

Abstract
Cholesterol conversion to bile acids is subject to a feedback regulatory mechanism by which bile acids down-regulate their own synthesis. This regulation occurs at the level of transcription of several genes encoding enzymes in the bile acid biosynthetic pathway. One of these enzymes is sterol 12α-hydroxylase/CYP8B1 (12α-hydroxylase), the specific enzyme required for cholic acid synthesis. The levels of this enzyme determine the ratio of cholic acid to chenodeoxycholic acid and thus the hydrophobicity of the circulating bile acid pool. Previous studies from this laboratory showed that fetoprotein transcription factor (FTF) is required for 12α-hydroxylase promoter activity and bile acid-mediated regulation. Here, we report that the short heterodimer partner (SHP) suppresses 12α-hydroxylase promoter activity via an interaction with FTF. Hepatic nuclear factor-4 (HNF-4) binds and activates the 12α-hydroxylase promoter and is required for 12α-hydroxylase promoter activity. Although HNF-4 interacts with SHP, it is not involved in SHP-mediated suppression of 12α-hydroxylase promoter activity. FTF and not HNF-4 is the factor involved in regulation of 12α-hydroxylase promoter activity by bile acids through its interaction with SHP. Finally, interaction of SHP with FTF displaces FTF binding to its sites within the 12α-hydroxylase promoter. These results provide insights into the mechanism of action of bile acid-mediated regulation of sterol 12α-hydroxylase transcription.
INTRODUCTION
Bile acid synthesis plays a crucial role in maintaining cholesterol homeostasis since it is responsible for the catabolism of >50% of body cholesterol. Bile acids also stimulate the excretion of excess hepatic cholesterol into bile. In the intestine, bile acids play an important role in the solubilization and absorption of fat-soluble vitamins and cholesterol. Thus, proper control of bile acid synthesis is important and is achieved through the regulation of several enzymes: cholesterol 7α-hydroxylase/CYP7A1 (7α-hydroxylase), the rate-limiting enzyme in the classic pathway (1); sterol 27-hydroxylase/CYP27, the first enzyme in the alternative pathway (2); and sterol 12α-hydroxylase/CYP8B1 (12α-hydroxylase), the specific enzyme for cholic acid synthesis that determines the ratio of cholic acid to chenodeoxycholic acid (CDCA) and thus the hydrophobicity of the circulating pool (3).
Bile acids negatively regulate transcription of the 7α-hydroxylase gene, which controls output from the classic pathway. Recent studies have delineated many of the factors involved in this regulation. Liver receptor homolog-1, also known as CYP7A promoter-binding factor (CPF), NR5A2 (4) and fetoprotein transcription factor (FTF) (Genome Database Nomenclature Committee) (5) have been proposed to be required for transcription of the 7α-hydroxylase gene (6). Bile acids activate transcription of the small heterodimer partner 1 (SHP) via binding of the hormone receptor farnesil X receptor (FXR) to its binding site in the SHP promoter. In turn, it has been proposed that SHP dimerizes with FTF and diminishes its activity on the 7α-hydroxylase promoter by mechanisms not yet well understood (7,8). An alternative mechanism has recently been proposed for this down-regulation of 7α-hydroxylase transcription that involves the c-Jun N-terminal kinase (JNK) (9). In this mechanism, the JNK pathway is activated by bile acids, which in turns activates c-Jun resulting in higher SHP transcription.
SHP is an orphan nuclear receptor originally identified on the basis of its interaction with the receptor CAR (10). It lacks a DNA-binding domain and interacts with several receptors, among them hepatic nuclear receptor-4 (HNF-4) (11), estrogen receptor (ER) (12) and retinoid X receptor α (RXR) (10). Upon interaction with these receptors, SHP acts to decrease transactivation by its partners in transient transfection assays. This inhibitory effect was first attributed to an inhibition of DNA binding by the SHP targets (10). Further studies, however, revealed that SHP carries a repression domain in its C-terminal domain (13) suggesting that SHP could inhibit transcription without interference with binding of its partner to the DNA. This alternative mechanism was confirmed by the demonstration that SHP is an inhibitor of ER transactivation, even though it does not prevent binding of ER to estrogen response elements (13).
Another enzyme in the bile acid biosynthetic pathway that is also regulated by bile acids is 12α-hydroxylase (3,14,15). We have recently shown that FTF is required for 12α-hydroxylase promoter activity (16). FTF binds to two sites within the 12α-hydroxylase promoter and both sites are required for both promoter activity and bile acid-mediated regulation. However, it has not been shown whether SHP is involved in this regulation, much less the mechanisms by which SHP suppresses FTF activity on bile acid regulated genes.
The two FTF sites in the 12α-hydroxylase promoter overlap with a direct repeat (DR-1) element that binds peroxisome proliferator-activated receptor α (PPARα) and mediates peroxisome proliferator activation of 12α-hydroxylase transcription (17). Interestingly, the 7α-hydroxylase promoter also contains a DR-1 element, which is a binding site for HNF-4 among other nuclear receptors. It has been shown that HNF-4 indeed does bind and activate the 7α-hydroxylase promoter (18). The existence of HNF-4 sites overlapping the FTF sites in both the 7α-hydroxylase and 12α-hydroxylase promoters together with the fact that SHP is a repressor of HNF-4 (11) raises the possibility that HNF-4 is involved in bile acid regulation of gene transcription through its interaction with SHP.
