


Abstract
Novel modified DNA duplexes with single bridging 5′-SS-monophosphoryldithio links [-OP(=O)-O–-SS-CH2-] were synthesized by autoligation of an oligonucleotide 3′-phosphorothioate and a 5′-mercapto-oligonucleotide previously converted to a 2-pyridyldisulfide adduct. Monophosphoryldisulfide link formation is not a stringent template-dependent process under the conditions used and does not require strong binding of the reactive oligomers to the complementary strand. The modified internucleotide linkage, resembling the natural phosphodiester bond in size and charge density, is stable in water, easily undergoes thiol–disulfide exchange and can be specifically cleaved by the action of reducing reagents. DNA molecules containing an internal -OP(=O)-O–-SS-CH2- bridge are stable to spontaneous exchange of disulfide-linked fragments (recombination) even in the single-stranded state and are promising reagents for autocrosslinking with cysteine-containing proteins. The chemical and supramolecular properties of oligonucleotides with 5′-sulfhydryl groups were further characterized. We have shown that under the conditions of chemical ligation the 5′-SH group of the oligonucleotide has a higher reactivity towards N-hydroxybenzotriazole-activated phosphate in an adjacent oligonucleotide than does the OH group. This autoligation, unlike disulfide bond formation, proceeds only in the presence of template oligonucleotide, necessary to provide the activated phosphate in close proximity to the SH-, OH- or phosphate function.
INTRODUCTION
Chemically reactive oligonucleotide duplexes that can spontaneously (auto)crosslink with nucleic acid-binding proteins are of interest as valuable tools in the study of a wide variety of biological processes and the molecular architecture of nucleic acid–protein complexes. These duplexes are also promising as inhibitors of biologically important proteins and enzymes because autocrosslinking might possibly be carried out inside intact cells. The introduction of a chemically active group at a selected position in the sugar–phosphate backbone of an oligonucleotide duplex was developed by Shabarova and co-workers (1,2). This original chemical approach involves automated synthesis of two oligonucleotides with modified 3′- and 5′-termini, followed by their chemical ligation to a complementary template. The terminal modifications determine the type of internucleotide linkage formed, while the sequences of the oligonucleotides and locations of reactive groups juxtapose the target amino acid in the protein recognition site. Using this approach, DNA and RNA duplexes carrying single tri-substituted pyrophosphate internucleotide linkages were prepared. They were shown to be reactive towards nucleophilic amino acids (Lys and His) and to permit nucleic acid–protein autocrosslinking (1–6). The characteristic properties of these substrate analogs are: (i) that the modified moiety does not significantly influence the thermal stability of the double helix and the binding affinity between nucleic acid and protein; (ii) that crosslinking of the reactive double-stranded oligonucleotides to the targeted enzyme active site or protein binding domain occurs under physiological conditions without any additional activation. Sets of DNA or RNA duplexes with chemically reactive internucleotide linkages were successfully used for affinity modification and to probe the nucleic acid-binding regions of restriction-modification enzymes (2,7), RNA-recognizing TAT peptide (4) and transcription factors NF-κB (3,5) and HNF1 (6).
Recently we reported hairpin DNA duplexes with diphosphoryldisulfide [-OP(=O)-O–-SS-O–-(O=)PO-] linkages in the backbone as promising reagents for autocrosslinking with protein cysteine residues (8). Here we present the synthesis and properties of DNA duplexes containing a single bridging 5′-SS-monophosphoryldithio link [-OP(=O)-O–-SS-CH2-] instead of a natural phosphodiester internucleotidic linkage. The 5′-SS-monophosphorodithio moiety mimics the phosphodiester linkage in size and charge density better than a diphosphoryldisulfide bridge. It also appears to be more effective in crosslinking with cysteine residues by thiol–disulfide interchange within a dsDNA–protein complex. Thiol–disulfide exchange, in which a thiolate acts as a nucleophile displacing a sulfur atom in an existing disulfide bond and forming a new bond with the other sulfur atom, was shown to be a prevalent reaction of ionized Cys residues (9). Successful intermolecular thiol–disulfide exchange was demonstrated earlier by Chu and Orgel (10). They developed a general method for combining a 2-pyridyldisulfide adduct, attached via a thio-ethylamino linker to the 5′-terminal phosphate group of any oligonucleotide, with a cysteine-containing protein. The reaction was non-specific and diffusion controlled. Directed disulfide bond formation between the active site cysteine and a thiol-containing analog of guanine within a methylguanine methyltransferase-modified oligonucleotide complex has also been described (11).
In the present study the 5′-SS-monophosphorodithio moiety was easily incorporated into the sugar–phosphate backbone of both hairpin and intermolecular DNA duplexes (Fig. 1) by reaction of a phosphorothioate at the 3′-end of one oligonucleotide with a thiopyridine derivative of 5′-mercapto-5′-deoxythymidine or cytidine in the other. The sulfur-containing oligonucleotide analogs ligated without added reagents (Fig. 2, reaction I). The participation of a 5′-mercapto group in chemical ligation of oligonucleotides resulting in 5′-S-phosphorothioester linkage formation was also studied (Scheme 1) and the results are discussed in terms of the theory of ‘hard’ and ‘soft’ ligands. The role of a complementary template in the ligation and recombination of thiol (disulfide)-containing oligonucleotides was also analyzed.
Figure 1.

