

Abstract
Smenamides are an intriguing class of peptide/polyketide molecules of marine origin showing antiproliferative activity against lung cancer Calu-1 cells at nanomolar concentrations through a clear pro-apoptotic mechanism. To probe the role of the activity-determining structural features, the 16-epi-analogue of smenamide A and eight simplified analogues in the 16-epi series were prepared using a flexible synthetic route. The synthetic analogues were tested on multiple myeloma (MM) cell lines showing that the configuration at C-16 slightly affects the activity, since the 16-epi-derivative is still active at nanomolar concentrations. Interestingly, it was found that the truncated compound 8, mainly composed of the pyrrolinone terminus, was not active, while compound 13, essentially lacking the pyrrolinone moiety, was 1000-fold less active than the intact substance and was the most active among all the synthesized compounds.
Keywords: smenamides, marine natural products, peptide/polyketide molecules, synthetic analogues, functional-analogues, antiproliferative activity, MM cell line
1. Introduction
Marine sponges, together with their symbiotic microorganisms, have proven to be a rich source of skeletally new substances [1,2,3], which have often inspired novel strategies in anticancer drug discovery. Targeted cancer therapies consist of “drugs” which interfere with specific molecules necessary for tumor growth and progression. A primary goal of these therapies is to fight cancer cells with more precision without hitting normal cells. These drugs are classified into monoclonal antibodies, directed against antigens expressed on the neoplastic cell surface, and small molecules, usually designed to interfere with protein targets [4].
Smenamides A (1) and B (2) (Figure 1) are highly functionalized peptide/polyketide substances isolated by our group in 2013 from the Caribbean sponge Smenospongia aurea [5]. They have proven to be interesting for their structural features, such as the unusual N-methylacetamide western terminus, the dolapyrrolidone eastern terminus, typical of dolastatin-15 (3), a potent antimitotic agent derived from Dolabella auricularia [6], and the chlorovinyl functional group, common to some cyanobacterial metabolites, such as jamaicamides (4–6, Figure 1), isolated from Lyngbiamajuscula [7]. The only difference between the two smenamides resides in the configuration of the C-13/C-15 double bond positioned close to the middle part of the polyketide portion of the molecule. It has been speculated that this could determine a different overall shape and, as a consequence, the different biological behavior observed for smenamides [5].
Figure 1.

Smenamide A (1) and B (2), dolastatin-15 (3), and jamaicamides (4–6). Configuration at C-16 in smenamide A as determined by synthesis [8].
Smenamides have proven to be active in blocking the proliferation of the Calu-1 cancer cell line at nanomolar concentrations, working, however, with a different mechanism of action. Smenamide A, more interestingly, acts with a clear pre-apoptotic mechanism proving to be the more promising as a lead compound. It is worth stating that the configuration at C-16 in both smenamides remained unassigned in the original study due to the limited amount of the natural substances available. In a recent study, a chiral protocol strategy aimed at the total synthesis of the smenamide family was designed, starting from commercially available S-citronellene, a cheap starting material [8]. Two stereoisomers of smenamide A, namely ent-smenamide A and 16-epi-smenamide A (7, Figure 2), were synthesized. This synthetic effort allowed us to determine the C-16 configuration of smenamide A as R (Figure 1), as well as to develop a flexible synthetic route towards this class of substances.
Figure 2.

16-epi-smenamide A (7) and its analogues 8–15. For structural comparison, numeration of analogues is in agreement with that of 16-epi-smenamide A.
In the present study, the antiproliferative activity of 16-epi-smenamide A has been evaluated on multiple myeloma (MM) cell lines. MM is a clonal plasma cell malignancy accounting for approximately 13% of all hematological cancers [9]. It originates from post-germinal centre B cells that accumulate somatic hypermutation and immunoglobulin heavy-chain class switching [8]. Several novel agents have been introduced into clinical practice but, after an initial response, most patients relapse or progress with a treatment-refractory disease [10]. For this reason, MM still proves to be incurable for most patients. In this scenario, it is necessary to develop new agents targeting novel pathways relevant for the MM cells, thereby increasing the range of available therapies.
In addition to 16-epi-smenamide A, the eight simplified synthetic analogues 8–15 (Figure 2) have also been synthesized. They were conceived as “functional-analogues” of smenamide A, incorporating some of the potential activity-determining structural features of the natural product. They were easily prepared thanks to the flexible nature of the previously developed synthetic route, with the aim of probing the importance of the main structural features of the smenamides, that is, the pyrrolinone, chlorovinyl and N-methylacetamido functional groups. In this paper, we illustrate a case-example of the application of this strategy to the design and study of functional-analogues of complex natural lead compounds.