In this study we show that SHP is involved in down-regulation of the 12α-hydroxylase promoter. Overexpression of SHP in HepG2 cells suppresses 12α-hydroxylase promoter activity. We also show that HNF-4 is required for 12α-hydroxylase promoter activity, but despite the interaction between HNF-4 and SHP, bile acids only suppress FTF-activated 12α-hydroxylase promoters. Additionally, we show that several SHP domains are required for that interaction, suggesting a complex mode of contact between FTF and SHP. Finally, we show that SHP prevents binding of FTF to its binding sites within the 12α-hydroxylase promoter, providing a mechanism of action for SHP-mediated suppression of 12α-hydroxylase transcription.
MATERIALS AND METHODS
Materials
Reagents used in DNA cloning and sequencing were from New England Biolabs, Boehringer Mannheim, US Biochemical Corp. and Gibco BRL. Common laboratory chemicals were from Fisher, Sigma or Bio-Rad. Oligonucleotides were prepared in the Medical College of Virginia DNA Synthesis Facility by the phosphoramidite method on an automated DNA synthesizer. The luciferase promoter-less vector, pGL3-Basic, was purchased from Promega. The mammalian two-hybrid vectors pVP16 and pM were purchased from Clontech. pCMX, a plasmid for expression in mammalian cells and in vitro, contains the CMV and T7 promoters, and TK-MH100×4-LUC, a plasmid that has four copies of the Gal4 DNA-binding domain in front of the luciferase reporter gene, were a gift from Dr Ronald M. Evans (The Salk Institute, La Jolla, CA). pCI/hFTF, an expression plasmid that contains the human FTF cDNA in the expression vector pCI (Promega), pCI/hFTFΔLBD and pCI/hFTFΔAF2 were a generous gift from Dr Bélanger (L’University Laval, Quebec, Canada). pCDM8/mSHP, an expression plasmid containing full-length mouse SHP, and all the other SHP plasmids used in this study were obtained from Dr Moore (Baylor College of Medicine, Houston, TX). pGL3-R12α-865 is a promoter reporter construct that contains 865 bp of the rat 12α-hydroxylase promoter in the pGL3 vector (16). The three 12α-hydroxylase promoter constructs used for the experiments shown in Figure 3A were made using a QuickChange site-directed mutagenesis kit (Stratagene) and the corresponding oligonucleotides. pGHR contains the rabbit growth hormone receptor cDNA in pCMV (19,20). Anti-HNF-4 antibodies were obtained from Santa Cruz Biotechnology Inc. and anti-FTF antibodies were a gift from Dr David W. Russell (Southwestern Medical Center, Dallas, TX) and were raised against a peptide corresponding to amino acids 180–197 of the DNA-binding domain.
Figure 3.


The 12α-hydroxylase DR-1 HNF-4-binding site is required for 12α-hydroxylase promoter activity. (A) Three 12α-hydroxylase promoter mutants were created as shown, with the mutated nucleotides in small capital letters. FTF sites are indicated by slashed boxes and the hexamers corresponding to the DR-1 site are shown by arrows. The wild-type 12α-hydroxylase promoter construct and the three mutants were transfected into HepG2 cells, incubated in the absence or presence of CDCA and relative promoter activity quantified as indicated in Materials and Methods. Data represent the means ± SD of four assays performed in duplicate. ND, not detectable (activity was ≤1.5-fold that obtained with the empty vector pGL3, whereas the wild-type 12α-hydroxylase promoter had 80-fold higher activity than pGL3). (B) Gel shift experiments were performed using the indicated probes and either in vitro synthesized FTF or HNF-4 proteins or in vitro produced GHR as a control. Arrows point to the retarded bands as well as the free probes.
General methods
Standard recombinant DNA procedures were carried out essentially as described (16).
Preparation of chimeric FTF expression plasmids
pCMX/FTF was prepared by inserting a 1600 nt SacII–SalI fragment from pCI/FTF into the BamHI site of pCMX. The FTF deletion plasmids were prepared by inserting the corresponding fragment, prepared by PCR with specific oligonucleotides, into its corresponding vector. pVP16-FTF was prepared by inserting the FTF cDNA into pVP16 (Clontech). Orientation and correct frame were confirmed by sequencing.
Transient transfection and luciferase assays
HepG2, Hep3B and CV-1 cells were obtained from the American Type Culture Collection. HepG2 and Hep3B cells were transfected by the calcium phosphate method using 2.0 µg total DNA. CV-1 cells were transfected with Lipofectin (Gibco) and 750 ng total DNA. HepG2 cells were transfected with 100 ng test plasmid, 5 ng pCMV-Gal [a plasmid containing the human cytomegalovirus (CMV) promoter in front of the bacterial β-galactosidase gene] to normalize for transfection efficiencies and the indicated amounts of wild-type pCDM8/mSHP. For SHP overexpression, CV-1 cells were transfected with 100 ng test plasmid, 10 ng pCMV-Gal, 100 ng pCI-FTF, an expression plasmid containing the FTF cDNA driven by the CMV promoter, and the indicated amounts of wild-type pCDM8/mSHP or the W160X mutant. For the mammalian two-hybrid system CV-1 cells were transfected with 100 ng TK-MH100×4-LUC, as a reporter plasmid, 25 ng pCMV-Gal and 325 ng each indicated hybrid. Hep3B cells were transfected with 250 ng test plasmid, 10 ng pCMV-Gal and 750 ng pCI/hFTF or the indicated deletion mutant. After 16 h the DNA was removed and, where indicated, 100 µM CDCA was added. Cells were harvested 48 h later and luciferase and β-galactosidase assays were performed with a kit from Tropix (Bedford, MA), according to the manufacturer’s protocol. Average values are for the number of experiments indicated.