Sequences of autoligation precursor DNA duplexes. The 3′- and 5′-terminal groups facing the nick are indicated. The numbers of oligonucleotides are given in parentheses. The recognition site for the transcription factor NF-κB is underlined.
Figure 2.

The mechanism of pyridyldisulfide-mediated autoligation of oligonucleotides resulting in monophosphoryldithio (reaction I) or 5′,5′-disulfide (reaction II) links. Abbreviations of modified oligonucleotides are indicated under the reaction products; i and j correspond to the number of oligonucleotides (see Fig. 1) linked by a disulfide bridge; T (C), thymine (cytosine) residues; B, any base.
MATERIALS AND METHODS
Oligonucleotide synthesis
Oligodeoxyribonucleotides were synthesized in a DNA synthesizer (Applied Biosystems 380B) using standard phosphoramidite chemistry. The 3′-terminal phosphorothioate groups required for the ligation reaction were introduced into the DNA strands by oxidation of the corresponding phosphite with a 1% solution of Beaucage thiolating reagent in acetonitrile as described (12). Oligodeoxyribonucleotides with terminal 5′-mercapto-5′-deoxythymidine were synthesized according to Burgin et al. (13) using 5′-tritylmercapto-5′-deoxythymidine-3′-O-(2-cyanoethyl-N,N-diisopropylamino)phosphite in the final coupling. In the case of the modified cytidine derivative, 5′-tritylmercapto-N4-benzoyl-2′,5′-dideoxycytidine-3′-O-(2-cyanoethyl-N,N-diisopropylamino)phosphite was prepared for the last coupling as described previously (14). 5′-Iodinated oligonucleotide was obtained as described (15). Removal of the trityl group by silver nitrate was done according to Sinha and Striepeke (16) with some modifications. Between 3 and 5 A260 units of 5′-tritylmercapto-oligonucleotide in 100 µl of water were mixed with 3 µl of 0.3 M silver nitrate. After 30 min, 20 µl of 0.1 M dithiothreitol (DTT) was added, then the mixture was vortexed and after 5 min was centrifuged for 10 min. The supernatant containing the thiolated oligonucleotide and excess DTT (excess DTT can be removed by ethyl acetate extraction) was placed in a separate tube, the precipitate was washed twice with 100 µl of water and the supernatants were combined. The oligonucleotide was isolated by standard ethanol precipitation. Gel-purified oligomers were kept in boiled water to avoid their oxidation to disulfide-linked dimers by dissolved oxygen. The small amounts of dimer generated by atmospheric oxygen were reduced to the monomer form with 0.05 M DTT (1 h, room temperature). After this procedure, sulfur-containing oligonucleotides were isolated by ethanol precipitation.
Non-enzymatic ligation reactions
The 5′-sulfhydryl oligonucleotide was converted in 95% yield to the 2-pyridyldisulfide adduct by treatment with 0.03 M 2,2′-dipyridyldisulfide in 2.5 mM Tris–HCl, pH 7.5, 50% dimethylformamide (DMF) for 3 h at room temperature and was isolated from the reaction mixture by ethanol precipitation. The other duplex-forming component(s) (Fig. 1) was then added to the 2-pyridyldisulfide adduct in 0.05 M MES, adjusted with NaOH to pH 7.0, 0.02 M MgCl2 (buffer 1); the DNA concentration was ∼1 mM per nucleotide residue. The equimolar mixture of components, which provides the 3′-phosphorothioate and 2-pyridyldisulfide moieties in close proximity (Fig. 2, reaction I), was heated to 60°C, slowly annealed and incubated overnight at 0°C.
5′-SS-5′-linked homodimers were obtained by mixing an oligomer containing a free 5′-sulfhydryl group with its 2-pyridyldisulfide adduct (Fig. 2, reaction II) in buffer 1; the reaction mixture was incubated overnight at 0°C.
N-hydroxybenzotriazole (HOBT) derivatives were obtained quantitatively by treatment of 3′-phosphorylated oligonucleotides with 2 M HOBT in 70% DMF as in Ivanovskaya et al. (17). The reactive DNA oligomer was hybridized on the complementary template with oligonucleotides carrying 5′-hydroxyl, 5′-sulfhydryl or 5′-phosphate groups (Scheme 1). The reaction mixtures in 0.4 M N-methylimidazole, 0.2 M NaCl, 0.12 M MgCl2, pH 8.0, (buffer 2) were heated to 60°C and slowly annealed. Oligonucleotide autoligation was performed at 4°C for 72 h.
The reaction of 5′-sulfhydryl oligomers with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was performed in 0.05 M MES, pH 6.0, 0.02 M MgCl2 at 4°C overnight. The concentration of oligonucleotides was 1 mM (per monomer); that of EDC was 0.3 M.
Reaction products were isolated by ethanol precipitation and taken up in 5 µl of loading buffer. Samples were heated to 95°C for 2 min prior to loading on a 20% polyacrylamide gel containing 7 M urea. Gels were visualized by UV shadowing. Aliquots of the reaction mixtures were analyzed quantitatively by ion exchange HPLC as described (8).
Characterization of the ligation products
After extraction from the gel slices with 10 mM Tris–HCl, pH 7.6, 0.3 M NaCl, 1 mM EDTA, ligation products with 5′-SS-monophosphoryldithio or 5′-SS-5′ disulfide internucleotide linkages were converted back to the initial oligonucleotides by 0.05 M DTT treatment (1 h, room temperature) and quantitatively analyzed by ion pair HPLC (8,18).