2. Results and Discussion
2.1. Compounds 7–15
16-epi-smenamide A (7, Figure 2) is the C-16 epimer of the natural smenamide A (1, Figure 1). It was synthesized starting from S-citronellene using the chiral protocol previously reported. 16-epi-smenamide A was tested on SKM-M1 and RPMI-8226 cells, two MM cell lines, showing it to be able to reduce cell viability in a dose-dependent way at nanomolar concentrations (see Section 2.2). We demonstrated that 16-epi-smenamide A, despite possessing the opposite configuration at C-16, retains the potent antiproliferative activity shown by the natural compound, smenamide-A, thus suggesting that this configuration does not affect the nature of its activity. Therefore, as a working hypothesis for the design of simplified analogues of 16-epi-smenamide A (7), this compound was hypothetically disconnected into two main building blocks, corresponding to the polyketide and the peptide moieties. To probe the importance of the main structural features of smenamides, eight “functional-analogues” of 16-epi-smenamide-A were prepared. In particular, the truncated compound 8, retaining the C1–C18 portion of smenamide A, was synthesized to investigate the role of the pyrrolinone moiety. Compounds 9–12, in turn, represent the simplified C15–C27 polyketide portion and retain only the chlorovinyl and N-methylacetamide functional groups. They also served to investigate the role of the geometric isomerism around the C20/C21 double bond. The modulation of the polarity within the 9/11 and 10/12 pairs was achieved by acetylation. Ester 13, only lacking the pyrrolinone moiety, was prepared to simulate the entire polyketide portion, while compound 14 and its acetyl-derivative 15 allowed us to investigate the role of the chlorine atom. In fact, it is well known that the presence of halogens in natural products is important for the modulation of the biological activity [11,12], as previously reported.
Thus, the activation of 2,4-dimethyl-2-pentenoic acid as the pentafluorophenylester (16) (Figure 3) and its subsequent coupling with the previously synthesized pyrrolinone subunit 17 [8], afforded compound 8 in an 85% yield.
Figure 3.

Preparation of pyrrolinone derivative 8.
Ketone 18 (Figure 4) is a versatile intermediate to access 16-epi-smenamide analogues. It was easily prepared from commercially available S-citronellene, as depicted in Figure 4, and used as the starting material to obtain the seven analogues 9–15 by the introduction of the chlorovinyl, methylene and α,β-unsaturated ethyl ester functionalities (Figure 5). Thus, the Wittig olefination of 18 gave the two isomeric chlorovinyl derivatives 19 and 20 in a 3:2 ratio in favor of 19, which could be separated by column chromatography.
Figure 4.

Synthesis of the ketone intermediate 18.
Figure 5.

Preparation of compounds 9–13.
Deprotection of both 19 and 20 with tetrabutylammonium fluoride (TBAF) in tetrahydrofuran (THF) afforded alcohols 9 and 10, respectively, whose acetylation with Ac2O/pyridine gave the corresponding acetyl derivatives 11 and 12, respectively. In order to introduce the α,β-unsaturated ethyl ester function, the oxidation of 9 was accomplished with the Ley-Griffith method [tetrapropylammonium perruthenate (TPAP) (cat)/N-Methylmorpholine N-oxide (NMO)] [13,14,15] to give aldehyde 21 that was used in the subsequent Wittig reaction without further purification. Finally, the reaction with Ph3P=CH(Me)-CO2Et led to ethyl ester 13 in a 70% yield.
The methylene derivatives 14 and 15 were prepared by Wittig olefination of 18 using methylenetriphenylphosphorane (Figure 6). In particular, the first obtained product 22 was deprotected with TBAF in THF to give the desired alcohol 14 whose acetylation with Ac2O/pyridine finally afforded the acetyl-derivative 15.
Figure 6.

Preparation of methylene derivatives 14 and 15.