In vitro transcription/translation and HepG2 nuclear extracts
Transcription/translation of cDNAs encoding FTF, SHP or the growth hormone receptor (GHR) as a control was performed using the TNT T7-coupled rabbit reticulocyte lysate system according to the manufacturer’s protocol (Promega). Proteins were synthesized with [35S]l-methionine and quantified using a Molecular Dynamics PhosphorImager. HepG2 nuclear extracts were prepared as indicated (16).
Electrophoretic mobility shift analysis and SDS–PAGE
DNA binding reactions contained 50 mM KCl, 20 mM Tris–HCl pH 8.0, 0.2 mM EDTA, 4% Ficoll, 1.0 µg poly(dI–dC), 4 µl of the translated protein and a 1500-fold molar excess of an irrelevant single-stranded DNA, in a final volume of 20 µl on ice. After 15 min incubation, 32 fmol 32P-labeled DNA probe (∼2 × 105 c.p.m.) were added. The probe used contained nucleotides –69 to –46 from the 12α-hydroxylase promoter. After incubation for 20 min on ice, samples were loaded onto a 4.5% polyacrylamide gel and subjected to electrophoresis at 4°C. Gels were dried and exposed to XAR-5 film (Kodak). For SDS–PAGE 1 µl of translated protein was loaded on a 12% SDS–acrylamide gel.
RESULTS
To determine whether SHP affects the activity of the 12α-hydroxylase promoter, we transfected increasing amounts of an expression plasmid containing the mouse SHP cDNA into HepG2 cells with a fixed amount of the 12α-hydroxylase promoter construct pGL3-R12α-865 (16). Figure 1 shows that SHP strongly suppresses 12α-hydroxylase promoter activity. The level of suppression was >5-fold when 200 ng SHP expression plasmid was transfected, comparable to the bile acid-mediated suppression observed in the same system (16).
Figure 1.

SHP represses the rat 12α-hydroxylase promoter–luciferase construct in HepG2 cells. HepG2 cells were co-transfected with the pGL3-R12α-865 construct and the indicated amounts of SHP expression plasmid. Data represent the means ± SD of three assays performed in duplicate.
The FTF sites within the 12α-hydroxylase promoter (16) overlap a DR-1 site that has recently been identified (17) as a functional PPAR-binding site. Since DR-1 is the binding site for HNF-4, a receptor known to interact with SHP (11), we decided to investigate whether HNF-4 could bind and activate the 12α-hydroxylase promoter. Moreover, we wanted to determine whether HNF-4 is involved in the suppression of 12α-hydroxylase promoter activity by bile acids. Figure 2A shows that both FTF and HNF-4 bind to the 12α-hydroxylase promoter, as demonstrated by supershift assays using HepG2 nuclear extracts and a specific HNF-4 antibody (lane 2). Anti-FTF antibodies abolished FTF binding and produced no supershifted band (lane 3), as expected, since the antibody was raised against a peptide corresponding to the DNA-binding domain of FTF (16). To establish the potential role of HNF-4 in 12α-hydroxylase promoter activity, we co-transfected the 12α-hydroxylase promoter construct pGL3-R12α-865 together with an expression plasmid with the HNF-4 cDNA in the presence and absence of the SHP expression plasmid. For recipient cells we used CV-1 cells, a non-liver cell line that has no 12α-hydroxylase activity because of the lack of liver-specific transcription factors. As a control, we used the FTF expression plasmid that we have already shown can activate the 12α-hydroxylase promoter (16). As seen in Figure 2B, HNF-4 is capable of activating the 12α-hydroxylase promoter almost as much as FTF (11- versus 14-fold). Most importantly, overexpression of SHP eliminated most of the FTF activation, but did not reduce the HNF-4-mediated activation of 12α-hydroxylase promoter activity.
Figure 2.
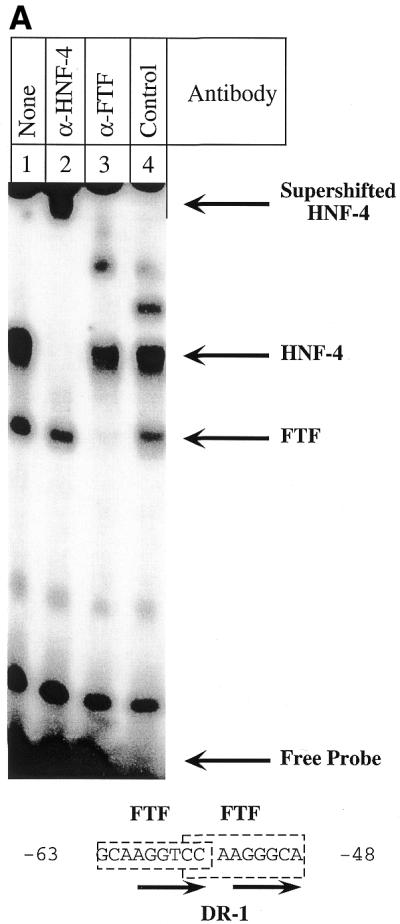

SHP represses FTF-dependent, but not HNF-4-dependent, activation of the rat 12α-hydroxylase promoter. (A) A gel shift experiment was performed as described in Materials and Methods, using 1 µg HepG2 nuclear extract and the indicated antibodies. The 12α-hydroxylase promoter sequence with the FTF sites (boxed) and the DR-1 site (arrows) highlighted is shown at the bottom. An irrelevant IgG was used as control antibody (lane 4). (B) CV-1 cells were co-transfected with the pGL3-R12α-865 construct and the indicated amounts of FTF or HNF-4 and/or SHP expression plasmids. Data represent the means ± SD of three assays performed in duplicate.