The P-S bond in 5′-S-bridging phosphothioester-linked DNA oligonucleotides was cleaved by treatment with 0.02 M silver nitrate at room temperature for 1.5 h in the dark. Then 0.1 M DTT was added and after 30 min the oligonucleotide material in the supernatant was precipitated with ethanol. The cleavage products were then analyzed by denaturing PAGE (20% polyacrylamide, 7 M urea, 1× TBE).
MALDY-TOF mass detection in a Vision 2000 spectrometer was used to characterize some of the oligonucleotide derivatives.
Thermal melting studies
Solutions for thermal melting studies contained a 1:1 ratio of the complementary strands (0.1 mM each per mononucleotide) in buffer 1. The samples were heated to 90°C and allowed to cool slowly to room temperature before melting. Melting studies were carried out in 1 cm path length quartz cells under a nitrogen atmosphere in a Hitachi 150-20 spectrophotometer equipped with a thermoprogrammer. Absorbance was monitored at 260 nm while the temperature was raised from 10 to 90°C at a rate of 0.5°C/min.
RESULTS AND DISCUSSION
Synthetic strategies to replace the bridging oxygen atom at a predetermined phosphorodiester linkage in an oligonucleotide with sulfur have been previously described (19,20). Bridging positions in the phosphate linkage are very important because they can make specific interactions with proteins and metals and act as leaving groups in various DNA cleavage reactions. More recently, it was shown that 5′-S-phosphorothioester linkages can be selectively introduced in DNA as a result of post-synthetic template-controlled oligonucleotide coupling when a 3′-phosphorothioate displaces a 5′-end tosylate or iodine moiety on a thymidine residue (21–23). Here we describe a novel sulfur-containing modification of the oligodeoxyribonucleotide sugar–phosphate backbone, a bridging 5′-SS-monophosphorodithio link, which may be useful as a reactive group in crosslinking reactions with protein cysteines. To produce this linkage, a pair of oligonucleotides is required in which one is terminated by a 3′-phosphorothioate and the other by a 5′-mercapto group. Oligodeoxyribonucleotides with a 5′-SH group located on the terminal thymidine or cytidine residue were synthesized according to Burgin et al. (13) and Sproat et al. (14), respectively, using standard phosphoramidite chemistry. Some modifications were introduced in the deprotection procedure (see Materials and Methods). 3′-Phosphorothioate groups, incorporated as described (12), are also required for ligation. Base-paired hairpin DNAs with nicks (I and II in Fig. 1) were used as promising constructs for template-directed chemical coupling of oligonucleotides, due to their increased thermal stability. A trinucleotide loop (AAA or GAA) helps in the formation of an extraordinarily stable DNA hairpin because of good stacking of the loop adenosine base on the closing A-A or G-A sheared pair (24). Indeed, duplex I demonstrated a melting profile with two cooperative transitions at 32 and 72°C, corresponding to separation of short oligomer 2 and denaturation of the self-folded oligomer 1, respectively (data not shown), which confirms the close proximity of the interacting groups at low temperatures. In addition, according to our recent findings, the specific secondary structure of hairpin DNA duplexes prevents recombination of disulfide-linked oligomers (8). Regular duplex III, consisting of three oligonucleotides (Fig. 1), was used to elucidate the role of the complementary template in ligation via -O-P(=O)-O–-SS-CH2- bridge formation and consequent spontaneous recombination of the reaction products. Duplex II contains the recognition site for the human DNA transcription factor NF-κB (underlined in Fig. 1), in which Cys59 was shown by X-ray analysis (25) to interact with the internucleotide phosphate group of the DNA substrate; duplexes I and III were used as model systems.