All synthesized compounds were tested on RPMI-8226 cell lines, as described in Section 2.2. Compound 8, lacking the great part of the polyketide moiety, was not active at all. As for the truncated polyketide compounds 9–15, it was shown that only compound 13, essentially lacking the pyrrolinone terminus, retained a certain degree of activity. In particular, a 1000-fold decreased EC50 value resulted, compared to the intact parent substance 7. Equally, neither alcohols 9 and 10 nor the corresponding acetates 11 and 12, not the dechlorinated analogues 14 and 15 showed significant activities. On the other hand, when the activity data of compounds 9–12 are compared with those of 13, it is evident that the α,β-unsaturated ethyl ester function plays a role in the activity. In addition, even if it seems that the pyrrolinone terminus does not represent a crucial functional part of the molecule, its absence reduces the activity of 13 suggesting that it, or the entire C1–C15 unsaturated moiety, may be equally important for the full activity of smenamides, conferring rigidity to the molecule, possibly needed to exert the activity. However, these data alone do not allow us to speculate about the importance of the chlorine atom as well as of the configuration of the C20/C21 double bond on the activity.
2.2. In Vitro Evaluation of Activity on Multiple Myeloma Cell Lines
In order to study the in vitro effects of 16-epi-smenamide A (7) and its synthetic analogues 8–15, MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] assays were performed on SKM-M1 and RPMI-8226, MM cell lines, to evaluate their effects on cell viability. Compound 7, tested at increasing concentrations (10–300 nM) for 48 h, was shown to reduce cell viability in both MM cell lines in a dose-dependent way (Figure 7). More than 50% of viability reduction was observed between 30 and 50 nM concentration. EC50 for compound 7 was calculated as 44 nM in SKM-M1 cells, and 24 nM in RPMI-8226 cells, after 48 h of treatment.
Figure 7.

Viability of SKM-M1 and RPMI-8226 multiple myeloma (MM) cell lines was evaluated by MTS assay after treatment with compound 7 at different concentrations (10, 30, 50, 100 and 300 nM) for 48 h. Results are expressed as percent of cell viability normalized to dimethylsulfoxide (DMSO)-treated control cells. The line-graphs represent average with standard deviation (SD) from three independent experiments.
Likewise, compound 8 was used to treat SKM-M1 and RPMI-8226 cell lines at 50 nM, 100 nM, 1 µM, 5 µM and 10 µM concentrations, for 24, 48 and 72 h. MTS assays showed that compound 8 had no effect on cell viability on SKM-M1 cell line and negligible effect on RPMI-8226 cell viability (Figure 8). For this compound, EC50 was not calculated.
Figure 8.

Cell viability was evaluated by MTS assay after treatment at different concentrations (50 nM, 100 nM, 1 µM, 5 µM, 10 µM) for 24, 48 and 72 h with compound 8 on SKM-M1 and RPMI-8226 cell lines. Results are expressed as percent of cell viability normalized to DMSO-treated cells. The line-graphs represent average with SD from three independent experiments.
Because compound 7 resulted more active on the RPMI-8226 cell line, its synthetic analogues 9–15 were tested on this cell line at increasing concentrations (50 nM, 100 nM, 1 µM, 5 µM) for all time points (24, 48 and 72 h). As shown in Figure 9, compound 9–12, 14 and 15 have negligible effect on RPMI-8226 cell viability; while compound 13 was able to reduce cell viability reaching 80% of reduction at 5 µM, after 72 h of treatment. EC50 of compound 13 at 72 h was calculated as 1.1 µM.
Figure 9.

Cell viability was evaluated by MTS assay after treatment at different concentrations (50 nM, 100 nM, 1 µM, 5 µM) for 24, 48 and 72 h with compounds 9–15 on RPMI-8226 cell line. Results are expressed as percent of cell viability normalized to DMSO-treated cells. The line-graphs represent average with SD from three independent experiments.
Further investigation of the cell death mechanism was carried out using compound 13 (at 1 and 5 µM) to treat RPMI-8226 cells. Control experiments were carried out with dimethylsulfoxide (DMSO) as vehicle control, or with untreated cells. After 72 h of treatment, Annexin-V fluorescein isothiocyanate (FITC)/propidium iodide (PI) analyses were performed to evaluate whether the cytotoxic activity of compounds 13 was related to apoptosis induction. Data obtained showed that a significant increase of apoptotic cells at both concentrations occurred when cells are treated with compound 13 (5% of increase at 1 µM respect to control (* p < 0.05) and 66% at 5 µM (*** p < 0.001) (Figure 10a,b)). Moreover, compound 13 was able to significantly decrease the number of cell in G0/G1 phase and increase those in S phase at both concentrations (Figure 10c,d).
Figure 10.

