

Abstract
Medullary thyroid cancer is a rare neuroendocrine tumor that arises the neural crest-derived parafollicular C cells and accounts for approximately 5% to 10% of thyroid cancers worldwide. These tumor can occur sporadically or as part of hereditary tumor syndromes, such as multiple endocrine neoplasia 2 and familial medullary thyroid cancer. The most common clinical presentation is a solitary thyroid nodule. The genetic defect in these disorders involves the RET proto-oncogene which is important for diagnosis of medullary thyroid cancer (including screening for hereditary medullary thyroid cancer) and for treatment guidance. This review summarizes the molecular basis and clinicopathologic features of medullary thyroid carcinoma.
Keywords: pathology competencies, organ system pathology, endocrine neoplasms, medullary thyroid carcinoma, cytologic diagnosis, molecular basis, clinical features, pathologic features
Primary Objective
Objective EN5.2: Medullary Thyroid Carcinoma: Describe the molecular basis and clinicopathologic features of medullary thyroid carcinoma.
Competency 2: Organ System Pathology; Topic Endocrine (EN); Learning Goal 5: Endocrine Neoplasms.
Patient Presentation
A 45-year-old woman presented to her endocrinologist with a single, gradually increasing nodule in her right neck. There was no relevant past medical or family history. She also mentioned having diarrhea intermittently. During her initial visit, the patient was in good condition, her blood pressure was 110/70 mm Hg, and her pulse was 75/minute and regular. Physical examination revealed a 2.0-cm firm mass with smooth borders on the right side of the thyroid that moved with swallowing. The rest of the examination was unremarkable. Ultrasonography of the thyroid revealed a 2.2-cm solid right thyroid nodule. The results of thyroid function tests were normal Thyroid-stimulating hormone (TSH) = 0.6 µIU/mL [range: 0.5-4.70 µIU/mL], T4 = 5.5 µg/dL [4.5-12.5 µg/dL], T3 = 115 ng/dL [80-200 ng/dL], and free T4 = 1.0 ng/dL [0.8-1.8 ng/dL]). However, the preoperative serum calcitonin value of 150 pg/mL (normal values: <8.8 pg/mL for men, <5.8 pg/mL for women; by immunochemiluminometric assay) and her serum calcium of 25 mg/dL (range: 8.5-10.5 mg/dL) were elevated. Fine-needle aspiration (FNA) biopsy of the nodule was performed.
Diagnostic Cytologic Findings
Microscopic examination of the FNA smears stained by Papanicolaou method showed loosely cohesive clusters and single plasmacytoid cells of variable sizes. The cells had eccentric nuclei with granular chromatin (“salt and pepper”) and abundant cytoplasm (Figure 1A and B). Occasional cells with nuclear enlargement were also seen (Figure 1). Immunohistochemical stains performed on the cellblock showed that the neoplastic cells were positive for calcitonin (Figure 2) and negative for thyroglobulin.
Figure 1.
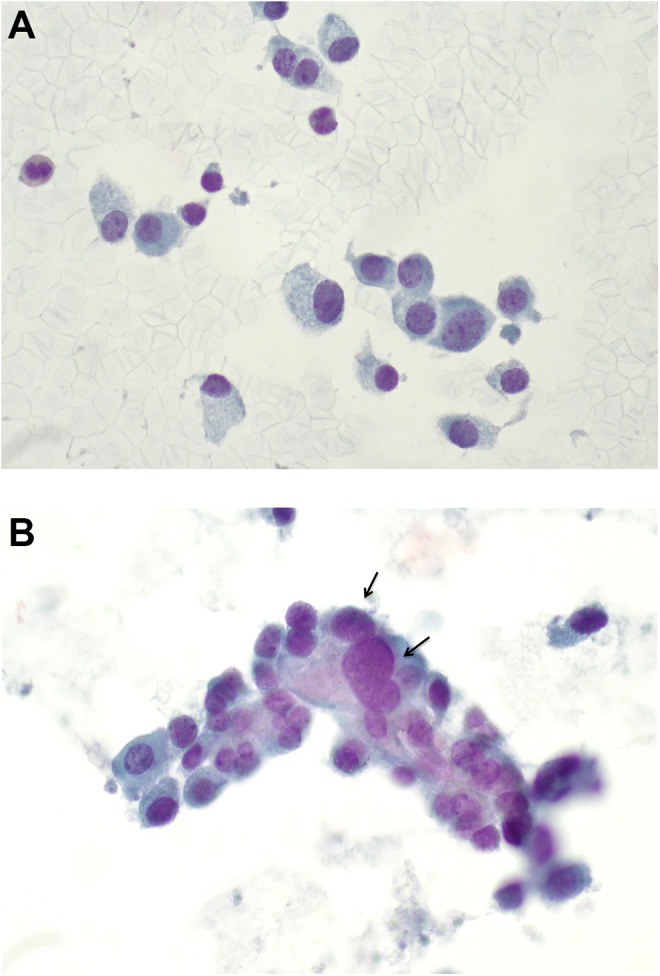
A, Smears show singly dispersed plasmacytoid (eccentric nuclei) cells of variable sizes, abundant amphophilic cytoplasm, granular chromatin, and inconspicuous nucleoli (PAP-stained, high power ×60 magnification). B, Smear shows a loosely cohesive cluster of neoplastic cells with occasional nuclear enlargement (arrows; PAP-stained, high power ×60 magnification).