In order to increase the selectivity of oligonucleotide coupling as against homodimer formation, phosphorothioate–disulfide exchange was used instead of direct oxidation of terminal sulfur-containing functions in duplexes I–III (Fig. 2, reaction I). Earlier we showed that a rather stable thiopyridine derivative of a phosphorothioate oligonucleotide (a disulfide) readily exchanges with the phosphorothioate group of another oligonucleotide molecule to give an autoligation product under mild conditions (8). Because oligomers carrying a 5′-mercapto function demonstrate a greater tendency to form disulfide-linked homodimers (Fig. 3, lane 5) than 3′-phosphorothioate oligonucleotides, oligonucleotides 2 and 4 were converted to 2-pyridyldisulfide adducts to prevent undesirable homodimerization. These adducts were easily prepared by treatment of oligonucleotides containing sulfhydryl groups with 2,2′-dipyridyldisulfide. They are stable under our chromatographic and electrophoretic conditions and can be preserved without decomposition in aqueous solutions at –10°C for at least several days. Efficient autoligation was shown to take place when oligomers hybridized to the complementary strand such that a phosphorothioate group and a 2-pyridyldisulfide moiety are brought into proximity (Fig. 2, reaction I). The reaction proceeded overnight at 0°C to give 80–90% yield of the ligation products in duplexes I–III: 1pSS2, 3pSS4 or 5pSS2, respectively (for abbreviations see Figs 1 and 2). As can be seen from the denaturing PAGE analysis, the coupling efficiency does not depend on the nature of the nucleotide residue (T or C) carrying the 5′-sulfhydryl group or its derivative (compare Fig. 4A and B, lanes 2). In the case of the three-component duplex III, the autoligation reaction was shown to occur very effectively even in the absence of complementary strand (6) and the yield of heterodimer 5pSS2 with a 5′-SS-monophosphoryldithio linkage approaches 90% (Fig. 3, lanes 2 and 3). In parallel, directed oligonucleotide homodimerization was carried out by incubation of a 1:1 mixture of oligomer 2 with a free sulfhydryl function and its 2-pyridyldisulfide adduct (Fig. 2, reaction II). The conversion of 2 to a homodimer with a 5′,5′-disulfide bridge was ∼95% (Fig. 4A, lane 3). As mentioned, the same product is formed spontaneously after storage of oligomer 2 in aqueous solution due to the strong tendency for oligonucleotide sulfhydryl groups to be oxidized by dissolved oxygen (Fig. 3, lane 5). The effectiveness of DTT in reducing the disulfide bond has been used as a criterion to identify 3′-pSS-5′ and 5′-SS-5′ linkages. The data from ion pair HPLC analysis confirmed that the product oligonucleotides described here were labile to DTT (Fig. 5A); the homogeneous peaks corresponding to hairpin heterodimer 1pSS2 (profile 1) and homodimer 25′-SS-5′2 (profile 2) were completely transformed to the starting oligonucleotides (profiles 3 and 4, respectively) after DTT treatment. Mass analysis of oligonucleotide 5pSS2, presented in Figure 5B, directly verified its identity and confirmed the nature of the newly formed linkage: the observed mass peak (6538.68) is very close to the theoretically expected mass (6534.37). A low level mass peak (3681.68) attributed to 5′-mercapto-oligodeoxyribonucleotide 2 was also identified (theoretical mass 3677.41).
Figure 3.
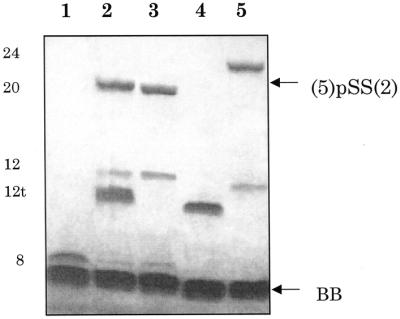
Denaturing PAGE analysis of autoligation of oligonucleotides 5 and 2 via a 5′-SS-monophosphoryldithio linkage (reaction I in Fig. 2) in the presence (lane 2) and absence (lane 3) of the complementary strand. For sequences see Figure 1, duplex III. Lane 4, template oligomer 6; lane 1, starting oligonucleotide 5; lane 5, starting oligomer 2 with a free 5′-sulfhydryl group and the product of its oxygen-induced dimerization. Bands were visualized by UV shadowing. The position of the ligation product and the chain lengths of oligonucleotides are indicated. For reaction conditions see Materials and Methods.
Figure 4.

Denaturing PAGE analysis of oligonucleotide autoligation via a 5′-SS-monophosphoryldithio linkage (reaction I in Fig. 2) into hairpin DNA duplexes I (A) and II (B). For sequences see Figure 1. Lanes 1, initial mixture of duplex-forming oligonucleotides; lanes 2, post-ligation mixture; lane 3, non-template-controlled homodimerization of oligonucleotide 2 via 5′,5′-disulfide bond formation (reaction II in Fig. 2). The positions of ligated products and the chain length of oligonucleotides are indicated. Bands were visualized by UV shadowing.
Figure 5.

Ion pair HPLC of DNA oligonucleotides containing single disulfide bridges in the sugar–phosphate backbone. Abbreviations of modified oligonucleotides are given above the profiles (for sequences and abbreviations see Figs 1 and 2). HPLC analysis was performed on a Waters chromatograph in an acetonitrile gradient (5–40%) in 48 mM potassium phosphate buffer, pH 7.0, containing 2 mM tetrabutyl ammonium dihydrophosphate as previously published (18). Elution rate 1 ml/min, 45°C.
The single-stranded ligation product 5pSS2 from system III with a monophosphorodithio (-P-SS-CH2-) internucleotide link appears not to undergo spontaneous disulfide exchange in water after removal of complementary template (6); it shows an individual peak in the HPLC profile (Fig. 5B) which corresponds to the heterodimer. In contrast, oligonucleotides 5 and 2, joined by a diphosphorydisulfide (-P-SS-P-) linkage, appear as a triplet on chromatograms (8). The ease of expulsion of a leaving group depends on its pKa value and on its state of protonation. In the case of a -P-SS-P- internucleotide bridge, the free phosphorothioate can attack any sulfur atom with equal probability, resulting in a mixture of homo- and heterodimers in a ratio close to the statistical expectation of 50% for a random distribution reaction (8). The apparent absence of disulfide bond exchange for the oligomer with a -P-SS-CH2- crosslink can be rationalized if one takes into account the different pKa values of oligonucleotide phosphorothioate (∼4.5) (26) and 5′sulfhydryl groups, which is expected to be close to the pKa value of Cys residues in proteins (8–9) (9). Obviously, oligonucleotide sulfhydryl groups are predominantly non-ionized and non-active at pH 5.5–6.0 (in pure water), whereas the phosphorothioate anion can act as a nucleophile, displacing the same phosphorothioate moiety due to its enhanced leaving group ability versus a 5′-sulfhydryl in an existing -P-SS-CH2- linkage.