Compound 13: cytofluorimetric evaluation of apoptosis/necrosis by the Annexin-V fluorescein isothiocyanate (FITC)/propidium iodide (PI) test (a,b) and cell cycle analysis by PI staining (c,d) on RPMI-8226 cell line, at 1 µM and 5 µM for 72 h. (a) Dot plots and (c) cell cycle histograms show a single representative experiment; (b,d) the bar-graphs represent average with S.D. (* p < 0.05, ** p < 0.01, *** p < 0.001).
3. Experimental Section
3.1. General Experimental Procedures
All reagents and anhydrous solvents were purchased (Aldrich and Fluka) at the highest commercial quality and used without further purification. Where necessary, flame-dried and argon-charged glassware was used. The reactions were monitored using thinlayer chromatography (TLC) carried out on precoated silica gel plates (Merck 60, F254, 0.25 mm thick). Merck silica gel (Kieselgel 40, particle size 0.063–0.200 mm) was used for the column chromatography. Na2SO4 was used as a drying agent for aqueous workup. Nuclear magnetic resonance (NMR) experiments were performed using Varian Unity Inova spectrometers at 400, 500, and 700 MHz in CDCl3. Proton chemical shifts were referenced to the residual CHCl3 signal (7.26 ppm). 13C-NMR chemical shifts were referenced to the solvent (77.0 ppm). Abbreviations for signal coupling are as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and b = broad. Optical rotations were measured using a JASCO P-2000 polarimeter at the sodium D line. High resolution mass spectra were recorded by infusion on a Thermo Linear Trap Quadrupole (LTQ) Orbitrap XL mass spectrometer equipped with an electrospray source in the positive mode using MeOH as the solvent.
See Supplementary Materials for all NMR spectra.
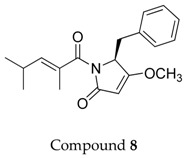
Compound 8. To a solution of 2,4-dimethyl-2-pentenoic acid (114 mg, 0.889 mmol) in EtOAc (4.0 mL), pentafluorophenol (188.2 mg, 1.02 mmol) and DCC (210.5 mg, 1.02 mmol) were added at 0 °C. The reaction mixture was stirred for 1 h at 0 °C and 3 h at room temperature and evaporated under reduced pressure to give 16 (185.9 mg, 0.632 mmol) that was used in the next step without further purification. 1H-NMR: (400 MHz, CDCl3): δ 6.90 (1H, d, J=9.75), 2.8–2.6 (1H, m), 1.95 (3H, s), 1.07 (6H, d, J = 6.6).
To a stirred solution of pyrrolinone 17 (126.6 mg, 0.624 mmol) [8] in THF (5.0 mL), nBuLi (0.390 mL, 0.632 mmol, 1.6 M soln in hexane) was added dropwise at −78 °C. After 15 min, a solution of pentafluorophenyl ester 16 (183.45 mg, 0.624 mmol) in THF (0.1 mL) was added via syringe. After 2 h, the reaction was quenched with a saturated aqueous NH4Cl solution (5 mL) and extracted with EtOAc (3 × 15 mL). The organic phase was washed with water (15 mL) and brine (15 mL), dried, and concentrated in vacuo. The crude was purified by preparative TLC (CHCl3/CH3OH, 98:2) to give 8 (166.2 mg, 0.530 mmol, 85%) as colourless oil. = +22.1 (c = 10, CHCl3); 1H-NMR: (400 MHz, CDCl3): δ 7.23–7.17 (3H, m, ArH), 7.0–6.9 (2H, m, ArH), 5.62 (1H, d, J = 9.47), 5.01–4.96 (1H, m), 4.84 (1H, s), 3.87 (3H, s, OCH3), 3.39 (1H, dd, J = 14.1, 5.4, Ha-7), 3.15 (1H, dd, J = 14.1, 2.0, Hb-7), 2.68–2.54 (1H, m), 1.8 (3H, s), 0.99 (6H, d, J = 6.5); 13C-NMR (100 MHz, CDCl3): δ 177.2, 171.3, 168.8, 145.2, 134.4, 129.8, 129.4, 128.1, 127.0, 94.8, 59.1, 58.3, 33.9, 27.4, 21.9, 21.5, 13.3; HRMS (ESI) m/z calcd. for C13H25ClNO2 [M + H]+ 262.1568, found 262.1566.