Figure 2.
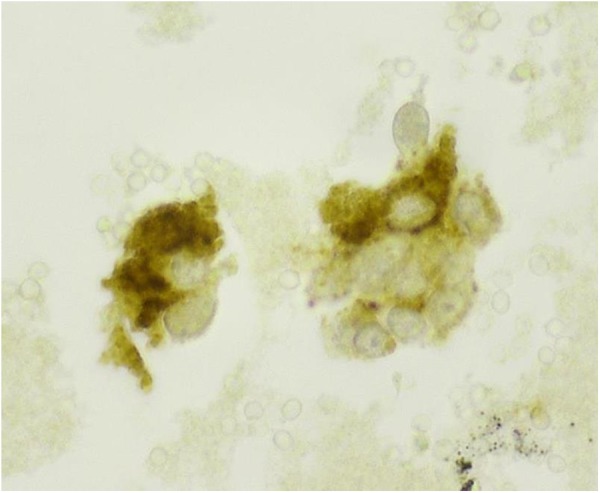
Immunohistochemical stain, performed on the cellblock, shows that the neoplastic cells are positive for calcitonin (cytoplasmic and granular staining; high power ×60 magnification).
Questions/Discussion Points
What Is Your Differential Diagnosis Based on the Clinical History and Cytologic Findings?
The differential diagnosis includes metastatic neuroendocrine carcinoma, poorly differentiated thyroid carcinoma (insular carcinoma), lymphoma, and medullary thyroid carcinoma. Based on the cytological features and immunohistochemical profile, a diagnosis of medullary thyroid carcinoma was rendered.
The patient was referred to a thyroid surgeon, and total thyroidectomy with cervical lymph node dissection was carried out. The tumor was 2.3 cm in greatest dimension with focal capsular invasion without any lymph node involvement. Pathologic findings in the thyroid gland were consistent with medullary thyroid carcinoma (Figure 3).
Figure 3.
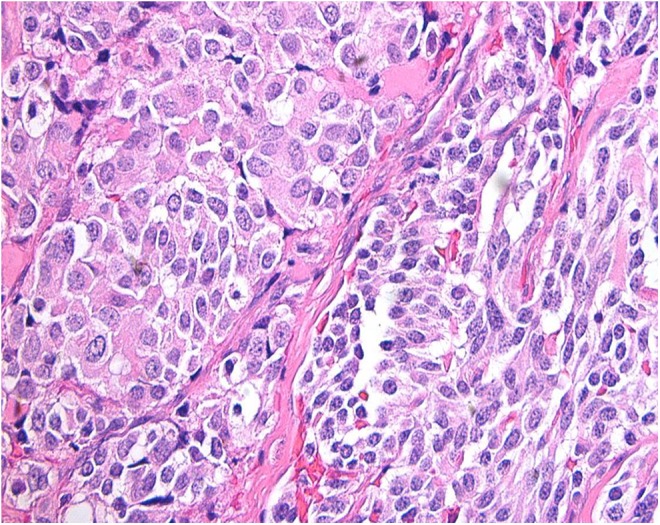
Section of the thyroid nodule showing nests of round and spindle cells outlined by fibrous tissue. The cells display granular cytoplasm and round nuclei with “salt and pepper” chromatin. (Hematoxylin and eosin stained slide, high power ×40 magnification).
What is Medullary Thyroid Carcinoma?