The 5′-SS-monophosphoryldithio linkage is stable for at least 3 days in aqueous buffer solutions at pH values of 5–8 at 37°C and causes insignificant destabilization of duplex DNA. The latter was tested in the context of duplex III carrying one disulfide linkage in the ‘upper’ strand and the unmodified complement (6). Thermal denaturation studies showed that Tm values were 59°C for the sulfur-containing duplex and 63°C for the unsubstituted duplex in buffer 1.
Additional efforts were made to study the ability of a 5′-sulfhydryl group to interact with an activated phosphate of an adjacent oligonucleotide. It is known that a ‘soft’ sulfur is usually a poor nucleophile at a ‘hard’ phosphorus (27). In contrast, a ‘soft’ tetrahedral carbon exhibits much more thiophilicity than phosphorus derivatives (28). Thus, for the displacement reaction on (R′O)2P(=O)X, ROH was found to be a better nucleophile than RSH by a factor of ∼30 (27). On the basis of the ‘softness–hardness’ principle, the position of attack was predicted for an ambident nucleophile, an oligonucleotide terminal phosphorothioate, on water-soluble carbodiimide (29,30). It was shown recently that an oligonucleotide 3′-phosphorothioate group can effectively displace a 5′-end tosylate or iodine, attacking the carbon atom (21–23). To test whether a 5′-S-phosphorothioester linkage can be formed between an oligonucleotide 5′-mercapto group and a 3′-phosphate activated by water-soluble carbodiimide, we treated nicked duplex I, where X = H and Y = OH, with EDC. However, instead of EDC-induced nick sealing, PAGE analysis showed a conjugate of 5′-mercapto-oligonucleotide 2 with EDC whose electrophoretic mobility was very different from the mobility of the expected 33-nt oligomer (Fig. 6, lane 3). The same product was found when oligonucleotide 1 with a 3′-terminal phosphate was excluded from the reaction mixture (Fig. 6, lane 2). The chemical behavior as well as electrophoretic and chromatographic mobilities of the unknown by-product allowed us to identify it as a stable S-alkylisothiourea derivative of oligonucleotide 2 with EDC (Scheme 2). Similar conjugates were demonstrated earlier for non-nucleotide analogs (31). Thus, it was established that the SH group of oligonucleotide 2 prefers to interact with the carbon center of EDC rather than with the activated phosphate of an adjacent oligonucleotide.
Figure 6.
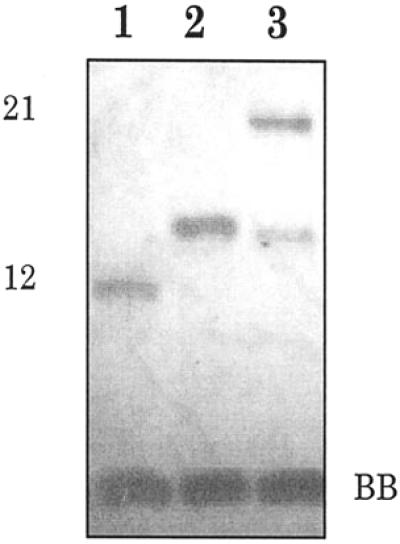
EDC-induced conversation of 5′-mercapto-oligodeoxyribonucleotide 2. Denaturing PAGE analysis of oligonucleotide 2 before (lane 1) and after EDC treatment (lane 2) and of an equimolar mixture of oligonucleotide 2 and oligonucleotide 1 with a 3′-terminal phosphate (duplex I in Fig. 1) after EDC treatment (lane 3). Reaction conditions are given in Materials and Methods. Bands were visualized by UV shadowing.
To enforce 5′-sulfhydryl group attack on the electrophilic terminal phosphate, we synthesized the HOBT derivative of 3′-phosphorylated oligonucleotide 1. The HOBT group is known to activate the phosphate center to nucleophilic attack (17). Comparative template-directed autoligation of three pairs of oligomers which differ in the 5′-terminal group facing the nick, SH, OH or phosphate (Scheme 1), and, consequently, in the nature of the newly formed internucleotide linkage, 5′-bridging phosphothioester, natural phosphodiester or pyrophosphate, respectively, was carried out in buffer 2 containing 0.4 M N-methylimidazole at 4°C. Simple qualitative inspection of a polyacrylamide gel for a given set of reactions showed (Fig. 7) that the yield of ligation product with a 5′-bridging phosphothioester linkage in duplex I (Scheme 1, reaction IIIa) is only a little less (∼80%) than that of the product with a pyrophosphate linkage (Scheme 1, reaction IIIc), which approaches 95%. Excess 2 is transformed to the 5′-SS-5′-linked homodimer, which appears as a slight middle band on a denaturing gel (Fig. 7, lane 3). In the case of a hydroxyl-containing oligomer (Scheme 1, reaction IIIb), the coupling yield is much lower (∼40%). The experiments with various terminal groups were carried out with both autoligation of a linear oligonucleotide to a self-folded hairpin oligomer (duplex I) and ligation of two adjacent linear oligonucleotides hybridized on the complementary template (duplex III). The results were similar: the reactivity of the SH group towards the activated phosphate of the adjacent oligonucleotide was higher than the reactivity of the hydroxyl group. No ligation product was observed in reactions IIIa–c in the absence of the complementary template (data not shown). A P-S link in the reaction product formed by SH-containing oligonucleotide 2 was confirmed by its selective cleavage with silver nitrate (Fig. 8).
