assembly at promoters of other RP genes, RPL2B and RPS11B.
To determine whether TOR similarly regulates transcriptional initiation at other RP genes, we analyzed the recruitment of the 19S proteasome subcomplex, NuA4 KAT, TBP, TAF1p, and RNA polymerase II to the promoters of two other RP genes, RPL2B and RPS11B, with or without rapamycin treatment. We found that the inhibition of TOR by rapamycin decreased the recruitment of the 19S proteasome subcomplex but not activator Rap1p to the UASs of RPL2B and RPS11B (Fig. 3A to C). Subsequently, the targeting of NuA4 KAT to the UASs of RPL2B and RPS11B was decreased following rapamycin treatment (Fig. 3B and C). Consistently, histone H4 acetylation at the RPL2B and RPS11B promoters was decreased following rapamycin inhibition of TOR (Fig. 3D and E). However, the inhibition of TOR by rapamycin did not alter the levels of histone H3-H4 tetramer at the promoters of RPL2B and RPS11B (Fig. 3D and E). Thus, similar to the results at RPS5, we found that TOR facilitated the recruitment of the 19S proteasome subcomplex but not activator Rap1p to the RPL2B and RPS11B UASs to enhance the targeting of NuA4 KAT and hence histone H4 acetylation. Consequently, PIC formation at the core promoters (i.e., recruitment of TBP, TAF1p, and RNA polymerase II) of these genes was impaired following rapamycin treatment (Fig. 4A and B). Consistently, transcription of RPL2B and RPS11B was decreased following inhibition of TOR by rapamycin (Fig. 4C). Together, our results support the idea that TOR facilitates the targeting of the 19S proteasome subcomplex to enhance the recruitment of NuA4 KAT and hence PIC formation (and consequently transcriptional initiation) at the promoters of the RP genes.
FIG 3.

TOR facilitates targeting of the 19S proteasome subcomplex to the UASs of RPL2B and RPS11B for enhanced recruitment of NuA4 KAT for histone H4 acetylation. (A) TOR facilitates the targeting of the 19S proteasome subcomplex to the UASs of RPL2B and RPS11B. A yeast strain expressing Myc-tagged Rpt6p was grown, cross-linked, and immunoprecipitated as for Fig. 1A. (B and C) Rapamycin inhibition of TOR does not alter the recruitment of activator Rap1p to the UASs of RPL2B and RPS11B but rather reduces the targeting of NuA4 KAT. (D and E) Histone H4 acetylation at the RPL2B and RPS11B promoters is impaired following rapamycin inhibition of TOR.
FIG 4.

TOR facilitates PIC formation at the RPS11B and RPL2B promoters to enhance transcription. (A and B) Inhibition of TOR by rapamycin reduces the recruitment of TBP, TAF1p, and Rpb1p to the core promoters of RPS11B and RPL2B. Yeast cells were grown as for Fig. 1A. (C) RT-PCR analysis. Transcription of the RPS11B and RPL2B genes is impaired following rapamycin inhibition of TOR.
NuA4 KAT does not regulate the recruitment of the 19S proteasome subcomplex to the RPS5, RPL2B, and RPS11B promoters.
The 19S proteasome subcomplex facilitates the targeting of NuA4 KAT to the activator Rap1p (1). Next, we analyzed whether NuA4 KAT has any effect on recruitment of the 19S proteasome subcomplex to the promoters of RPS5, RPL2B, and RPS11B. For this purpose, we tagged the Rpt2p component of the 19S proteasome subcomplex with the Myc epitope in the Esa1p wild-type (WT) and ts mutant strains and then performed the ChIP assay at the promoters of the RP genes. We found that the recruitment of the 19S proteasome subcomplex to these promoters was not altered in the esa1-ts mutant strain (Fig. 5A to C). On the other hand, the 19S proteasome subcomplex promotes the recruitment of NuA4 KAT to the promoters of these genes (1). These results support the idea that the 19S proteasome subcomplex facilitates the targeting of NuA4 KAT to activator Rap1p, while NuA4 KAT is dispensable for recruitment of the 19S proteasome complex. Further, we found that the 19S proteasome subcomplex did not interact genetically with NuA4 KAT, as the deletion of EAF1 (an integral component of NuA4 KAT [36]) in the sug2-ts mutant (Sug2p or Rpt4p ATPase component of the 19S proteasome subcomplex) does not reverse the growth defect of the sug2-ts mutant at the nonpermissive temperature (Fig. 5D). Consistently, Lee et al. (37) have also found that the 19S proteasome subcomplex does not physically interact with NuA4 KAT.
FIG 5.

NuA4 KAT is dispensable for recruitment of the 19S proteasome subcomplex to the RPS5, RPL2B, and RPS11B promoters. (A to C) ChIP analysis of Rpt2p at the UASs of RPS5, RPL2B, and RPS11B. Yeast cells expressing Myc-tagged Rpt2p were grown in YPD up to an OD600 of 0.9 and then switched to 37°C for 1 h prior to cross-linking. (D) Growth analysis of wild-type, eaf1Δ, sug2-ts (or rpt4-ts), and eaf1Δsug2-ts (or eaf1Δrpt4-ts) strains in the solid YPD medium at 23°C, 30°C, and 37°C.
TOR differentially regulates PIC formation at the promoters of the non-RP genes.
The results for the above-mentioned RP genes delineate a new TOR-mediated stimulatory network for PIC formation and hence transcriptional initiation. To test whether TOR also promotes PIC formation at the non-RP genes, we next analyzed the recruitment of TBP and RNA polymerase II, two important components of the PIC, to the core promoters of the non-RP genes, ADH1, PHO84, and PGK1, in the presence and absence of rapamycin. We found that the recruitment of TBP and RNA polymerase II to the ADH1, PHO84, and PGK1 promoters was decreased following rapamycin treatment (Fig. 6A and B). As a control, we showed that the recruitment of TBP and RNA polymerase II to the core promoter of a rapamycin-independent gene, ACT1 (a non-RP gene), was not impaired following rapamycin inhibition of TOR (Fig. 6A and B). Thus, like the results for the RP genes, TOR facilitates PIC formation at the non-RP genes. Consistently, transcription of these genes was also decreased following rapamycin inhibition of TOR (Fig. 6C). However, transcription of the rapamycin-nonresponsive ACT1 gene was not altered following rapamycin treatment (Fig. 6C). Thus, our results support the idea that the inhibition of TOR by rapamycin impairs PIC formation (and hence transcription) at ADH1, PHO84, and PGK1. Therefore, like the results for the RP genes, TOR promotes transcriptional initiation of ADH1, PHO84, and PGK1. Intriguingly, we found that SED1 (a non-RP gene), which is involved in formation of the cell wall, is upregulated in response to rapamycin treatment (Fig. 7). We found that the inhibition of TOR by rapamycin enhances the recruitment of TBP and RNA polymerase II to the promoter of SED1, thus implying a repressive role for TOR in PIC formation (Fig. 7A and B). Consistently, transcription of SED1 was enhanced following rapamycin inhibition of TOR (Fig. 7C). Therefore, in addition to its stimulatory function, TOR also represses PIC formation (and hence transcription). SED1 is a diauxic shift gene (38). When cells are grown in dextrose-containing growth medium, they metabolize dextrose predominantly by glycolysis and produce ethanol (39). Cells enter diauxic shift when dextrose becomes limiting and metabolism is switched from glycolysis to aerobic utilization of ethanol (39). Thus, diauxic shift is characterized by slow growth and a metabolic switch from glycolysis to ethanol utilization (39). Previous studies have demonstrated that diauxic-shift genes are upregulated in response to rapamycin treatment (40). Thus, transcription of SED1 (as a diauxic-shift gene) was increased following rapamycin inhibition of TOR (Fig. 7C). Further, our results reveal that such enhanced transcription following rapamycin inhibition of TOR is mediated via facilitation of PIC formation (Fig. 7A and B). Thus, in addition to its role in stimulating PIC formation (and transcriptional initiation), TOR also represses transcription via impaired PIC formation. Further, TOR does not have an effect on the PIC formation (and transcription) at ACT1 (Fig. 7). Therefore, TOR differentially regulates PIC formation and hence transcriptional initiation or activation.