To further test whether FTF is the factor required for bile acid-mediated regulation of 12α-hydroxylase expression, we used Hep3B cells, a hepatoma cell line in which the 12α-hydroxylase promoter is poorly expressed. Consistent with the notion that expression of FTF is needed for both activity and bile acid-mediated regulation of 12α-hydroxylase, CDCA did not regulate 12α-hydroxylase promoter activity when transfected in the absence of other expression plasmids (Table 1). However, overexpression of FTF increased 12α-hydroxylase promoter activity 7.7-fold and provided sensitivity to CDCA, indicating that FTF is limiting in H3B cells. In contrast, HNF-4 overexpression also activated 12α-hydroxylase promoter activity 2-fold, but showed no regulation by CDCA. To further prove that expression of intact FTF is required for bile acid-mediated regulation, we overexpressed two FTF deletion plasmids. pCI-FTFΔLBD contains a FTF cDNA with the ligand-binding domain deleted. The ligand-binding domain is known to be required for interaction of other nuclear receptors with different coactivators or corepressors. pCI-FTFΔLBD activated the 12α-hydroxylase promoter but did not support regulation by bile acids, suggesting that the FTF ligand-binding domain is involved in the interaction with SHP to provide regulation. pCI-FTFΔAF-2 is a FTF mutant with the C-terminal activation domain deleted. Overexpression of this mutant decreased 12α-hydroxylase activity to undetectable levels, presumably by competing out endogenous FTF by the inert mutant.
Table 1. Overexpression of FTF or FTFΔLBD increases 12α-hydroxylase promoter–luciferase construct activity in Hep3B cells.
| Expression plasmid | Promoter activity | Regulation (%) | |
| |
–CDCA |
+CDCA |
|
| Control (GHR) | 100 | 102 ± 15 | 100 (3) |
| Wild-type FTF | 769 ± 362 | 432 ± 161 | 58 ± 8 (4)a |
| HNF-4 | 199 ± 34 | 276 ± 96 | 131 ± 32 (3)b |
| FTFΔLBD | 296 ± 137 | 316 ± 155 | 106 ± 10 (3)b |
| FTFΔAF-2 | ND | ND | ND |
pCMV-GHR, as a control, pCI-FTF, pCI-HNF-4, pCI-FTFΔLBD or pCI-FTFΔAF-2 was co-transfected into Hep3B cells together with pGL3-R12α-865. Cells were treated with or without 100 µM CDCA, harvested and analyzed for luciferase and β-galactosidase activities as described in Materials and Methods. The data were normalized to the activity produced by construct pGL3-R12α-865 co-transfected with the control plasmid pCMV-GHR in the absence of CDCA and represent the averages of (n) experiments ± SD. ND, not detectable.
aDiffers from the control (P < 0.05).
bDoes not differ from the control (P > 0.1).
To further evaluate the specific roles of FTF and HNF-4 in both 12α-hydroxylase promoter activity as well as for its bile acid-mediated regulation, we attempted to create 12α-hydroxylase promoters lacking either a HNF-4 site or both FTF sites previously characterized (located between nucleotides –63 and –48) and transfected them into HepG2 cells that were incubated with and without CDCA to study both promoter activity and bile acid regulation (Fig. 3A). FTF and HNF-4 binding to these mutants was tested by mobility shift analysis using in vitro made proteins (Fig. 3B). Wild-type 12α-hydroxylase binds both in vitro produced proteins, although with very different affinities (Fig. 3B, lanes 2 and 3). HNF-4 binds to the wild-type sequence, although with lower affinity than HNF-4 from HepG2 cells (compare Fig. 3B, lane 3 with Fig. 2A, lane 1). We then created two 12α-hydroxylase promoter constructs with the HNF-4 site mutated. In one construct (Fig. 3A, 12α-hydroxylase 5′-flanking DR-1 mutant) the three nucleotides located 5′ of the two DR-1 repeats were mutated to maintain FTF but not HNF-4 binding, since these nucleotides are known to be required for PPARα binding to the DR-1 site (17). This mutant promoter had no detectable activity (Fig. 3A), presumably due to lack of HNF-4 binding (Fig. 3B, lane 6). FTF binds to this mutant essentially as to the wild-type (Fig. 3B, lanes 5 and 2). The second mutant that we created, 12α-hydroxylase FTF consensus, contained one FTF consensus site instead of two. We used the rat 7α-hydroxylase promoter FTF site (21) and the 3′ repeat was mutated (Fig. 3A) so that the 12α-hydroxylase HNF-4 site was destroyed. This promoter mutant had no detectable activity (Fig. 3A) and, as expected, it is capable of binding FTF but not HNF-4 (Fig. 3B, lanes 8 and 9). In an attempt to create a 12α-hydroxylase promoter mutant with a HNF-4 site but no FTF sites, we mutated the 12α-hydroxylase FTF/HNF-4 site to the apoprotein CIII C3P site (22,23) to create 12α-hydroxylase apoCIII C3P HNF-4 (Fig. 3A). This promoter construct showed approximately one-third of the wild-type promoter activity and was slightly less regulated by CDCA than the wild-type promoter (Fig. 3A). As expected, this mutant binds HNF-4 with greater affinity than the wild-type 12α-hydroxylase site (Fig. 3B, lane 12). Unexpectedly, this mutant still binds FTF (Fig. 3B, lane 11). Close examination of the sequence revealed that it contains a reversed FTF site with 8 of 9 nt conserved (Fig. 3A). Multiple attempts to create a mutant that would bind HNF-4 but not FTF based either on the 12α-hydroxylase FTF/HNF-4 site or the apoprotein CIII C3P site were unsuccessful.