Figure 7.
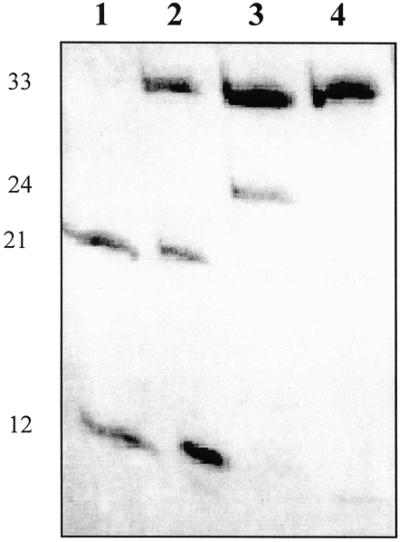
Comparative nick sealing in duplex I via nucleophilic attack of 5′-hydroxyl (lane 2), 5′-sulfhydryl (lane 3) or 5′-phosphate groups (lane 4) on a N-hydroxybenzotriazole-activated 3′-phosphate (Scheme 1). Lane 1, the initial oligonucleotides. The chain length of oligonucleotides is indicated. The bands on the denaturing polyacrylamide gel were visualized by UV shadowing.
Figure 8.
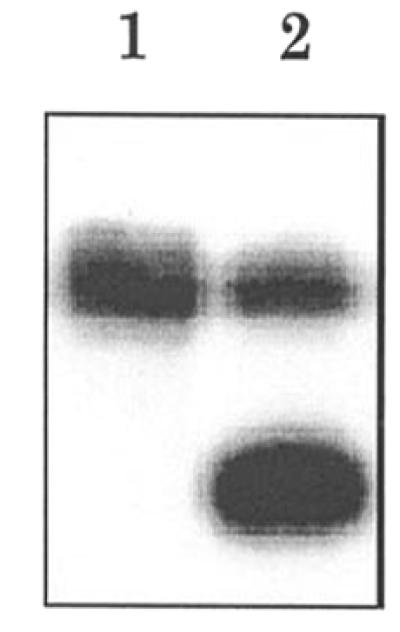
Cleavage of the P-S linkage. Autoradiogram of denaturing PAGE analysis of oligonucleotide d(TTGCGTA-GpST-GACTGCACGGT), containing a 5′-bridging thioester at the position denoted by pS, before (lane 1) and after (lane 2) silver nitrate treatment.


According to the literature, an ‘inverted’ reactivity of oligonucleotide sulfhydryl and hydroxyl groups takes place in some enzyme-catalyzed phosphoryl transfer reactions. It is known that the key reaction of protein tyrosine phosphatases (PTPs), which catalyze hydrolysis of phosphotyrosine-containing substrates, is nucleophilic attack by the sulfhydryl group of the active site Cys on the phosphorus atom (32–36). It has been shown for several enzymes of this family that a single oxygen for sulfur substitution (Cys→Ser) leads to a total loss of enzymatic activity, although the inactive mutant protein remains capable of binding substrate (32,33). According to X-ray data (34,35), the cysteine thiol group is ideally positioned to act as a nucleophile on the tyrosine phosphate moiety. It is noteworthy that the pKa value of the active site Cys in PTPs is 4.7, indicating that the Cys is a thiolate anion at physiological pH (37). Considerable evidence exists for the involvement of a His residue in stabilizing the thiolate anion in the active sites of PTPs (32–34,37) and other enzymes (38), for example by forming an ion pair with the reactive thiol (37). It was shown, in particular, that the apparent thiol pKa value and reactivity were very dramatically altered when the adjacent invariant His was changed to Asp or Ala (37).
Thus, in contrast to many reactions where thiolate nucleophiles were remarkably resistant to attack at the electrophilic phosphate center, in the systems under investigation here a 5′-sulfhydryl was more competent in ligation than a 5′-hydroxyl. An effective way was found to introduce a 5′-bridging phosphothioester linkage at a pre-selected position in DNA at a T or C residue. This approach is more universal than the phosphorothioate–iodine (or tosylate) autoligation reaction, which restricts the modified linkage to near T residues only (21–23). It is worth mentioning that direct solid phase synthesis of DNA oligonucleotides with a single bridged phosphorothioate at an adenosine residue has also been described (39).
CONCLUSION
The present results show that a 5′-SS-bridging monophosphoryldithio link can be conveniently incorporated into DNA duplexes at T or C residues by chemical coupling of an oligonucleotide 3′-phosphorothioate and a 5′-mercapto-oligodeoxyribonucleotide converted to a 2-pyridyldisulfide adduct. The modified linkage introduces little disturbance of the DNA structure. It is reasonably stable in water at 37°C and can selectively interact with reducing reagents carrying thiol groups. As a potential tool for inhibition of cellular proteins and enzymes, 5′-SS-monophosphoryldithio-containing DNA duplexes offer several advantages over previously described single modified constructs with diphosphoryldisulfide linkages: (i) -P-SS-CH2- linkages mimic natural phosphodiester bonds in size and charge density better than -P-SS-P- bridges; (ii) oligonucleotides carrying a -P-SS-CH2- moiety are stable to spontaneous disulfide bond exchange even in the single-stranded state; (iii) due to the different pKa values of phosphorothioate and sulfhydryl groups, it might be possible to predict the direction of attack of the Cys residue on the modified linkage within specific protein–dsDNA complexes.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Elena Romanova for synthesis of oligonucleotides and Dr Vadim Tashlitsky for assisting in ion pair HPLC. This work was supported by RFBR grant no. 00-04-48260 and Foundation for Scientific School Support (grant no. 00-15-97944).