FIG 6.

TOR enhances PIC formation (and hence transcription) at the promoters of the ADH1, PHO84, and PGK1 genes. (A) TOR facilitates the recruitment of TBP to the core promoters of the ADH1, PHO84, and PGK1 genes. Yeast cells were grown as for Fig. 1A. (B) TOR facilitates recruitment of Rpb1p to the core promoters of the ADH1, PHO84, and PGK1 genes. (C) RT-PCR analysis. Transcription of the ADH1, PHO84, and PGK1 genes is impaired following rapamycin inhibition of TOR.
FIG 7.
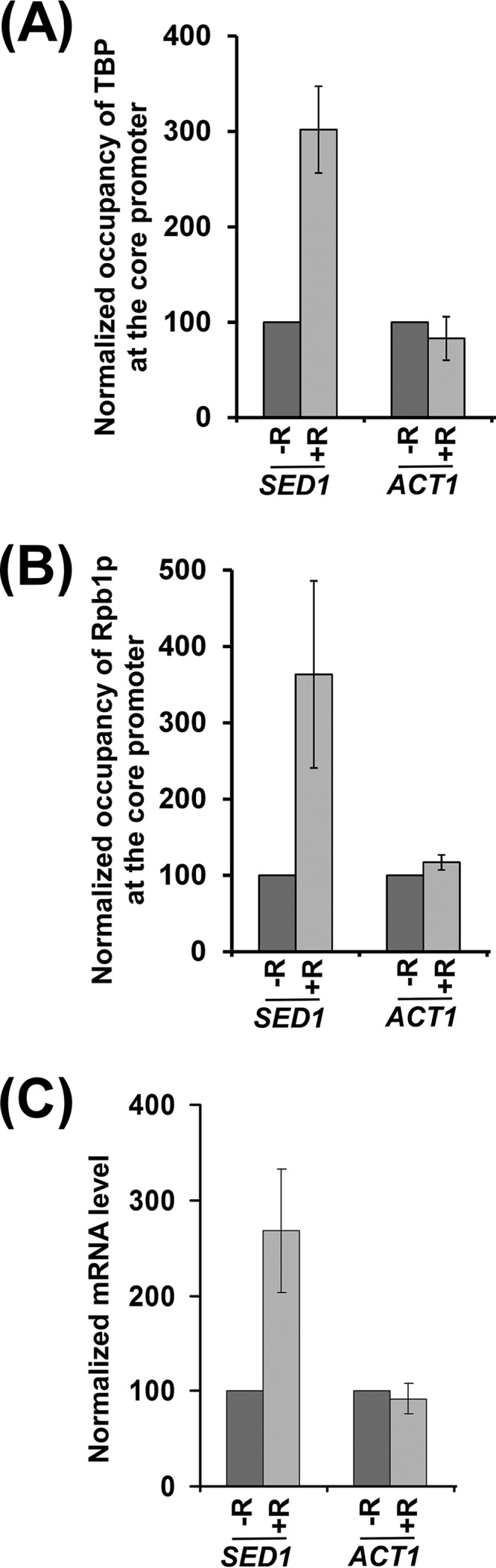
TOR represses PIC formation (and hence transcription) at the promoter of the SED1 gene. (A) TOR represses the recruitment of TBP to the SED1 core promoter. Yeast cells were grown as for Fig. 1A. (B) TOR represses recruitment of Rpb1p to the SED1 core promoter. (C) RT-PCR analysis. Transcription of the SED1 gene is increased following rapamycin inhibition of TOR.
DISCUSSION
Previous studies (1, 41, 42) demonstrated NuA4 KAT as the target of activator Rap1p at the promoters of the RP genes for transcriptional initiation or activation. Recently, we have demonstrated that Rap1p-NuA4 KAT interaction is enhanced by the 19S proteasome subcomplex in a proteolysis-independent manner (1). Consistently, previous studies (14, 37, 43) also demonstrated the stimulatory role of the 19S proteasome subcomplex in the interaction between activator Gal4p and coactivator SAGA to enhance transcriptional initiation of the Gal4p-regulated genes. Likewise, we have also recently demonstrated the stimulatory role of the 19S proteasome subcomplex in targeting SAGA and NuA4 KAT to the UAS of a high-affinity inorganic phosphate (Pi) transporter gene, PHO84, in the presence and absence of Pi in the growth media, respectively (44). Although our recent studies (1) implied a role for the 19S proteasome subcomplex in enhancing the interaction between activator Rap1p and coactivator NuA4 KAT at the UASs of the RP genes, TOR has also been found to enhance the targeting of NuA4 KAT to the UASs of the RP genes (1, 2, 5, 6). In agreement with this, transcription of the RP genes is decreased in response to TOR inhibition by rapamycin (or nutrient deprivation) as revealed by genome-wide expression analysis (40, 45–49). However, it is not clear how TOR facilitates the targeting of NuA4 KAT to the promoters of the RP genes (and hence transcription). It is quite likely that TOR directly regulates the association of NuA4 KAT with the promoters of the RP genes or indirectly via regulation of the recruitment of the 19S proteasome subcomplex (since the 19S proteasome subcomplex promotes the targeting of NuA4 KAT to the activator Rap1p [1]). If the recruitment of NuA4 KAT but not the 19S proteasome subcomplex is impaired following rapamycin inhibition of TOR, it would imply that TOR regulates the targeting of NuA4 KAT to the UASs of the RP genes independently of the 19S proteasome subcomplex. To address this, we analyzed recruitment of the 19S proteasome subcomplex and NuA4 KAT to the UASs of the representative set of RP genes, RPS5, RPL2B, and RPS11B, following inhibition of TOR by rapamycin. We found that the recruitment of the 19S proteasome subcomplex to the UASs of the RPS5, RPL2B, and RPS11B genes was impaired following rapamycin inhibition of TOR (Fig. 1A and 3A). Similarly, the recruitment of NuA4 KAT to the UASs of these RP genes was decreased following rapamycin treatment (Fig. 1C and 3B and C). Since the 19S proteasome subcomplex is involved in promoting the targeting of NuA4 KAT to the UASs of the RP genes (1), our results support the idea that TOR facilitates the targeting of the 19S proteasome subcomplex, which subsequently enhances the recruitment of NuA4 KAT, thus deciphering a novel regulatory network of the recruitment of NuA4 KAT to the UASs of the RP genes. Consistently, transcription of RPS5, RPL2B, RPS11B and other RP genes was impaired following rapamycin inhibition of TOR (Fig. 2B and 4C) (40, 45, 46, 48). Likewise, transcription of the RP genes is also impaired following nutrient or amino acid starvation (40). In agreement, cellular growth, protein synthesis, and ribosome biogenesis are impaired following rapamycin inhibition of TOR or nutrient deprivation in yeast (2, 4, 47, 49, 50). Likewise, mammalian transcription, cellular growth or ribosome biogenesis are also enhanced by TOR (2, 32, 33, 51). Further, both 19S proteasome subcomplex and NuA4 KAT promoted RP gene transcription and cellular growth (Fig. 2D) (1, 42, 52–54). Thus, our results reveal a physiologically relevant novel regulatory network for recruitment of NuA4 KAT (and hence PIC formation and transcription) by TOR at the promoters of the RP genes.