Medullary thyroid cancer (MTC) is a rare neuroendocrine tumor that arises from C cells (formerly called parafollicular cells) which are derived from the neural crest. Medullary thyroid cancer accounts for approximately 5% to 10% of thyroid cancers worldwide, and approximately 1% to 2% of thyroid cancer in United States.1 The C cells are located throughout the thyroid gland, but they are predominant at the junction of the upper third and lower two-thirds of each lobe, which is where the majority of MTCs are found. C cells secrete a variety of peptides and hormones, and MTC is characterized by the secretion of calcitonin, which is used as a diagnostic and prognostic marker in MTC.2
How Does Medullary Thyroid Cancer Manifest?
Most medullary thyroid carcinomas are sporadic. However, approximately 20% to 25% of cases are familial and are usually a component of multiple endocrine neoplasia (MEN) syndrome 2A or 2B or present as pure familial MTC (FMTC) syndrome.3
Sporadic Medullary Thyroid Cancer
Sporadic MTC accounts for approximately 75% to 80% of all cases of medullary thyroid cancer. There are typically unilateral with no associated endocrinopathies. The typical age of presentation is in the fourth and sixth decades of life, more commonly affecting women in a 3:2 ratio. The most common clinical presentation of sporadic MTC is that of a solitary thyroid nodule (35%-50%) or enlarged lymph node (up to 70%).
The tumors are generally unilateral and tend to arise in the posterior thyroid. In some patients, MTC presents with symptoms suggesting invasion of the surrounding structures (dysphagia, hoarseness, and/or respiratory difficulty) or can present with distant metastases at the time of diagnosis. The most common locations for metastatic MTC are the mediastinum, liver, lungs, and bone.1-3
Systemic symptoms may occur due to hormonal secretion by the tumor. Tumor secretion of calcitonin, calcitonin gene-related peptide, or other substances (calcitonin, prostaglandins, serotonin, or vasoactive intestinal polypeptide) can cause diarrhea or facial flushing in patients with advanced disease.1,3
Hereditary (Familial) Medullary Thyroid Cancer
Multiple Endocrine Neoplasia 2A (Sipple Syndrome)
Multiple endocrine neoplasia 2A syndrome or Sipple syndrome is the most common form of MEN 2 syndrome, accounting for 75% of hereditary MTC. It is associated with MTC, pheochromocytoma (∼50%), and hyperparathyroidism (∼20%). This syndrome is inherited in an autosomal dominant manner, affecting males and females equally. Peak incidence of syndromic MTC is in the 30s, ranging from adolescence to early adulthood.3,4
Multiple Endocrine Neoplasia 2B
Multiple endocrine neoplasia 2B accounts for 8% to 15% of all patients with MEN 2. It is associated with MTC and pheochromocytoma, and an unusual physical appearance, characterized by mucosal ganglioneuromas and marfanoid habitus (long limbs, hyperlaxity, and arachnodactyly). Inheritance is autosomal dominant as in MEN 2A, again equally affecting males and females.3 Patients with MEN 2B present MTC early in life, diagnosed in infancy or early childhood before their 30s.3 Almost 100% of patients with MEN 2B develop MTC with the disease having a more aggressive course.
Familial Medullary Carcinoma Without Associated Endocrinopathies
The FMTC category of MTC is the least aggressive and is defined as MTC without other hereditary endocrine tumors. Similar to other types of thyroid cancers, the peak incidence is between the ages of 40 and 50 years.3
What Are the Genetic Features of Medullary Thyroid Cancer?
The genetic defect in these disorders involves the RET proto-oncogene on chromosome 10q11.2. RET is a single-pass transmembrane receptor belonging to the tyrosine kinase superfamily. The germ line RET mutations lead to the activation of major intracellular pro-oncogenic pathways (eg, RAS/MAPK, JUN kinase, PI3K/AKT, and nuclear factor-κB), that is central for the development of sporadic and hereditary MTC. Currently known RET mutations account for about 95% of MEN 2A and 85% of FMTC families. Somatic RET point mutations have been identified in about 50% of patients with sporadic MTC.1-4
Mutations involving the RET proto-oncogene are also seen in other malignant and nonmalignant diseases. Chromosomal translocations activating RET proto-oncogene can occur in 20% to 30% of patients with papillary thyroid carcinoma, and can also be seen, but less frequently, in patients with lung adenocarcinoma and chronic myelomonocytic leukemia. Inactivating RET mutations occur in patients with hereditary and sporadic Hirschsprung disease (HD).5
Describe the Pathological Features of Medullary Thyroid Cancer
Grossly, sporadic or hereditary MTC present as a well-demarcated, solid, white-gray/yellow, firm, and gritty nodule of variable size. Sporadic MTC tumors typically present as a single and unilateral nodule. Hereditary MTC are usually bilateral, multicentric, single, or multiple nodules. Although the tumors in sporadic and hereditary MTC are sharply circumscribed, they are not encapsulated. Medullary thyroid cancer typically is the first neoplasm observed in both MEN 2A and MEN 2B syndromes. Both sporadic and FMTC arise at the junctions of the upper and middle thirds of the lateral lobes.3
The histopathology is quite variable and numerous histological subtypes are described. The characteristic features of MTC, in both sporadic and familial, are sheets, nests, or trabecular arrangement of cells which can vary in shape, round, polygonal, spindle, or giant cells, all with varying amount of granular cytoplasm. Tumor cells usually have uniform, round to oval nuclei with granular or punctate chromatin, also referred to as “salt and pepper” chromatin. Fibrous or amyloid stroma can be present. The hereditary MTC is usually preceded by “C-cell hyperplasia” (CCH) and usually occurs in the upper two-thirds of the thyroid adjacent to a MTC or may be seen in asymptomatic carriers of RET mutation. Multiple CCH foci are a hallmark of MEN 2 syndromes and FMTC syndrome. The presence of neoplastic CCH foci is considered a paradigm of a genetically determined condition.1-4
How Is Medullary Thyroid Cancer Diagnosed?