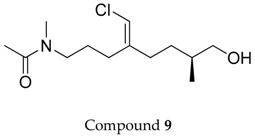
A mixture of compounds 19 and 20 was prepared as previously described [8]. Pure 19 and 20 were obtained by silica gel chromatography (hexane-EtOAc, 1:2). Deprotection of 19, as reported [5] afforded alcohol 9 as colourless oil. = −63.4 (c = 1.5, CHCl3); 1H-NMR: (400 MHz, CDCl3, mixture of rotamers): δ 5.86 (0.4H, s, vinyl proton), 5.82 (0.6H, s, vinyl proton), 3.46 (2H, t, J = 5.3), 3.42–3.24 (2H, m’s), 2.99 (1.8H, s, H3-27), 2.89 (1.2H, s, H3-27), 2.27–2.02 (7H, overlapped signals including two singlets at 2.09 and 2.07 for H3-26), 1.78–1.52 (4H, m), 1.30–1.15 (1H, m), 0.93, 0.91 (overall 3H, overlapped d’s, both J = 6.0, H3-17); 13C-NMR (100 MHz, CDCl3): δ 170.6, 170.4, 142.0, 141.3, 113.2, 112.6, 67.8, 67.7, 50.5, 47.3, 36.1, 35.2, 33.2, 32.3, 32.2, 31.1, 31.0, 27.4, 27.3, 25.8, 24.6, 21.9, 21.2, 16.44, 16.38; HRMS (ESI) m/z calcd. for C13H25ClNO2 [M + H]+ 262.1568, found 262.1566.
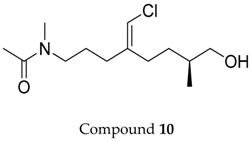
To a solution of 20 (3.9 mg, 0.008 mmol) in THF (0.6 mL), TBAF (0.012 mL, 0.012 mmol, 1.0 M solution in THF) was added at 0 °C. The reaction mixture was allowed to reach room temperature and stirred for 1 h. Then, the reaction was quenched with a satd. aq. solution of NH4Cl (0.5 mL). The phases were separated, and the aqueous layer was extracted with EtOAc (3 × 3 mL). The combined organic phases were dried and evaporated in vacuo. The crude was subjected to High Performance Liquid Chromatography (HPLC) separation [column Ascentis Si (Supelco), 25 cm × 4.6 mm, 5 µm; eluent: n-hexane/isopropanol 7:3, flow rate 1 mLmin−1] to give alcohol 10 (1.0 mg, 48%, tR = 14.5 min) as colourless oil. = +12.1 (c = 0.13; CHCl3); 1H-NMR (400 MHz, CDCl3, mixture of rotamers): δ 5.83 (0.4H, s, vinyl proton), 5.81 (0.6H, s, vinyl proton), 3.50 (2H, bt, J = 5.7), 3.34, 3.26 (1H each, both t, J = 7.5, H2-24), 2.98 (1.8H, s, H3-27), 2.92 (1.2H, s, H3-27), 2.31–2.17 (2H, m), 2.11–2.05 (5H, overlapped signals including a singlet at 2.08 for H3-26); 1.75–1.53 (4H, overlapped multiplets); 1.29–1.19 (1H, m), 0.98, 0.96 (overall 3H, overlapped doublets, both J = 6.1, H3-17); 13C-NMR (100 MHz, CDCl3): δ 170.7, 170.5, 142.0, 141.4, 113.0, 112.5, 67.9, 50.2, 47.4, 36.25, 36.0, 35.7, 35.6, 33.2, 32.2, 31.7, 30.33, 30.28, 27.6, 27.5, 26.2, 25.2, 22.0, 16.4; HRMS (ESI) m/z calcd. for C13H25ClNO2 [M + H]+ 262.1568; found 262.1566.