How does TOR promote the targeting of the 19S proteasome subcomplex to the UASs of the RP genes? It may be likely that TOR is involved in phosphorylating some component(s) of the 19S proteasome subcomplex to regulate its function in promoting recruitment of NuA4 KAT to the UASs of the RP genes. In support of this possibility, recent studies (55, 56) demonstrated the phosphorylation of several components (e.g., Rpn1p, Rpt2p, Rpt3p, Rpt5p, and Rpt6p) of the 19S proteasome subcomplex. Such phosphorylation has been implicated in facilitating the ATPase activity of the 19S proteasome subcomplex and its assembly (55, 57) and hence function (since previous studies demonstrated the role of the 19S ATPases in stimulation of the interaction between activator and coactivator [1, 14, 37, 44]). Intriguingly, changes of phosphorylation of several components (e.g., Rpt1p, Rpt2p, Rpt3p, Rpt5p, and Rpn1p) of the 19S proteasome subcomplex were observed following rapamycin inhibition of TOR (58). Taken together, these studies support the TOR regulation of phosphorylation of the 19S proteasome subcomplex and its function. Thus, TOR-mediated phosphorylation of the 19S proteasome subcomplex is likely to promote the targeting of NuA4 KAT. Indeed, recruitment of NuA4 KAT to the UASs of the RP genes was significantly impaired following TOR inhibition by rapamycin (Fig. 1C and 3B and C). Thus, our results establish an important regulatory network for the recruitment of NuA4 KAT to the UASs of the RP genes by TOR. Subsequently, NuA4 KAT acetylates histone H4 at the promoter for PIC formation to initiate transcription (Fig. 1I, 2A and B, 3D and E, and 4). These results demonstrate how TOR facilitates PIC formation and hence transcriptional initiation of the RP genes via the 19S proteasome subcomplex. However, TOR regulation of the phosphorylation of the 19S proteasome subcomplex and its ATPase activity as well as assembly to promote its targeting to the UASs of the RP genes remains to be elucidated.
Although our results demonstrate a new TOR-mediated stimulatory pathway of PIC formation (and hence transcription) at the promoters of the RP genes via the 19S proteasome subcomplex, the transcriptional initiation of the RP genes is further controlled by other TOR-mediated factors (e.g., Fhl1p, Ifh1p, Crf1p, and Sfp1p) as described above in the introduction. Thus, greater defects in PIC formation at the promoters of the RP genes are likely to be observed following rapamycin inhibition of TOR. Indeed, we observed a greater defect in PIC formation in comparison to the 19S proteasome subcomplex at the promoters of the RP genes following the inhibition of TOR by rapamycin (Fig. 1A, 2A, 3A, and 4A and B). Thus, TOR regulates PIC formation at the promoters of the RP genes via distinct mechanisms. While other pathways were known from previous studies (5–10, 59), we decipher here a novel TOR-mediated transcriptional stimulatory mechanism of the RP genes via the 19S proteasome subcomplex.
Like the RP genes, non-RP genes are transcriptionally stimulated by TOR, as revealed by genome-wide expression analysis following rapamycin inhibition of TOR or nutrient deprivation (40). However, it is not clear how TOR promotes transcription of the non-RP genes. To address this, we analyzed PIC formation at the promoters of a representative set of non-RP genes—ADH1, PGK1, and PHO84—that are transcriptionally stimulated by TOR (40). We found that PIC formation at ADH1 and PGK1 (glycolytic genes) is enhanced by TOR, and hence transcription of these genes is impaired following TOR inhibition by rapamycin (Fig. 6). These genes are not regulated by NuA4 KAT (42). Thus, TOR appears to regulate PIC formation independently of NuA4 KAT, unlike the results for the RP genes. Likewise, TOR facilitates the PIC formation and hence transcription initiation at another non-RP gene, PHO84 (which is independent of NuA4 KAT in yeast extract-peptone-dextrose [YPD] [44]), involved in Pi transport across cell membrane (Fig. 6). Thus, like the results for the RP genes, TOR facilitates PIC formation (and hence transcription) at the non-RP genes.
Since 19S proteasome subcomplex promotes the targeting of the coactivator SAGA (14, 37, 44), PIC formation at the SAGA-dependent genes would also be impaired following TOR inhibition by rapamycin (as TOR promotes phosphorylation of the 19S proteasome subcomplex and its function [55, 57, 58]). Indeed, we found that PIC formation at the promoter of the SAGA-regulated, but NuA4 KAT-independent, ADH1 gene (42) was decreased following TOR inhibition by rapamycin (Fig. 6A and B), and the 19S proteasome subcomplex promotes the recruitment of SAGA to ADH1 (37). Likewise, PHO84 is dependent on SAGA (which is facilitated by the 19S proteasome subcomplex for recruitment [44]) but not NuA4 KAT for PIC formation and transcriptional initiation in the presence of Pi in the growth medium or YPD (44) and is stimulated by TOR for PIC formation and hence transcription (Fig. 6). Similarly, PGK1 has also been implicated to be SAGA dependent (60), but independent of NuA4 KAT and TFIID (26, 42), and is upregulated by TOR for PIC formation and transcription (Fig. 6). Thus, like NuA4 KAT-regulated genes, SAGA-dependent genes are stimulated by TOR. SAGA, or NuA4 dependency of a gene is dictated by the coactivator specificity of the activator, which binds to the particular DNA sequence in a gene-specific manner (11).