To additionally evaluate the effect of SHP on regulation of the 12α-hydroxylase promoter, we overexpressed wild-type SHP and a deletion mutant that lacks the C-terminus of the SHP protein, SHPΔ160X(1–159) (11), in CV-1 cells. The C-terminus of SHP contains a domain that is required for repression of nuclear receptors, such as HNF-4. Figure 4 shows that SHP suppressed FTF-dependent activation of the 12α-hydroxylase promoter in a dose-dependent manner. However, when the SHP deletion mutant was used, no suppression was observed.
Figure 4.

SHP repression of FTF-dependent activation of the rat 12α-hydroxylase promoter in CV-1 cells requires the SHP repression domain. CV-1 cells were co-transfected with the 12α-hydroxylase promoter construct and the indicated amounts of FTF and either wild-type SHP or SHPW160(1–159) (11), a SHP cDNA with the suppression domain deleted, as indicated in the figure. Data represent the means ± SD of three assays performed in duplicate.
It has been previously shown that FTF and SHP interact in vivo (7,8) but the domains required for that interaction are unknown. To characterize the potential distinctive domain(s) required for this interaction, we performed two-hybrid assays in mammalian cells. We co-expressed a VP16–FTF fusion construct and several GAL4–SHP fusion constructs, as shown in Figure 5. When a GAL4 fusion construct was used that contained the full-length SHP cDNA, an interaction was observed as indicated by an 8-fold activation when compared with the empty pVP16 vector. However, deletion of any SHP domain eliminated that activation. As a control we used the same GAL4–SHP constructs with a pVP16-HNF-4 plasmid. GAL4–SHP constructs that contained the putative interaction domain were capable of interaction with HNF-4 as described (11), suggesting that the modes of interaction of SHP with HNF-4 and FTF are different.
Figure 5.

FTF requires an intact SHP protein for the interaction between them in vivo. pVP16-FTF and previously described pGAL4-SHP mutants (12) were used in a mammalian two-hybrid assay in CV-1 cells. Five hundred nanograms of each pVP16-FTF, or pVP16 as a control, and each pGAL4-SHP construct were transfected together with 100 ng thymidine kinase promoter–luciferase reporter plasmid containing four GAL4-binding sites and pCMV-gal as a control for transfection efficiency. As a positive control, pVP16-HNF-4 was used with each pGAL4-SHP plasmid. The x-axis shows the different GAL4–SHP mutants used and the numbers correspond to the SHP amino acid number included in each construct. The y-axis represents the fold activation compared with the activity of an empty pVP16 vector. The scheme at the bottom represents the GAL4–SHP fusion protein showing the different SHP domains and the amino acid numbers that expand each domain. Data represent the means ± SD of three assays performed in duplicate.
To gain further insight into the mechanism of action of SHP repression on the 12α-hydroxylase promoter, we performed gel shift experiments using in vitro produced FTF in the absence or presence of SHP. Figure 6 shows that SHP prevents binding of FTF to the 12α-hydroxylase promoter in a linear fashion to the point that FTF binding is essentially eliminated (lanes 2–5). We included [35S]methionine in the synthesis reactions, in order to quantify actual protein synthesis and to ensure that the diminished binding was not due to less FTF synthesized. Although FTF synthesis was slightly diminished when the SHP plasmid was included in the in vitro synthesis reaction (lanes 7–10), FTF binding decreased to a much greater degree.
Figure 6.

FTF binding to the 12α-hydroxylase promoter is prevented by SHP. Gel shift experiments (lanes 1–5) using the 12α-hydroxylase promoter probe and SDS–acrylamide gels (lanes 6–10) were performed as described in Materials and Methods, using in vitro synthesized FTF and SHP proteins or in vitro produced GHR as a control. The number of micrograms refers to the amount of the corresponding plasmid used in the protein synthesis reaction. The GHR plasmid was also used to bring the amount of T7 promoter-containing plasmids to a total of 1 µg. The wild-type FTF/HNF-4 site from the 12α-hydroxylase promoter was used as probe for the gel shift with FTF alone (lane 2) or with different amounts of SHP (lanes 3–5). Protein samples used for the gel retardation were analyzed in a SDS gel (lanes 6–10).
DISCUSSION
In this study we present evidence that SHP inhibits 12α-hydroxylase expression through the FTF orphan receptor and not through HNF-4, in spite of HNF-4 being an activator of 12α-hydroxylase transcription that is required for 12α-hydroxylase promoter activity and of HNF-4 being capable of interacting with SHP. We also provide data that show that SHP prevents binding of FTF to the 12α-hydroxylase promoter site. Finally, we have determined the FTF and SHP domains that are responsible for interaction between these two proteins.