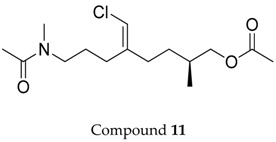
To a stirred solution of alcohol 9 (1.4 mg, 0.005 mmol) in pyridine (0.6 mL), excess acetic anhydride (0.4 mL) was added at rt. After 2 h the reaction mixture was evaporated under reduced pressure. The crude was subjected to HPLC separation [column Ascentis Si (Supelco), 25 cm × 4.6 mm, 5 µm; eluent: n-hexane/isopropanol 75:25, flow rate 1 mLmin−1] to give acetyl derivative 11 as a colourless oil (1.5 mg, 0.0047 mmol, 95%). = +5.1 (c = 0.12, CHCl3); 1H-NMR: (400 MHz, CDCl3, mixture of rotamers): δ 5.87 (0.4H, s, vinyl proton), 5.82 (0.6H, s, vinyl proton), 3.98–3.85 (2H, m), 3.39 (1.2H, t, J = 6.7, H2-24), 3.29 (0.8H, t, J = 6.7, H2-24), 3.00 (1.8H, s, H3-27), 2.93 (1.2H, s, H3-27), 2.27–2.03 (10H, overlapped signals including singlets at 2.10, 2.09 and 2.07 for acetates), 1.80–1.54 (4H, m), 1.57–1.47 (1H, m), 1.31–1.21 (1H, m), 0.95, 0.93 (overall 3H, overlapped d’s, both J = 6.0, H3-17); 13C-NMR (100 MHz, CDCl3): δ 141.9, 141.7, 141.3, 141.0, 113.5, 112.7, 68.92, 68.83, 50.5, 47.2, 36.0, 33.2, 32.12, 32.11, 32.09, 32.08, 31.31, 31.29, 31.27, 31.26, 27.45, 27.40, 27.38, 25.8, 24.7, 21.2, 20.9, 16.7; HRMS (ESI) m/z calcd. for C15H27ClNO3 [M + H]+ 304.1674, found 304.1669.
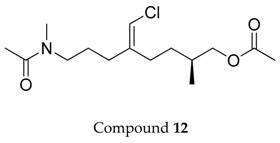
To a stirred solution of alcohol 10 (1.2 mg, 0.004 mmol) in pyridine (0.5 mL), excess acetic anhydride (0.4 mL) was added at room temperature. After 2 h the reaction mixture was evaporated under reduced pressure. The crude was subjected to HPLC separation [column Ascentis Si (Supelco), 25 cm × 4.6 mm, 5 µm; eluent: n-hexane/isopropanol 75:25, flow rate 1 mLmin−1] to give acetyl derivative 12 as colourless oil (1.0 mg, 0.003 mmol, 75%). = +12.88 (c = 0.06; CHCl3); 1H-NMR: (500 MHz, CDCl3, mixture of rotamers): δ 5.83 (0.4H, s, vinyl proton), 5.82 (0.6H, s, vinyl proton), 3.99–3.88 (2H, m), 3.34 (1.2H, t, J = 7.6, H2-24), 3.26 (0.8H, t, J = 7.6, H2-24), 2.98 (1.8H, s, H3-27), 2.91 (1.2H, s, H3-27), 2.27–2.20 (3H, m,), 2.10–2.03 (7H, overlapped signals including singlets at 2.08, 2.07 and 2.06 for acetates), 1.85–1.45 (5H, m), 1.32–1.23 (1H, m), 0.99, 0.98 (overall 3H, overlapped d’s, both J = 6.0, H3-17); 13C-NMR (100 MHz, CDCl3): δ 171.4, 171.3, 170.6, 141.7, 141.0, 113.24, 112.7, 112.6, 69.0, 68.9, 50.2, 47.2, 36.2, 33.2, 32.44, 32.40, 31.7, 30.5, 27.5, 27.4, 26.1, 25.2, 22.0, 21.1 16.7; HRMS (ESI) m/z calcd. for C15H27ClNO3 [M + H]+ 304.1674; found 304.1671.
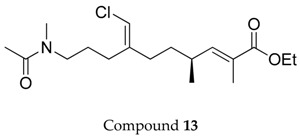
Compound 13 was prepared from alcohol 9 as previously described [8]. = +127.4 (c = 0.5, CHCl3); IR (neat) νmax: 2957, 2927, 2858, 1707, 1651, 1596, 1459, 1424, 1373, 1262, 1122 cm−1; 1H-NMR (400 MHz, CDCl3, mixture of rotamers): δ 6.49 (1H, d, J = 10.1, H-15), 5.82 (0.5H, s, vinyl proton), 5.76 (0.5H, s, vinyl proton), 4.18 (2H, q, J = 7.0, OCH2CH3), 3.37, 3.27 (1H each, both t, J = 7.6, H2-24), 2.99 (1.5H, s, H3-27), 2.91 (1.5H, s, H3-27), 2.46 (1H, m, H-16), 2.18 (2H, m), 2.09 (1.5H, s, H3-26), 2.08 (1.5H, s, H3-26), 2.01 (2H, t, J = 8.6), 1.83 (1.5H, d, J = 1.2, H3-14), 1.82 (1.5H, d, J = 1.2, H3-14), 1.30 (3H, t, J = 7.0, OCH2CH3), 1.02 (1.5H, d, J = 6.6, H3-17), 1.00 (1.5H, d, J = 6.6, H3-17); 13C-NMR (100 MHz, CDCl3) δ 170.5, 170.3, 168.3, 168.2, 146.9, 146.6, 141.6, 140.8, 132.1, 132.0, 131.94, 131.91, 128.5, 128.4, 127.2, 127.0, 113.4, 112.7, 60.6, 60.5, 50.4, 47.1, 36.0, 34.7, 34.6, 33.1, 32.7, 27.4, 27.3, 25.7, 24.6, 21.9, 21.3, 20.01, 19.98, 14.3, 12.63, 12.61; HRMS (ESI) m/z calcd. for C18H30ClNNaO3 [M + Na]+ 366.1812; found 366.1802.