Intriguingly, previous genome-wide studies revealed a repressive role of TOR in transcription (40). One such gene is SED1, which is involved in cell wall formation. Recent studies have implicated SED1 as a diauxic-shift gene (38). Diauxic-shift genes are generally upregulated by rapamycin inhibition of TOR (40). Although TOR has been shown to repress transcription of SED1, it is not clear how TOR suppresses transcription. Here we show that TOR represses PIC formation at the SED1 promoter (Fig. 7A and B). Consistently, transcription of SED1 is elevated when TOR is inactivated by rapamycin (Fig. 7C). Thus, our results demonstrate that the diauxic-shift gene SED1 is transcriptionally upregulated by rapamycin inhibition of TOR at the level of PIC formation. However, it remains to be further elucidated as to how TOR represses PIC formation.
In summary, we show here that TOR enhances the targeting of the 19S proteasome subcomplex to the promoters of the RP genes. The 19S proteasome subcomplex subsequently facilitates the targeting of the NuA4 KAT to the promoters of the RP genes for histone H4 acetylation in formation of the PIC and hence transcriptional initiation (Fig. 8). Thus, our results decipher a new regulatory pathway of RP gene expression (and hence ribosome biogenesis) by TOR. Since ribosome biogenesis is correlated with translation (and thus cellular growth and function), our results point to a new regulatory mechanism of TOR in cellular functions and growth via 19S proteasome subcomplex-mediated transcriptional activation of the RP genes.
FIG 8.

Schematic diagram showing TOR-mediated RP gene activation. Rap1p interacts with the 19S proteasome subcomplex (19S regulatory particle [RP]) and recruits it to the UAS of the RP gene. The 19S RP subsequently promotes the targeting of NuA4 to the activator Rap1p. NuA4 facilitates recruitment of TFIID (transcription factor IID, which is composed of TBP and a set of TBP-associated factors [TAFs]) toward the formation of the PIC, which includes RNA polymerase II for transcriptional initiation. TOR promotes the targeting of the 19S RP to the activator and hence facilitates recruitment of NuA4 and subsequent PIC formation and transcriptional initiation. ORF, open reading frame. +, stimulation.
MATERIALS AND METHODS
Plasmids.
Plasmid pFA6a-13Myc-KanMX6 (61) was used for genomic tagging of Rpt2p, Rpt6p, and Esa1p by the Myc epitope. Plasmid pFA6a-3HA-His3MX6 (61) was used for genomic hemagglutinin (HA) epitope tagging of Rpt6p. Plasmid pRS406 was used to delete EAF1 by PCR-based gene disruption.
Yeast strains.
The yeast (S. cerevisiae) strain bearing a ts mutation in Rpt4p (rpt4-ts or sug2-13; Sc677) and its isogenic WT equivalent (Sc599) were obtained from the Kodadek and Johnston laboratories (52). Multiple Myc epitope tags were added at the original chromosomal loci of RPT2, RPT6, and ESA1 in the WT strain (Sc599 [52]) to generate PSY17 (Rpt2p-Myc [14]), PSY18 (Rpt6p-Myc [14]), and BUY12 (Esa1p-Myc [1]). Multiple HA epitope tags were added at the original chromosomal locus of RPT6 in W303a to generate BUY4 (Rpt6p-HA). The esa1-ts mutant (LPY3291) and WT (LPY3498) strains were obtained from the Pillus laboratory (62). Multiple Myc epitope tags were added at the original chromosomal locus of RPT2 in the Esa1p WT strain (LPY3498 [62]) and esa1-ts mutant (LPY3291 [62]) to generate the BUY21 and BUY22 strains, respectively. The EAF1 gene was deleted in Sug2p (or Rpt4p) WT (PSY17 [14]) and ts mutant (Sc677) strains to generate the BUY14 and BUY64 strains, respectively.
Growth medium.
For studies with RPS5, RPL2B, RPS11B, ADH1, PHO84, PGK1, ACT1, and SED1, yeast cells were grown in YPD up to an optical density at 600 nm (OD600) of 0.9 at 30°C and then treated with 100 nM rapamycin (Sigma) for next 30 min prior to formaldehyde-based in vivo cross-linking or harvesting for RNA analysis.
ChIP assay.
The ChIP assay for TBP, Rpb1p, TAF1p and histone H4 acetylation was performed as described previously (1, 63–70). For ChIP analysis of Myc-tagged Esa1p, the ChIP protocol was modified as described previously (1, 67). Briefly, a total of 800 μl of lysate was prepared from 100 ml of yeast culture. Following sonication, 400 μl of ysate was used for each immunoprecipitation (using 10 μl of anti-Myc antibody and 100 μl of protein A/G plus agarose beads from Santa Cruz Biotechnology, Inc.), and immunoprecipitated DNA sample was dissolved in 10 μl Tris-EDTA (TE; pH 8.0), of which 1 μl was used for PCR analysis (a total of 23 cycles). In parallel, PCR analysis for input DNA was performed using 1 μl of DNA that was prepared by dissolving purified DNA from 5 μl of lysate in 100 μl TE (pH 8.0). The ChIP analysis for histone H3 was performed as described previously (71–73). For ChIP analysis of Myc-tagged Rpt2p and Rpt6p, the ChIP protocol was modified as described previously (1). Serial dilutions of input and immunoprecipitated DNA samples were used to assess the linear range of PCR amplification as described previously (63, 65). The data presented here are within the linear range of PCR analysis. The primer pairs used for PCR analysis were as follows: RPS5 (upstream activation sequence [UAS]), 5′-AGAAACAATGAACAGCCTTGAGTTCT-3′ and 5′-GCAGGGCCATTCTCATCTGA-3′; for RPS5 (core promoter [core]), 5′-GGCCAACTTCTACGCTCACGTTAG-3′ and 5′-CGGTGTCAGACATCTTTGGAATGGTC-3′; for RPL2B (UAS), 5′-TACCGATTACCAAGTTTTCAGACTA-3′ and 5′-AATTCCTTCTTTTTCTCCCTAGCGG-3′; for RPL2B (core), 5′-TGGTGGATTCTGCTCTGGAAACTAT-3′ and 5′-CTTTGTGGTTTCTTGGTGAGTTTAT-3′; for RPS11B (UAS), 5′-GATATACACAAGAATTTCTGGAAGA-3′ and 5′-CACTTCCTCATTTCACAAAGACACT-3′; for RPS11B (core), 5′-AAGTCCAATAGCTTTACGTTTCCCT-3′ and 5′-CTTTTTCCCTGGCTTGATACGTTTC-3′; for ACT1 (core), 5′-AACCGTTTTGAAACCAAACTCGCCT-3′ and 5′-TTCTTGGTTTGAGTAGAAAGGGGAA-3′; for ADH1 (core), 5′-GGTATACGGCCTTCCTTCCAGTTAC-3′ and 5′-GAACGAGAACAATGACGAGGAAACAAAAG-3′; for PGK1 (core), 5′-GAATCGTGTGACAACAACAGCCTG-3′ and 5′-CTTGCATTGACCAATTTATGC-3′; for PHO84 (core), 5′-GATCCACTTACTATTGTGGCTCGT-3′ and 5′-GTTTGTTGTGTGCCCTGGTGATCT-3′; and for SED1 (core), 5′-TAGTACGGTATGGCTCCCAGTC-3′ and 5′-GGTGAGATAATACGATAATGGAG-3′.