Recent studies have implicated SHP as the factor responsible for bile acid-mediated suppression of 7α-hydroxylase transcription (7,8), the rate-limiting enzyme in the classic pathway for bile acid synthesis. These studies used mammalian two-hybrid systems and GST pull-down assays to demonstrate that FTF and SHP interact both in vivo and in vitro. They also showed that overexpression of SHP can override FTF-mediated transactivation of 7α-hydroxylase promoter activity when both FTF and SHP expression plasmids were co-transfected with a 7α-hydroxylase promoter construct in a non-liver cell line (7,8). In the present studies we have shown that SHP overturns FTF-mediated activation of 12α-hydroxylase when co-transfected in a non-liver cell line (Figs 2 and 4). SHP is also capable of suppressing the 12α-hydroxylase promoter in HepG2 cells without the need for overexpression of co-transfected FTF cDNA (Fig. 1).
SHP is a repressor of several nuclear receptors, such as RXR (10), ER (12) and HNF-4 (11). Of particular interest is the interaction between HNF-4 and SHP, because HNF-4 has been shown to activate the 7α-hydroxylase promoter (18) through binding to a DR-1 element that overlaps the FTF site (6,21). Interestingly, the 12α-hydroxylase promoter also has a DR-1 element overlapping the FTF sites recently localized in our laboratory (16), and this site binds PPARα, another nuclear receptor (17). These observations raised the question as to whether HNF-4 could be the major transcription factor that interacts with SHP and mediates transcriptional regulation of the 7α-hydroxylase and 12α-hydroxylase genes by bile acids. To date, HNF-4 has not been eliminated as a major transcription factor mediating repression of 7α-hydroxylase and 12α-hydroxylase by bile acids. Indeed, we show that HNF-4 binds to the 12α-hydroxylase promoter, as demonstrated by supershift assays (Figs 2A and 3B). HNF-4 also activates the 12α-hydroxylase promoter in a non-liver cell line (Fig. 2B, lane 4), but overexpression of SHP cannot override this activation (Fig. 2A, lane 5). Additionally, the experiments shown in Figure 3 demonstrate the key roles of both HNF-4 and FTF in the activity of the 12α-hydroxylase promoter. Indeed, 12α-hydroxylase promoter mutants that lack HNF-4 binding had no detectable activity.
FTF binding is also important for 12α-hydroxylase promoter activity, although not as much as HNF-4. A 12α-hydroxylase mutant that binds HNF-4 with much higher activity than the wild-type promoter and binds FTF with about half the affinity of the wild-type (12α-hydroxylase apoCIII C3P HNF-4) had about one-third of the wild-type promoter activity (Fig. 3A). This promoter mutant is regulated by bile acids slightly less than the wild-type promoter, probably because of less FTF binding (Fig. 3B, lane 11). If HNF-4 were implicated in bile acid-mediated regulation of the 12α-hydroxylase promoter, the 12α-hydroxylase apoCIII C3P HNF-4 mutant would be expected to be more regulated by bile acids than the wild-type promoter, not less. Unfortunately we have not been able to create a mutant that would bind HNF-4 but not FTF, probably because of homology between the FTF and HNF-4 binding sites. Such a mutant would be useful to further rule out any involvement of HNF-4 in the regulation of 12α-hydroxylase promoter activity by bile acids and support the overexpression experiments shown in Figure 2B and Table 1. As we demonstrated earlier, FTF also activates the 12α-hydroxylase promoter in CV-1 cells (16) but, in contrast to HNF-4 activation, this FTF-mediated activation can be overridden by overexpression of SHP (Fig. 2B, lanes 2 and 3). Together these results show that FTF is the nuclear receptor that interacts with SHP to provide suppression of 12α-hydroxylase. How both FTF and HNF-4 can bind to their overlapping sites so that 12α-hydroxylase expression is sustained by HNF-4 and regulated by the interaction between FTF and SHP remains to be studied. It is important to point out that the promoter of the 7α-hydroxylase gene, a gene whose expression is coordinately regulated with 12α-hydroxylase by bile acids (15), also contains an overlapping FTF site and DR-1 element (21).
We used Hep3B cells to further demonstrate that FTF is the factor that interacts with SHP to mediate regulation of 12α-hydroxylase transcription by bile acids (Table 1). Hep3B cells have low 12α-hydroxylase promoter activity that does not respond to CDCA (Table 1), most likely because FTF and HNF-4 are expressed at low levels (24). This is analogous to the lack of bile acid-mediated regulation of 7α-hydroxylase expression in the absence of a FXR expression plasmid in HepG-2 cells (25) and in primary hepatocyes when they are transfected with relatively high amounts of 7α-hydroxylase promoter plasmid (21). In those cases the lack of regulation was overcome by transfecting a FXR expression plasmid. In Hep3B cells overexpression of FTF not only activated the 12α-hydroxylase promoter but also provided regulation by bile acids (Table 1). Regulation of the 12α-hydroxylase promoter by bile acids is lower in Hep3B cells than that seen in HepG2 cells (16), probably due to a relatively low expression of SHP in Hep3B cells as compared with HepG2 cells. Overexpression of SHP provides further suppression of 12α-hydroxylase in Hep3B cells (data not shown), although this was further studied in CV-1 cells (Fig. 4). On the other hand, HNF-4 activation of the 12α-hydroxylase promoter was not regulated by bile acids, supporting the notion that FTF is the factor that interacts with SHP to mediate regulation by bile acids.