References
- 1.Kuznetsova S.A., Ivanovskaya,M.G. and Shabarova,Z.A. (1990) Chemical reactions in double-stranded nucleic acids. IX. Directed introduction of substituted pyrophosphate bonds into DNA structure. Bioorg. Khim., 16, 219–225. [PubMed] [Google Scholar]
- 2.Purmal A.A., Shabarova,Z.A. and Gumport,R.I. (1992) A new affinity reagent for the site-specific covalent attachment of DNA to active site nucleophiles: application to the EcoRI and RsrI restriction and modification enzymes. Nucleic Acids Res., 20, 3713–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kubareva E.A., Fedorova,O.A., Gottikh,M.B., Tanaka,H., Malvy,C. and Shabarova,Z.A. (1996) NF-kappa B p50 subunit cross-linking to DNA duplexes containing a monosubstituted pyrophosphate internucleotide bond. FEBS Lett., 381, 35–38. [DOI] [PubMed] [Google Scholar]
- 4.Naryshkin N.A., Farrow,M.A., Ivanovskaya,M.G., Oretskaya,T.S., Shabarova,Z.A. and Gait,M.J. (1997) Chemical cross-linking of the human immunodeficiency virus type 1 Tat protein to synthetic models of the RNA recognition sequence TAR containing site-specific trisubstituted pyrophosphate analogues. Biochemistry, 36, 3496–3505. [DOI] [PubMed] [Google Scholar]
- 5.Kozlov I.A., Kubareva,E.A., Ivanovskaya,M.G. and Shabarova,Z.A. (1997) Design of new reagents on the base of DNA duplexes for irreversible inhibition of transcription factor NF-kappa B. Antisense Nucleic Acid Drug Dev., 7, 279–289. [DOI] [PubMed] [Google Scholar]
- 6.Kuznetsova S.A., Clusel,C., Urgarte,E., Elias,I., Vasseur,M., Blumenfeld,M. and Shabarova,Z.A. (1996) Crosslinking of double-stranded oligonucleotides containing O-methyl-substituted pyrophosphate groups to the HNF1 transcription factor in nuclear cell extract. Nucleic Acids Res., 24, 4783–4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheflyan G.Ya., Kubareva,E.A., Kuznetsova,S.A., Karyagina,A.S., Nikolskaya,I.I., Gromova,E.S. and Shabarova,Z.A. (1996) Cross-linking of SsoII restriction endonuclease to cognate and non-cognate DNAs. FEBS Lett., 390, 307–310. [DOI] [PubMed] [Google Scholar]
- 8.Dolinnaya N., Metelev,V., Oretskaya,T., Tabatadze,D. and Shabarova,Z. (1999) Hairpin-shaped DNA duplexes with disulfide bonds in sugar-phosphate backbone as potential DNA reagents for crosslinking with proteins. FEBS Lett., 444, 285–290. [DOI] [PubMed] [Google Scholar]
- 9.Bulaj G., Kortemme,T. and Goldenberg,D.P. (1998) Ionization–reactivity relationships for cysteine thiols in polypeptides. Biochemistry, 37, 8965–8972. [DOI] [PubMed] [Google Scholar]
- 10.Chu B.C.F. and Orgel,L.E. (1988) Ligation of oligonucleotides to nucleic acids or proteins via disulfide bonds. Nucleic Acids Res., 16, 3671–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paalman S.R., Noll,D.M. and Clarke,N.D. (1997) Formation of a covalent complex between methylguanine methyltransferase and DNA via disulfide bond formation between the active site cysteine and a thiol-containing analog of guanine. Nucleic Acids Res., 25, 1795–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Metelev V.G. and Oretskaya,T.S. (1999) Synthesis of oligodeoxyribonucleotides modified at the terminal phosphorothioate group. Bioorg. Khim., 25, 490–494. [Google Scholar]
- 13.Burgin A.B.Jr, Huizenga,B.N. and Nash,H.A. (1995) A novel suicide substrate for DNA topoisomerases and site-specific recombinases. Nucleic Acids Res., 23, 2973–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sproat B.S., Beijer,B., Rider,P. and Neuner,P. (1987) The synthesis of protected 5′-mercapto-2′,5′-dideoxyribonucleoside-3′-O-phosphoramidites; uses of 5′-mercaptooligodeoxyribonucleotides. Nucleic Acids Res., 15, 4837–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garegg P.J., Regberg,T., Stawinski,J. and Stromberg,R. (1987) A phosphorus nuclear magnetic resonance spectroscopic study of the conversion of hydroxy groups into iodo groups in carbohydrates using the iodine-triphenylphosphine-imidazole reagent. J. Chem. Soc. Perkin Trans., II, 271–274. [Google Scholar]
- 16.Sinha N.D. and Striepeke,S. (1991) Oligonucleotides with reporter groups attached to the 5′-terminus. In Eckstein,F. (ed.), Oligonucleotides and Analogues. IRL Press, Oxford, New York, pp. 185–210.