Autoradiograms were scanned and quantitated by the National Institutes of Health Image 1.62 program. Immunoprecipitated DNA was quantitated as the ratio of immunoprecipitate to input and represented as a ChIP signal. The average ChIP signal of the biologically independent experiments is reported with standard deviation (SD) (Microsoft Excel).
Preparation of total RNA.
Total RNA was prepared from yeast cell culture as described previously (63, 74–76). Briefly, 10 ml of yeast culture was harvested and suspended in 100 μl of RNA preparation buffer (500 mM NaCl, 200 mM Tris-HCl, 100 mM Na2EDTA, and 1% SDS) along with 100 μl of phenol-chloroform-isoamyl alcohol and a 100-μl equivalent of glass beads (acid washed; Sigma). Subsequently, yeast cell suspension was vortexed at maximum speed (10 in a VWR minivortexer; catalog no. 58816-121) five times (30 s each). After vortexing, 150 μl of RNA preparation buffer and 150 μl of phenol-chloroform-isoamyl alcohol were added to the yeast cell suspension, followed by vortexing for 30 s at maximum speed on a VWR minivortexer. The aqueous phase was collected for isolation of total RNA by precipitation with ethanol.
RT-PCR analysis.
Reverse transcription-PCR (RT-PCR) analysis was performed as described previously (74, 77, 78). Briefly, RNA was treated with RNase-free DNase (M610A; Promega) and then reverse transcribed into cDNA using oligo(dT) as described in the protocol supplied by Promega (A3800). PCR was performed using synthesized first strand as the template and the primer pairs targeted to the open reading frames (ORFs) of the RPS5, RPL2B, RPS11B, ACT1, ADH1, PGK1, PHO84, and SED1 genes. RT-PCR products were separated by 2.2% agarose gel electrophoresis and visualized by ethidium bromide staining. The average signal for the biologically independent RT-PCR experiments is reported with SD (Microsoft Excel). The primer pairs used in the PCR analysis of cDNAs were as follows: for ADH1, 5′-CGGTAACAGAGCTGACACCAGAGA-3′ and 5′-ACGTATCTACCAACGATTTGACCC-3′; for RPS5, 5′-AGGCTCAATGTCCAATCATTGAAAG-3′ and 5′-CAACAACTTGGATTGGGTTTTGGTC-3′; for ACT1, 5′-TCCACCACTGCTGAAAGAGAAATTG-3′ and 5′-AATAGTGATGACTTGACCATCTGGA-3′; for RPL2B, 5′-GTGCTTTCCACAAGTACAGATTGAA-3′ and 5′-TTTGACCAGAAACGGCACCTCTAGA-3′; for RPS11B, 5′-GCACCGTACCATTGTCATCAGAAGA-3′ and 5′-GGTCTACATTGACCAACGGTAACAA-3′; for PHO84, 5′-TCTGCAGACATTTTGGTCAATGGAA-3′ and 5′-AAACGTTTTTGGAACCGGCATAAC-3′; for PGK1, 5′-AGACGAAGTTGTCAAGAGCTCTGC-3′ and 5′-GAAAGCAACACCTGGCAATTCCT-3′; and for SED1, 5′-GAATCTAAGGGCACTACCACC-3′ and 5′-AAGCACCTGGAACGACGACGACGTT-3′.
Whole-cell extract preparation and Western blot analysis.
For analysis of global levels of Rpt6p, Rpt2p, and Esa1p with or without rapamycin treatment, yeast cells were grown in YPD up to an OD600 of 0.9 and then treated with 100 nM rapamycin (Sigma) for next 30 min prior to harvesting for Western blot analysis. The harvested cells were lysed and sonicated to prepare the whole-cell extract with solubilized chromatin following the protocol as described previously for the ChIP assay (1, 63–70). The whole-cell extract was run on an SDS-polyacrylamide gel and then analyzed by Western blotting. The anti-Myc (9E10; Santa Cruz Biotechnology) and antiactin (A2066; Sigma) antibodies against Myc-tagged Rpt6p, Myc-tagged Rpt2p, Myc-tagged Esa1p, and actin were used in the Western blot analysis.
Co-IP assay.
The coimmunoprecipitation (Co-IP) assay was performed as described previously (1, 69). Briefly, the yeast strain carrying HA-tagged Rpt6p was grown in YPD up to an OD600 of 0.9 and then treated with rapamycin (100 nM) for 30 min prior to cross-linking by formaldehyde. The whole-cell extract was prepared by lysing and sonicating the cross-linked yeast cells as described for the ChIP assay. Immunoprecipitation was performed using an anti-Rap1p antibody (SC-6663; Santa Cruz Biotechnology, Inc.) against Rap1p activator and protein A/G plus agarose beads. After immunoprecipitation, the agarose beads were washed as in the ChIP assay. The washed A/G plus agarose beads were boiled in the SDS-PAGE loading buffer, and then the supernatant was used for SDS–PAGE and Western blot analysis. An anti-HA–horseradish peroxidase (HRP) antibody (H6533-1VL; Sigma-Aldrich) against HA-tagged Rpt6p was used in the Western blot analysis.
Human cell culture, RNA isolation, cDNA preparation, and RT-PCR.
Proliferating human embryonic kidney (HEK293) cells (ATCC) were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS; HyClone) in a humidified CO2 incubator at 37°C, according to the standard protocol. When the cells reached 80% confluence, they were treated with rapamycin (20 nM) or an equal volume of dimethyl sulfoxide (DMSO) for 30 min prior to RNA isolation. RNA was isolated from cells with Ribozol RNA extraction reagent (VWR). Following treatment with DNase (Promega), 2 μg of total RNA was reversed transcribed with MultiScribe MuLV reverse transcriptase (Applied Biosystems) to obtain cDNA. PCR was performed using cDNA as the template, and the primer pairs targeted to the ORFs of the RPS5, TSR2, and MGP genes. RT-PCR products were separated by 2.2% agarose gel electrophoresis and visualized by ethidium bromide staining. The average signal for the biologically independent RT-PCR experiments is reported with SD (Microsoft Excel). The primer pairs used in the PCR analysis of cDNAs were as follows: for RPS5, 5′-GGACTGTGAGACGACAGGC-3′ and 5′-TCAGCAATGGTCTTAATGTTCCG-3′; for TSR2, 5′-CATGCGCAATGCTGACTTGG-3′ and 5′-CCTCTGGAAGTGGTGGAACA-3′; and for MGP, 5′-TTTGTGTTATGAATCACATGAAAGC-3′ and 5′-GTGGACAGGCTTAGAGCGTT-3′.