Overexpressing two FTF mutants provided preliminary characterization of the FTF domains involved in the activation or bile acid-mediated regulation of 12α-hydroxylase. FTFΔAF-2, a mutant that has the activation domain AF-2 deleted, did not activate the 12α-hydroxylase promoter. In fact, this mutant eliminated even the low basal activity, acting as a dominant negative mutant similar to observations with the hepatitis virus core promoter (24). Another FTF mutant with its ligand-binding domain deleted (FTFΔLBD) mediated transactivation of the 12α-hydroxylase promoter. However, bile acids could not regulate its activity, suggesting that the FTF ligand-binding domain acts as an interacting domain with SHP (Table 1).
An important issue that has yet to be explored is the molecular mechanism involved in the suppression of FTF transactivation by SHP. Different mechanisms have been elucidated for the interaction of SHP with other nuclear receptors. It has been shown that SHP contains a repressor domain in its C-terminal region that could function on RXR, ER and HNF-4 (11–13). Our results support the functionality of the SHP repressor domain on transactivation of the 12α-hydroxylase promoter by FTF since a SHP mutant with no suppressor domain did not prevent FTF-mediated activation of the 12α-hydroxylase promoter in CV-1 cells (Fig. 4). However, the mechanism of action for SHP suppression appears to be different on FTF transactivation than on other nuclear receptors. First, there is no distinctive domain in SHP sufficient for interaction with FTF. Two-hybrid experiments in mammalian cells show that a fully intact SHP protein is required for its interaction with FTF (Fig. 5). In contrast, only amino acids 92–148 in the SHP protein are required for the interaction with RXR or HNF-4 (11,12). This discrepancy was confirmed by carrying out control experiments in parallel with the same SHP mutants and HNF-4. These results corroborate a different interaction between SHP and FTF, and SHP and HNF-4 (Fig. 5).
Our studies add FTF, to the list of nuclear receptors whose binding to its DNA target is prevented by SHP. Interaction between SHP and FTF prevents binding of FTF to its binding site within the 12α-hydroxylase promoter (Fig. 6). This observation is similar to the effect of SHP on binding of the RAR–RXR heterodimer to its recognition site (10), but different to the lack of effect of SHP on binding of ER to its DNA element (13). It needs to be investigated whether inhibition of FTF binding by SHP is a general mechanism for bile acid-regulated genes, such as 7α-hydroxylase, and whether this prevention of binding of FTF to its target DNA is the only mechanism of action for the repression of FTF transactivation by SHP. In other receptors, such as HNF-4, SHP represses receptor-mediated transactivation via two separate steps, i.e. competition with coactivators and a direct effect of its transcriptional repressor function. It is possible that SHP represses FTF transactivation by more than one mechanism.
In summary, these studies show the key roles of HNF-4 and FTF in expression of the 12α-hydroxylase gene. They also add further support to FTF being the SHP target involved in bile acid-mediated regulation of gene transcription and rule out HNF-4 as such a target, at least for the 12α-hydroxylase gene. In addition, our studies provide important insights into the mechanisms of action of FTF and SHP on the regulation of gene transcription by bile acids.
Acknowledgments
ACKNOWLEDGEMENTS
Stacy Beck provided invaluable technical help. We thank Dr Luc Bélanger for suggestions and for plasmids pCI-FTF, pCI-FTFΔLBD and pCI-FTFΔAF-2, Drs David Moore and Yoon-Kwang Lee for SHP plasmids and Dr David W. Russell for anti-FTF antibodies. We are grateful to Dr Hylemon for critical comments and support. We also thank Dr Wells for discussions and critical review of the manuscript. This research was supported in part by American Heart Association Grant-in-Aid 9950042N and National Institutes of Health Grants DK44218 and DK 38030.
References
- 1.Russell D.W. (1999) Nuclear orphan receptors control cholesterol catabolism. Cell, 97, 539–542. [DOI] [PubMed] [Google Scholar]
- 2.Javitt N.B. (1994) Bile acid synthesis from cholesterol: regulatory and auxiliary pathways. FASEB J., 8, 1308–1311. [DOI] [PubMed] [Google Scholar]
- 3.Eggertsen G., Olin,M., Andersson,U., Ishida,H., Kubota,S., Hellman,U., Okuda,K.-I. and Björkhem,I. (1996) Molecular cloning and expression of rabbit sterol 12α-hydroxylase. J. Biol. Chem., 271, 32269–32275. [DOI] [PubMed] [Google Scholar]
- 4.Nuclear Receptors Nomenclature Committee (1999) A unified nomenclature system for the nuclear receptor superfamily. Cell, 97, 161–163. [DOI] [PubMed] [Google Scholar]