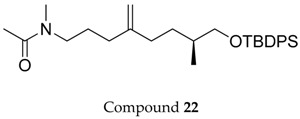
To a stirred suspension of methylenetriphenylphosphorane (6.6 mg, 0.024 mmol) in THF (0.5 mL), nBuLi (0.015 mL, 0.024 mmol, 1.6 M sol. in hexane) was added dropwise at 0 °C under argon. After 30 min at 0 °C, a solution of ketone 18 (5.5 mg, 0.012 mmol) in dry THF (0.3 + 0.3 mL rinse) was added, and the mixture was allowed to reach room temperature. After 4 h, the reaction was quenched with a saturated aqueous NH4Cl solution (2 mL) and extracted using Et2O (3× 5 mL). The organic phase was washed with brine, dried, and evaporated under reduced pressure. The crude was purified by preparative TLC (chloroform/methanol 95:5) affording compound 22 colourless oil (4.5 mg, 0.096 mmol, 80%) as a colourless oil. 1H-NMR (400 MHz, CDCl3, mixture of rotamers): δ 7.66 (4H, J = 6.9, ArH), 7.44–7.35 (6H, m, ArH), 4.76 (0.5H, s, methylene proton), 4.72 (0.5H, s, methylene proton), 4.71 (1H, s, methylene protons), 3.53–3.44 (2H, m), 3.34, 3.23 (1H each, both t, J = 7.6, H2-24), 2.96 (1.5H, s, H3-27), 2.90 (1.5H, s, H3-27), 2.07 (3H, s, H3-26), 2.05–1.92 (4H, m), 1.74–1.56 (4H, m), 1.32–1.17 (1H, m), 1.05 (9H, s, C(CH3)3), 0.93 (3H, d, J = 6.5, H3-17); 13C-NMR (100 MHz, CDCl3): δ 170.4, 149.2, 148.4, 135.6, 134.0, 133.9, 129.52, 129.48, 109.5, 108.9, 68.8, 68.7, 50.5, 47.4, 36.1, 35.4, 33.45, 33.38, 33.2, 32.8, 31.2, 29.7, 26.9, 26.1, 25.3, 21.9, 21.2, 19.3, 16.7; HRMS (ESI) m/z calcd. for C29H43NO2Si [M + H]+ 466.3136; found 466.3124.
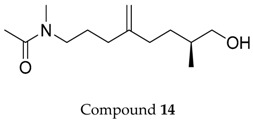
To a solution of 22 (5.4 mg, 0.012 mmol) in THF (0.8 mL), TBAF (0.017 mL, 0.017 mmol, 1.0 M solution in THF) was added at 0 °C. The reaction mixture was allowed to reach rt and stirred for 1 h. Then, the reaction was quenched with a satd. aq. solution of NH4Cl (1 mL). The phases were separated, and the aqueous layer was extracted with EtOAc (3 × 5 mL). The combined organic phases were dried and evaporated in vacuo. The crude was subjected to HPLC separation [column Ascentis Si (Supelco), 25 cm × 4.6 mm, 5 µm; eluent: ethyl acetate/isopropanol 9:1, flow rate 1 mLmin−1] to give alcohol 14 (1.9 mg, 0.008 mmol, 70%) as colourless oil. = +7.24 (c = 0.07; CHCl3); 1H-NMR (400 MHz, CDCl3, mixture of rotamers): δ 4.79 (0.5H, s, methylene proton), 4.75 (1.5H, bs, methylene protons), 3.54–3.43 (2H, m), 3.41–3.30 (1H, m, H2-24), 3.27 (1H, t, J=7.4, H2-24), 2.99 (1.5H, s, H3-27), 2.92 (1.5H, s, H3-27), 2.12 1.98 (overall 7H, including singlets at 2.09 and 2.07 for H3-26), 1.75–1.50 (4H, m), 1.32–1.19 (1H, m), 0.95, 0.93 (overall 3H, overlapped d’s, J = 6.5, H3-17); 13C-NMR (100 MHz, CDCl3): δ 170.6, 150.8, 148.8, 148.3, 109.6, 109.2, 68.14, 68.10, 50.4, 47.5, 36.3, 35.4, 33.4, 33.2, 33.1, 33.0, 32.1, 31.08, 31.03, 29.7, 26.0, 25.1, 21.3, 16.6, 16.5; HRMS (ESI) m/z calcd. for C13H26NO2 [M + H]+ 228.1958; found 228.1956.