ACKNOWLEDGMENTS
We thank Michael R. Green for anti-TBP and anti-TAF1 antibodies and Loraine Pillus, Thomas Kodadek, and Stephen Johnston for yeast strains. We also thank Judy Davie and Abhinav Adhikari at Southern Illinois University School of Medicine for reagents and technical assistance in carrying out experiments with the HEK293 cell line.
The work in the Bhaumik laboratory was supported by the grants from National Institutes of Health (1R15GM088798-01 and 2R15GM088798-02), American Heart Association (15GRNT25700298), and Southern Illinois University School of Medicine.
REFERENCES
- 1.Uprety B, Lahudkar S, Malik S, Bhaumik SR. 2012. The 19S proteasome subcomplex promotes the targeting of NuA4 HAT to the promoters of ribosomal protein genes to facilitate the recruitment of TFIID for transcriptional initiation in vivo. Nucleic Acids Res 40:1969–1983. doi: 10.1093/nar/gkr977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayer C, Grummt I. 2006. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 25:6384–6391. doi: 10.1038/sj.onc.1209883. [DOI] [PubMed] [Google Scholar]
- 3.Warner JR. 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24:437–440. doi: 10.1016/S0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 4.Schmelzle T, Hall MN. 2000. TOR, a central controller of cell growth. Cell 103:253–262. doi: 10.1016/S0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 5.Rohde J, Cardenas ME. 2003. The tor pathway regulates gene expression by linking nutrient sensing to histone acetylation. Mol Cell Biol 23:629–635. doi: 10.1128/MCB.23.2.629-635.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humphrey EL, Shamji AF, Bernstein BE, Schreiber SL. 2004. Rpd3p relocation mediates a transcriptional response to rapamycin in yeast. Chem Biol 11:295–299. doi: 10.1016/j.chembiol.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Martin DE, Soulard A, Hall MN. 2004. TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL. Cell 119:969–979. doi: 10.1016/j.cell.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 8.Jorgensen P, Rupes I, Sharom JR, Schneper L, Brouch JR, Tyers M. 2004. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev 18:2491–2505. doi: 10.1101/gad.1228804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schawalder SB, Kabani M, Howald I, Choudhury U, Werner M, Shore D. 2004. Growth-regulated recruitment of the essential yeast ribosomal protein gene activator Ifh1. Nature 432:1058–1061. doi: 10.1038/nature03200. [DOI] [PubMed] [Google Scholar]
- 10.Rudra D, Zhao Y, Warner JR. 2005. Central role of Ifh1p-Fhl1p interaction in the synthesis of yeast ribosomal proteins. EMBO J 24:533–542. doi: 10.1038/sj.emboj.7600553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhaumik SR. 2011. Distinct regulatory mechanisms of eukaryotic transcriptional activation by SAGA and TFIID. Biochim Biophys Acta 1809:97–108. doi: 10.1016/j.bbagrm.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhaumik SR, Malik S. 2008. Diverse regulatory mechanisms of eukaryotic transcriptional activation by the proteasome complex. Crit Rev Biochem Mol Biol 43:419–433. doi: 10.1080/10409230802605914. [DOI] [PubMed] [Google Scholar]
- 13.Frankland-Searby S, Bhaumik SR. 2012. The 26S proteasome complex: an attractive target for cancer therapy. Biochim Biophys Acta 1825:64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malik S, Shukla A, Sen P, Bhaumik SR. 2009. The 19S proteasome subcomplex establishes a specific protein interaction network at the promoter for stimulated transcriptional initiation in vivo. J Biol Chem 284:35714–35724. doi: 10.1074/jbc.M109.035709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sen R, Malik S, Frankland-Searby S, Uprety B, Lahudkar S, Bhaumik SR. 2014. Rrd1p, an RNA polymerase II-specific prolyl isomerase and activator of phosphoprotein phosphatase, promotes transcription independently of rapamycin response. Nucleic Acids Res 42:9892–9907. doi: 10.1093/nar/gku703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Douville J, David J, Lemieux KM, Gaudreau L, Ramotar D. 2006. The Saccharomyces cerevisiae phosphatase activator RRD1 is required to modulate gene expression in response to rapamycin exposure. Genetics 172:1369–1372. doi: 10.1534/genetics.105.046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poschmann J, Drouin S, Jacques PE, El Fadili K, Newmarch M, Robert F, Ramotar D. 2011. The peptidyl prolyl isomerase Rrd1 regulates the elongation of RNA polymerase II during transcriptional stresses. PLoS One 6:e23159. doi: 10.1371/journal.pone.0023159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altaf M, Auger A, Monnet-Saksouk J, Brodeur J, Piquet S, Cramet M, Bouchard N, Lacoste N, Utley RT, Gaudreau L, Côté J. 2010. NuA4-dependent acetylation of nucleosomal histones H4 and H2A directly stimulates incorporation of H2A.Z by the SWR1 complex. J Biol Chem 285:15966–15977. doi: 10.1074/jbc.M110.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durant M, Pugh BF. 2007. NuA4-directed chromatin transactions throughout the Saccharomyces cerevisiae genome. Mol Cell Biol 27:5327–5335. doi: 10.1128/MCB.00468-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobson RH, Ladurner AG, King DS, Tjian R. 2000. Structure and function of a human TAF(II)250 double bromodomain module. Science 288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- 21.Ladurner AG, Inouye C, Jain R, Tjian R. 2003. Bromodomains mediate an acetyl-histone encoded antisilencing function at heterochromatin boundaries. Mol Cell 11:365–376. doi: 10.1016/S1097-2765(03)00035-2. [DOI] [PubMed] [Google Scholar]
- 22.Matangkasombut O, Buratowski S. 2003. Different sensitivities of bromodomain factors 1 and 2 to histone H4 acetylation. Mol Cell 11:353–363. doi: 10.1016/S1097-2765(03)00033-9. [DOI] [PubMed] [Google Scholar]
- 23.Pamblanco M, Poveda A, Sendra R, Rodriguez-Navarro S, Perez-Ortin JE, Tordera V. 2001. Bromodomain factor 1 (Bdf1) protein interacts with histones. FEBS Lett 496:31–35. doi: 10.1016/S0014-5793(01)02397-3. [DOI] [PubMed] [Google Scholar]
- 24.Matangkasombut O, Buratowski RM, Swilling NW, Buratowski S. 2000. Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev 14:951–962. [PMC free article] [PubMed] [Google Scholar]