Fine needle aspiration cytology is a widely utilized tool for the diagnosis of thyroid lesions with a high degree of sensitivity, specificity, and diagnostic accuracy. It is a simple, rapid, and cost-effective test that can effectively distinguish between neoplastic and nonneoplastic lesions of the thyroid. Fine-needle aspiration can effectively triage patients with thyroid nodules as to who require surgery and who do not.6,7
In the year 2007, the National Cancer Institute (NCI), Bethesda, Maryland, organized the NCI Thyroid Fine Needle Aspiration State-of-the-Science Conference, and an initiative was undertaken to publish an atlas and guidelines using a standardized nomenclature for the interpretation of thyroid FNAs, known as the Bethesda system for reporting thyroid cytopathology. The atlas describes 6 diagnostic categories of lesions: Nondiagnostic/unsatisfactory, benign, atypical follicular lesion of undetermined significance, “suspicious” for follicular neoplasm, suspicious for malignancy, and malignant. The 6 diagnostic categories of the Bethesda system have individual implied risks of malignancy that influence management paradigms.7
The diagnosis of MTC is usually made after FNA biopsy. The patients typically present with high calcitonin serum levels that can also be helpful for diagnosis. Fine-needle aspiration smears of MTC nodules show variable cytologic features. The classic smear pattern is usually cellular, yielding tumor cells that are dispersed and are characterized by eccentric nuclei (plasmacytoid cell pattern), granular chromatin (neuroendocrine-type), and inconspicuous nucleoli in a relatively clean background. The cytoplasm of the tumor cells is faintly granular but may show conspicuous red granules. Deposition of amorphous, glassy, eosinophilic material consistent with amyloid, can be seen in the background, and sometimes is confused with colloid material.
Depending on the specific cytomorphology of the tumor, a number of differential diagnoses may arise and immunohistochemical staining may be helpful. The small cell pattern may be mistaken for a malignant lymphoma (CD45 positive), poorly differentiated thyroid carcinoma (thyroglobulin positive), or metastatic small-cell carcinoma (calcitonin negative) among others. When MTC is suspected on cytological examination, immunohistochemical staining for calcitonin is important for making a diagnosis of MTC.
The 2015 American Thyroid Association (ATA) guidelines for MTC management recommend that all cases of MTC, either sporadic or inherited, and patients with CCH, should be analyzed for germ line mutations in the RET proto-oncogene. RET molecular diagnosis is considered the gold standard for the recognition of patients at risk of MTC in MEN 2 families, and the recommended method of initial testing for patients with MEN 2 is either a single or a multi-tiered analysis to detect RET mutations in exon 10 (codons 609, 611, 618, and 620), exon 11 (codons 630 and 634), and exons 8, 13, 14, 15, and 16.5
The ATA also recommends that genetic counseling and genetic testing should be offered in patients with sporadic MTC found to have a RET mutation; in first-degree relatives of patients with proven hereditary MTC; in parents whose infants or young children have the classic phenotype of MEN 2B; in infants or young children with HD and RET germ line mutations, and in adults with MEN 2A who have symptoms suggestive of HD.5
Describe the Treatment Options for Medullary Thyroid Cancer
Standard treatment for MTC requires surgical removal of the thyroid with regional lymph node dissection. Unlike other thyroid malignancies, MTCs are resistant to radioactive iodine therapy because they do not concentrate radioactive iodine. The extent of the surgical resection and lymph node dissection is determined by the size of the primary tumor, the extent of nodal, and distant metastases. In the presence of advanced metastatic disease, a more palliative approach is recommended, and aggressive neck surgery is typically not performed to improve quality of life.