- 17.Ivanovskaya M.G., Gottikh,M.B., Shabarova,Z.A. and Prokofiev,M.A. (1987) Active ethers of oligonucleotides—a new type of phosphorylating agents in aqueous medium. Dokl. Akad. Nauk SSSR, 295, 477–481. [Google Scholar]
- 18.Tashlitsky V.N. and Oretskaya,T.S. (1997) Optimization of conditions for ion-pair HPLC of oligonucleotides. Bioorg. Khim., 23, 732–741. [PubMed] [Google Scholar]
- 19.Cook A.F. (1970) Nucleoside S-alkyl phosphorothioates. IV. Synthesis of nucleoside phosphorothioate monoesters. J. Am. Chem. Soc., 92, 190–195. [Google Scholar]
- 20.Rybakov V.N., Rivkin,M.I. and Kumarev,V.P. (1981) Some substrate properties of analogues of oligothymidylates with p-s-C5′ bonds. Nucleic Acids Res., 9, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrlein M.K., Nelson,J.S. and Letsinger,R.L. (1995) A covalent lock for self-assembled oligonucleotide conjugates. J. Am. Chem. Soc., 117, 10151–10152. [Google Scholar]
- 22.Xu Y. and Kool,E.T. (1997) A novel 5′-iodonucleoside allows efficient nonenzymatic ligation of single-stranded and duplex DNAs. Tetrahedron Lett., 38, 5595–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y. and Kool,E.T. (1998) Chemical and enzymatic properties of bridging 5′-S-phosphorothioester linkages in DNA. Nucleic Acids Res., 26, 3159–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou S.-H., Zhu,L., Gao,Z., Cheng,J.-W. and Reid,B.R. (1996) Hairpin loops consisting of single adenine residues closed by sheared A·A and G·G pairs formed by the DNA triplets AAA and GAG: solution structure of the d(GTACAAAGTAC) hairpin. J. Mol. Biol., 264, 981–1001. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh G., Duyne,G.V., Ghosh,S. and Sigler,P.B. (1995) Structure of NF-κB p50 homodimer bound to a κB site. Nature, 373, 303–310. [DOI] [PubMed] [Google Scholar]
- 26.Frey P.A. and Sammons,R.D. (1985) Bond order and change localization in nucleoside phosphorothioates. Science, 228, 541–546. [DOI] [PubMed] [Google Scholar]
- 27.Pearson R.G. (1966) Acids and bases. Science, 151, 172–177. [DOI] [PubMed] [Google Scholar]
- 28.Connolly B.A. (1987) Solid phase 5′-phosphorylation of oligonucleotides. Tetrahedron Lett., 28, 463–466. [Google Scholar]
- 29.Metelev V.G., Borisova,O.A., Dolinnaya,N.G. and Shabarova,Z.A. (1999) Oligodeoxyribonucleotides with internucleotidic or terminal phosphorothioate groups: different pathways in the reaction with water-soluble carbodiimide. Nucl. Nucl., 18, 2711–2719. [Google Scholar]
- 30.Michalski J., Reimschüssel,W. and Kaminski,R. (1978) Organic monopyrophosphates. Russian Chem. Rev., XLVII, 814–820. [Google Scholar]
- 31.Williams A. and Ibrahim,I.T. (1981) Carbodiimide chemistry: recent advances. Chem. Rev., 81, 589–636. [Google Scholar]
- 32.Guan K. and Dixon,J.E. (1991) Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J. Biol. Chem., 266, 17026–17030. [PubMed] [Google Scholar]
- 33.Barford D., Flint,A.J. and Tonks,N.K. (1994) Crystal structure of human protein tyrosine phosphatase 1B. Science, 263, 1397–1403. [PubMed] [Google Scholar]
- 34.Stuckey J.A., Schubert,H.L., Fauman,E.B., Zhang,Z.-Y., Dixon,J.E. and Saper,M.A. (1994) Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 Å and the complex with tungstate. Nature, 370, 571–575. [DOI] [PubMed] [Google Scholar]
- 35.Su X.-D., Taddei,N., Stefani,M., Ramponi,G. and Nordlund,P. (1994) The crystal structure of a low-molecular-weight phosphotyrosine protein phosphatase. Nature, 370, 575–578. [DOI] [PubMed] [Google Scholar]
- 36.Denu J.M., Lohse,D.L., Vijayalakshmi,J., Saper,M.A. and Dixon,J.E. (1996) Visualization of intermediate and transition-state structures in protein-tyrosine phosphatase catalysis. Proc. Natl Acad. Sci. USA, 93, 2493–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z.-Y. and Dixon,J.E. (1993) Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry, 32, 9340–9345. [DOI] [PubMed] [Google Scholar]
- 38.Polgár L. and Csoma,C. (1987) Dissociation of ionizing groups in the binding cleft inversely controls the endo- and exopeptidase activities of cathepsin B. J. Biol. Chem., 262, 14448–14453. [PubMed] [Google Scholar]
- 39.Kuimelis R.G. and McLaughlin,L.W. (1995) Cleavage properties of an oligonucleotide containing a bridged internucleotide 5′-phosphorothioate RNA linkage. Nucleic Acids Res., 23, 4753–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]