- 25.Shen WC, Bhaumik SR, Causton HC, Simon I, Zhu X, Jennings EG, Wang TH, Young RA, Green MR. 2003. Systematic analysis of essential yeast TAFs in genome-wide transcription and preinitiation complex assembly. EMBO J 22:3395–3402. doi: 10.1093/emboj/cdg336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li XY, Bhaumik SR, Green MR. 2000. Distinct classes of yeast promoters revealed by differential TAF recruitment. Science 288:1242–1244. doi: 10.1126/science.288.5469.1242. [DOI] [PubMed] [Google Scholar]
- 27.Mencía M, Moqtaderi Z, Geisberg JV, Kuras L, Struhl K. 2002. Activator-specific recruitment of TFIID and regulation of ribosomal protein genes in yeast, Mol Cell 9:823–833. [DOI] [PubMed] [Google Scholar]
- 28.Li XY, Bhaumik SR, Zhu X, Li L, Shen WC, Dixit B, Green MR. 2002. Selective recruitment of TAFs by yeast upstream activating sequences: implications for eukaryotic promoter structure. Curr Biol 12:1240–1244. [DOI] [PubMed] [Google Scholar]
- 29.Garbett KA, Tripathi MK, Cencki B, Layer JH, Weil PA. 2007. Yeast TFIID serves as a coactivator for Rap1p by direct protein-protein interaction. Mol Cell Biol 27:297–311. doi: 10.1128/MCB.01558-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Layer JH, Miller SG, Weil PA. 2010. Direct transactivator-transcription factor IID (TFIID) contacts drive yeast ribosomal protein gene transcription. J Biol Chem 285:15489–15499. doi: 10.1074/jbc.M110.104810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li XY, Virbasius A, Zhu X, Green MR. 1999. Enhancement of TBP binding by activators and general transcription factors. Nature 399:605–609. doi: 10.1038/21232. [DOI] [PubMed] [Google Scholar]
- 32.Jimenez RH, Lee JS, Francesconi M, Castellani G, Neretti N, Sanders JA, Sedivy J, Gruppuso PA. 2010. Regulation of gene expression in hepatic cells by the mammalian Target of Rapamycin (mTOR). PLoS One 5:e9084. doi: 10.1371/journal.pone.0009084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang BT, Ducker GS, Barczak AJ, Barbeau R, Erle DJ, Shokat KM. 2011. The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proc Natl Acad Sci U S A 108:15201–15206. doi: 10.1073/pnas.1103746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akcakanat A, Zhang L, Tsavachidis S, Meric-Bernstam F. 2009. The rapamycin-regulated gene expression signature determines prognosis for breast cancer. Mol Cancer 8:75. doi: 10.1186/1476-4598-8-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng T, Golub TR, Sabatini DM. 2002. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol Cell Biol 22:5575–5584. doi: 10.1128/MCB.22.15.5575-5584.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uprety B, Sen R, Bhaumik SR. 2015. Eaf1p is required for recruitment of NuA4 in targeting TFIID to the promoters of the ribosomal protein genes for transcriptional initiation in vivo. Mol Cell Biol 35:2947–2964. doi: 10.1128/MCB.01524-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee D, Ezhkova E, Li B, Pattenden SG, Tansey WP, Workman JL. 2005. The proteasome regulatory particle alters the SAGA coactivator to enhance its interactions with transcriptional activators. Cell 123:423–436. doi: 10.1016/j.cell.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 38.Li L, Miles S, Melville Z, Prasad A, Bradley G, Breeden LL. 2013. Key events during the transition from rapid growth to quiescence in budding yeast require posttranscriptional regulators. Mol Biol Cell 24:3697–3709. doi: 10.1091/mbc.e13-05-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galdieri L, Mehrotra S, Yu S, Vancura A. 2010. Transcriptional regulation in yeast during diauxic shift and stationary phase. OMICS 14:629–638. doi: 10.1089/omi.2010.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardwick JS, Kuruvilla FG, Tong JK, Shamji AF, Schreiber SL. 1999. Rapamycin-modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proc Natl Acad Sci U S A 96:14866–14870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joo YJ, Kim JH, Kang UB, Yu MH, Kim J. 2011. Gcn4p-mediated transcriptional repression of ribosomal protein genes under amino-acid starvation. EMBO J 30:859–872. doi: 10.1038/emboj.2010.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reid JL, Iyer VR, Brown PO, Struhl K. 2000. Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol Cell 6:1297–1307. doi: 10.1016/S1097-2765(00)00128-3. [DOI] [PubMed] [Google Scholar]
- 43.Kwak J, Workman JL, Lee D. 2011. The proteasome and its regulatory roles in gene expression. Biochim Biophys Acta 1809:88–96. doi: 10.1016/j.bbagrm.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Ferdoush J, Sen R, Kaja A, Barman P, Bhaumik SR. 2018. Two distinct regulatory mechanisms of transcriptional initiation in response to nutrient signaling. Genetics 208:191–205. doi: 10.1534/genetics.117.300518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powers T, Walter P. 1999. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol Biol Cell 10:987–1000. doi: 10.1091/mbc.10.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cardenas ME, Cutler NS, Lorenz MC, Di Como CJ, Heitman J. 1999. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev 13:3271–3279. doi: 10.1101/gad.13.24.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Preiss T, Baron-Benhamou J, Ansorge W, Hentze MW. 2003. Homodirectional changes in transcriptome composition and mRNA translation induced by rapamycin and heat shock. Nat Struct Biol 10:1039–1047. doi: 10.1038/nsb1015. [DOI] [PubMed] [Google Scholar]
- 48.Xiao L, Grove A. 2009. Coordination of ribosomal protein and ribosomal RNA gene expression in response to TOR signaling. Curr Genomics 10:198–205. doi: 10.2174/138920209788185261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.González A, Ruiz A, Casamayor A, Ariño J. 2009. Normal function of the yeast TOR pathway requires the type 2C protein phosphatase Ptc1. Mol Cell Biol 29:2876–2888. doi: 10.1128/MCB.01740-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cohen R, Engelberg D. 2007. Commonly used Saccharomyces cerevisiae strains (e.g. BY4741, W303) are growth sensitive on synthetic complete medium due to poor leucine uptake. FEMS Microbiol Lett 273:239–243. [DOI] [PubMed] [Google Scholar]
- 51.Laplante M, Sabatini DM. 2013. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 126:1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Russell SJ, Johnston SA. 2001. Evidence that proteolysis of Gal4 cannot explain the transcriptional effects of proteasome ATPase mutations. J Biol Chem 276:9825–9831. doi: 10.1074/jbc.M010889200. [DOI] [PubMed] [Google Scholar]