Prophylactic thyroidectomy is recommended in patients with germ line RET mutations, and the timing of surgery depends on the type of MEN syndrome and the RET mutation, but it is usually recommended before the onset of clinically significant disease.
For patients with metastatic MTC, treatment depends on the extent of metastases. Surgery may be an option for surgically resectable tumor lesion to relieve tumor burden. Conventional chemotherapy has limited efficacy in patients with MTC. A new class of therapies targeting the RET receptor tyrosine kinase family has been developed because of its role in the pathogenesis of MTC. Several clinical trials have been developed involving tyrosine kinase inhibitors (such as sorafenib, sunitinib, vandetanib, motesanib, and cabozantinib) with the rationale that they can block the molecular pathways involved in MTC. Some of these treatments (like vandetanib and cabozantinib) have been approved by the US Food and Drug Administration for the treatment of adults with symptomatic or progressive metastatic MTC.2,8
What Are the Features That Drive Prognosis in Patients With Medullary Thyroid Cancer?
The size of the primary tumor and the presence of nodal and distant metastasis are important factors predicting survival in patients with MTC. When the tumor is localized to the thyroid gland, the 10-year survival rate is approximately 75% to 95%. Patients with regional lymph node disease have a 5-year overall survival rate of 75.5%. Distant metastases at initial diagnosis is associated with a poor prognosis, with a 10-year survival rate of <50%.2,8 Younger age (<40 years old) at the time of diagnosis is associated with better prognosis.
Factors that may also predict a poor prognosis includes:
Calcitonin doubling times less than 6 to 12 months is associated with poor survival, while doubling times >24 months are associated with a very favorable prognosis.
Specific germ line mutations in RET predict the aggressiveness of the tumor (MEN 2B have worse a prognosis).
Recurrent disease develops in approximately 50% of patients with MTC. Calcitonin levels are very sensitive for detecting either residual or recurrent disease.
Teaching Points
Medullary thyroid cancers are rare tumors that arise from the neural crest–derived parafollicular C cells.
They occur sporadically or as part of hereditary tumor syndromes (MEN 2 and FMTC).
A solitary thyroid nodule is the most common presentation of sporadic MTC (in 75%-95% of patients). In most patients, the disease involves regional lymph nodes at the time of diagnosis.
Genetic testing for RET mutations is important for diagnosis of MTC (including screening for hereditary MTC) and for treatment guidance. Therefore, germ line RET testing is recommended in all patients with newly diagnosed CCH or apparently sporadic MTC.
Due to limited adjuvant treatment options, adequate initial surgical management is essential to the successful treatment of MTC.
New molecular targeted treatment options are available in cases of distant or recurrent disease not amenable to surgery.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Tuttle M, Ross DS, Mulder JE. Medullary thyroid cancer: clinical manifestations, diagnosis, and staging. Up to Date. September 27, 2017. Accessed November 11, 2017 https://www.uptodate.com/contents/medullary-thyroid-cancer-clinical-manifestations-diagnosis-and-staging
- 2. Roy M, Chen H, Sippel RS. Current understanding and management of medullary thyroid cancer. Oncologist. 2013;18:1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nose V. Familial thyroid cancer: a review. Modern Pathol. 2011;24:S19–S33. [DOI] [PubMed] [Google Scholar]
- 4. Ball DW. Medullary thyroid cancer: monitoring and therapy. Endocrinol Metab Clin North Am. 2007;36:823–837, viii. doi:10.1016/j.ecl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wells SA, Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25:567–610. doi:10.1089/thy.2014.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Renuka IV, Saila Bala G, Aparna C, Kumari R, Sumalatha K. The bethesda system for reporting thyroid cytopathology: interpretation and guidelines in surgical treatment. Indian J Otolaryngol Head Neck Surg. 2012;64:305–311. doi:10.1007/s12070-011-0289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mondal SK, Sinha S, Basak B, Roy DN, Sinha SK. The Bethesda system for reporting thyroid fine needle aspirates: a cytologic study with histologic follow-up. J Cytol. 2013;30:94–99. doi:10.4103/0970-9371.112650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dhakshinamoorthy G, Paulson E, Duran C, Cabanillas M, Busaidy N, Charnsangavej C. Current update on medullary thyroid carcinoma. Am J Roentgenol. 2013;201:W867–W876. [DOI] [PubMed] [Google Scholar]