- 5.Galarneau L., Pare,J.F., Allard,D., Hamel,D., Levesque,L., Tugwood,J.D., Green,S. and Belanger,L. (1996) The alpha1-fetoprotein locus is activated by a nuclear receptor of the Drosophila FTZ-F1 family. Mol. Cell. Biol., 16, 3853–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nitta M., Ku,S., Brown,C., Okamoto,A.Y. and Shan,B. (1999) CPF: an orphan nuclear receptor that regulates liver-specific expression of the human cholesterol 7α-hydroxylase gene. Proc. Natl Acad. Sci. USA, 96, 6660–6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodwin B., Jones,S.A., Price,R.R., Watson,M.A., McKee,D.D., Moore,L.B., Galardi,C., Wilson,J.G., Lewis,M.C., Roth,M.E. et al. (2000) A regulatory cascade of the nuclear receptors FXR, SHP-1 and LRH-1 represses bile acid biosynthesis. Mol. Cell, 6, 517–526. [DOI] [PubMed] [Google Scholar]
- 8.Lu T.T., Makishima,M., Repa,J.J., Schoonjans,K., Kerr,T.A., Auwerx,J. and Mangelsdorf,D.J. (2000) Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell, 6, 507–515. [DOI] [PubMed] [Google Scholar]
- 9.Gupta S., Stravitz,R.T., Dent,P. and Hylemon,P.B. (2001) Down-regulation of cholesterol 7α-hydroxylase (CYPA1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J. Biol. Chem., 276, 15816–15822. [DOI] [PubMed] [Google Scholar]
- 10.Seol W., Choi,H.S. and Moore,D.D. (1996) An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science, 272, 1336–1339. [DOI] [PubMed] [Google Scholar]
- 11.Lee Y.K., Dell,H., Dowhan,D.H., Hadzopoulou-Cladaras,M. and Moore,D.D. (2000) The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression. Mol. Cell. Biol., 20, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seol W., Chung,M. and Moore,D.D. (1997) Novel receptor interaction and repression domains in the orphan receptor SHP. Mol. Cell. Biol., 17, 7126–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seol W., Hanstein,B., Brown,M. and Moore,D.D. (1998) Inhibition of estrogen receptor action by the orphan receptor SHP (short heterodimer partner). Mol. Endocrinol., 12, 1551–1557. [DOI] [PubMed] [Google Scholar]
- 14.Andersson U., Yang,Y.Z., Bjorkhem,I., Einarsson,C., Eggertsen,G. and Gafvels,M. (1999) Thyroid hormone suppresses hepatic sterol 12alpha-hydroxylase (CYP8B1) activity and messenger ribonucleic acid in rat liver: failure to define known thyroid hormone response elements in the gene. Biochim. Biophys. Acta, 1438, 167–174. [DOI] [PubMed] [Google Scholar]
- 15.Vlahcevic Z.R., Eggerten,G., Björkhem,I., Hylemon,P.B., Redford,K. and Pandak,W.M. (2000) Regulation of sterol 12α-hydroxylase and cholic acid biosynthesis in the rat. Gastroenterology, 118, 599–607. [DOI] [PubMed] [Google Scholar]
- 16.del Castillo-Olivares A. and Gil,G. (2000) α1-Fetoprotein transcription factor is required for the expression of sterol 12α-hydroxylase, the specific enzyme for cholic acid synthesis. J. Biol. Chem., 275, 17793–17799. [DOI] [PubMed] [Google Scholar]
- 17.Hunt M.C., Yang,Y.Z., Eggertsen,G., Carneheim,C.M., Gafvels,M., Einarsson,C. and Alexson,S.E. (2000) The peroxisome proliferator-activated receptor alpha (PPARalpha) regulates bile acid biosynthesis. J. Biol. Chem., 275, 28947–28953. [DOI] [PubMed] [Google Scholar]
- 18.Stroup D. and Chiang,J.Y. (2000) HNF4 and COUP-TFII interact to modulate transcription of the cholesterol 7alpha-hydroxylase gene (CYP7A1). J. Lipid Res., 41, 1–11. [PubMed] [Google Scholar]
- 19.Subramanian A., Teixeira,J., Wang,J. and Gil,G. (1995) A STAT factor mediates the sexually dimorphic regulation of hepatic cytochrome P450 3A10/lithocholic acid 6β-hydroxylase gene expression by growth hormone. Mol. Cell. Biol., 15, 4672–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian A., Wang,J. and Gil,G. (1998) STAT-5 and NF-Y are involved in expression and growth hormone-mediated sexually dimorphic regulation of cytochrome P450 3A10/lithocholic acid 6β-hydroxylase. Nucleic Acids Res., 29, 2173–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.del Castillo-Olivares A. and Gil,G. (2000) Role of FXR and FTF on the bile acid-mediated suppression of cholesterol 7α-hydroxylase transcription. Nucleic Acids Res., 28, 3587–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leff T., Reue,K., Melian,A., Culver,H. and Breslow,J.L. (1989) A regulatory element in the ApoCIII promoter that directs hepatic specific transcription binds to proteins in expressing and nonexpressing cell types. J. Biol. Chem., 264, 16132–16137. [PubMed] [Google Scholar]
- 23.Ladias J.A., Hadzopoulou-Cladaras,M., Kardassis,D., Cardot,P., Cheng,J., Zannis,V. and Cladaras,C. (1992) Transcriptional regulation of human apolipoprotein genes ApoB, ApoCIII and ApoAII by members of the steroid hormone receptor superfamily HNF-4, ARP-1, EAR-2 and EAR-3. J. Biol. Chem., 267, 15849–15860. [PubMed] [Google Scholar]
- 24.Gilbert S., Galarneau,L., Lamontagne,A., Roy,S. and Belanger,L. (2000) The hepatitis B virus core promoter is strongly activated by the liver nuclear receptor fetoprotein transcription factor or by ectopically expressed steroidogenic factor 1. J. Virol., 74, 5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makishima M., Okamoto,A.Y., Repa,J.J., Tu,H., Learned,R.M., Luk,A., Hull,M.V., Lustig,K.D., Mangelsdorf,D.J. and Shan,B. (1999) Identification of a nuclear receptor for bile acids. Science, 284, 1362–1365. [DOI] [PubMed] [Google Scholar]