- 53.Ginsburg DS, Govind CK, Hinnebusch AG. 2009. NuA4 lysine acetyltransferase Esa1 is targeted to coding regions and stimulates transcription elongation with Gcn5. Mol Cell Biol 29:6473–6487. doi: 10.1128/MCB.01033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knutson BA, Hahn S. 2011. Domains of Tra1 important for activator recruitment and transcription coactivator functions of SAGA and NuA4 complexes. Mol Cell Biol 31:818–831. doi: 10.1128/MCB.00687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kikuchi J, Iwafune Y, Akiyama T, Okayama A, Nakamura H, Arakawa N, Kimura Y, Hirano H. 2010. Co- and post-translational modifications of the 26S proteasome in yeast. Proteomics 10:2769–2779. doi: 10.1002/pmic.200900283. [DOI] [PubMed] [Google Scholar]
- 56.Marquez-Lona EM, Torres-Machorro AL, Gonzales FR, Pillus L, Patrick GN. 2017. Phosphorylation of the 19S regulatory particle ATPase subunit, Rpt6, modifies susceptibility to proteotoxic stress and protein aggregation. PLoS One 12:e0179893. doi: 10.1371/journal.pone.0179893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Im E, Chung KC. 2016. Precise assembly and regulation of 26S proteasome and correlation between proteasome dysfunction and neurodegenerative diseases. BMB Rep 49:459–473. doi: 10.5483/BMBRep.2016.49.9.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iesmantavicius V, Weinert BT, Choudhary C. 2014. Convergence of ubiquitylation and phosphorylation signaling in rapamycin-treated yeast cells. Mol Cell Proteomics 13:1979–1992. doi: 10.1074/mcp.O113.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wade JT, Hall DB, Struhl K. 2004. The transcription factor Ifh1 is a key regulator of yeast ribosomal protein genes. Nature 432:1054–1058. doi: 10.1038/nature03175. [DOI] [PubMed] [Google Scholar]
- 60.Huisinga KL, Pugh BF. 2004. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol Cell 13:573–585. doi: 10.1016/S1097-2765(04)00087-5. [DOI] [PubMed] [Google Scholar]
- 61.Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pingle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961. doi:. [DOI] [PubMed] [Google Scholar]
- 62.Clarke AS, Lowell JE, Jacobson SJ, Pillus L. 1999. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol Cell Biol 19:2515–2526. doi: 10.1128/MCB.19.4.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Durairaj G, Sen R, Uprety B, Shukla A, Bhaumik SR. 2014. Sus1p facilitates pre-initiation complex formation at the SAGA-regulated genes independently of histone H2B de-ubiquitylation. J Mol Biol 426:2928–2941. doi: 10.1016/j.jmb.2014.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malik S, Bagla S, Chaurasia P, Duan Z, Bhaumik SR. 2008. Elongating RNA polymerase II is disassembled through specific degradation of its largest but not other subunits in response to DNA damage in vivo. J Biol Chem 283:6897–6905. doi: 10.1074/jbc.M707649200. [DOI] [PubMed] [Google Scholar]
- 65.Bhaumik SR, Green MR. 2002. Differential requirement of SAGA components for recruitment of TATA-box-binding protein to promoters in vivo. Mol Cell Biol 22:7365–7371. doi: 10.1128/MCB.22.21.7365-7371.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bhaumik SR, Green MR. 2003. Interaction of Gal4p with components of transcription machinery in vivo. Methods Enzymol 370:445–454. doi: 10.1016/S0076-6879(03)70038-X. [DOI] [PubMed] [Google Scholar]
- 67.Shukla A, Stanojevic N, Duan Z, Sen P, Bhaumik SR. 2006. Ubp8p, a histone deubiquitinase whose association with SAGA is mediated by Sgf11p, differentially regulates lysine 4 methylation of histone H3 in vivo. Mol Cell Biol 26:3339–3352. doi: 10.1128/MCB.26.9.3339-3352.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bhaumik SR, Raha T, Aiello DP, Green MR. 2004. In vivo target of a transcriptional activator revealed by fluorescence resonance energy transfer. Genes Dev 18:333–343. doi: 10.1101/gad.1148404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Malik S, Chaurasia P, Lahudkar S, Durairaj G, Shukla A, Bhaumik SR. 2010. Rad26p, a transcription-coupled repair factor, is recruited to the site of DNA lesion in an elongating RNA polymerase II-dependent manner in vivo. Nucleic Acids Res 38:1461–1477. doi: 10.1093/nar/gkp1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sen R, Lahudkar S, Durairaj G, Bhaumik SR. 2013. Functional analysis of Bre1p, an E3 ligase for histone H2B ubiquitylation, in regulation of RNA polymerase II association with active genes and transcription in vivo. J Biol Chem 288:9619–9633. doi: 10.1074/jbc.M113.450403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Durairaj G, Chaurasia P, Lahudkar S, Malik S, Shukla A, Bhaumik SR. 2010. Regulation of chromatin assembly/disassembly by Rtt109p, a histone H3 Lys56-specific acetyltransferase, in vivo. J Biol Chem 285:30472–30479. doi: 10.1074/jbc.M110.113225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malik S, Bhaumik SR. 2012. Rad26p, a transcription-coupled repair factor, promotes the eviction and prevents the reassociation of histone H2A-H2B dimer during transcriptional elongation in vivo. Biochemistry 51:5873–5875. doi: 10.1021/bi3005768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malik S, Chaurasia P, Lahudkar S, Uprety B, Bhaumik SR. 2012. Rad26p regulates the occupancy of histone H2A-H2B dimer at the active genes in vivo. Nucleic Acids Res 40:3348–3363. doi: 10.1093/nar/gkr1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Durairaj G, Lahudkar S, Bhaumik SR. 2014. A new regulatory pathway of mRNA export by an F-box protein, Mdm30. RNA 20:133–142. doi: 10.1261/rna.042325.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peterson CL, Kruger W, Herskowitz I. 1991. A functional interaction between the C-terminal domain of RNA polymerase II and the negative regulator SIN1. Cell 64:1135–1143. doi: 10.1016/0092-8674(91)90268-4. [DOI] [PubMed] [Google Scholar]
- 76.Lahudkar S, Durairaj G, Uprety B, Bhaumik SR. 2014. A novel role for Cet1p mRNA 5-triphosphatase in promoter proximal accumulation of RNA polymerase II in Saccharomyces cerevisiase. Genetics 196:161–176. doi: 10.1534/genetics.113.158535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Struhl K. Current protocols in molecular biology. Wiley, New York, NY. [Google Scholar]
- 78.Malik S, Durairaj G, Bhaumik SR. 2013. Mechanisms of antisense transcription initiation from the 3′ end of theGAL10 coding sequence in vivo. Mol Cell Biol 33:3549–3567. doi: 10.1128/MCB.01715-12. [DOI] [PMC free article] [PubMed] [Google Scholar]