To a stirred solution of alcohol 14 (1.5 mg, 0.006 mmol) in pyridine (0.2 mL), excess acetic anhydride (0.2 mL) was added at rt. After 2 h the reaction mixture was evaporated under reduced pressure. The crude was subjected to HPLC separation [column Ascentis Si (Supelco), 25 cm × 4.6 mm, 5 µm; eluent: n-hexane/isopropanol 75:25, flow rate 1 mLmin−1] to give acetyl derivative 15 as a colourless oil (1.0 mg, 0.004 mmol, 62%). = +13.63 (c = 0.07; CHCl3); 1H-NMR (400 MHz, CDCl3, mixture of rotamers): δ 4.78 (0.5H, s, methylene proton), 4.75 (0.5H, s, methylene proton), 4.74 (1H, s, methylene protons), 3.99–3.84 (2H, m), 3.36, 3.26 (1H each, both t, J = 7.6, H2-24), 2.98 (1.5H, s, H3-27), 2.92 (1.5H, s, H3-27), 2.12–1.96 (10H, overlapped signals including singlets at 2.09, 2.08 and 2.06 for acetates) 1.82–1.60 (4H, m), 1.34–1.22 (1H, m), 0.95, 0.94 (overall 3H, overlapped d’s, J = 6.5, H3-17); 13C-NMR (175 MHz, CDCl3): δ 170.42, 171.36, 170.6, 141.7, 141.05, 113.24, 112.6, 112,65, 69.0, 68.9, 50.2, 47.2, 36.2, 33.2, 32.43, 32.40, 31.9, 31.7, 30.5, 27.5, 27.4, 26.1, 25.2, 22.0, 21.4, 21.1, 16.7; HRMS (ESI) m/z calcd. for C15H27NO3 [M + H]+ 270.2063; found 270.2061.
3.2. Biological Activity
3.2.1. Cell Lines and Chemical
Human MM cell lines, SKM-M1 and RPMI-8226, were cultured in RPMI 1640 (Gibco, Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS. Gibco, Life Technologies, Carlsbad, CA, USA), 1% of penicillin-streptomycin (Gibco) at 37 °C and 5% CO2.
All chemical compounds were dissolved in DMSO (Sigma Aldrich, St. Louis, MO, USA) and diluted in FBS for cell treatments.
3.2.2. Cell Viability
SKM-M1 and RPMI-8226 cell lines were seeded into 96-well plates (3 × 104 cells/100 μL) and incubated with all compounds at increasing concentrations for different time points. In particular, compound 7 was used at concentrations 10–300 nM for 48 h; compound 8 at 50–10 µM for 24, 48 and 72 h; compounds 9–15 at 50–5 µM for 24, 48 and 72 h. Cells treated with the DMSO vehicle were used as control. Cell viability was determined using the CellTiter 96 Aqueous One Solution assay kit (MTS, Promega, Madison, WI, USA). The optical density was measured at 492 nm by plate reader (Das srl, Rome, Italy). Cellular viability was calculated as percentage of viable cells compared with DMSO control. All experiments were conducted in triplicate. EC50 values were obtained by GraphPad Prism (GraphPad Prism, San Diego, CA, USA).
3.2.3. Functional Tests
RPMI-8226 cell line was treated with 1 μM and 5 μM of compound 13 or with DMSO vehicle or not treated for 72 h (cell density 3×105 cells/mL) and used in:
- Apoptosis assay
Apoptosis of RPMI-8226 was evaluated by cytometric analysis of Annexin V and PI-stained cells using fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection kit I (Becton Dickinson, BD, Franklin, NJ, USA) [16]. Samples were prepared following the manufacturer’s instructions; stained cells were acquired using NAVIOS flow cytometer (Beckman Coulter, Brea, CA, USA) and analyzed by Kaluza software (Beckman Coulter). 10,000 events were acquired for each samples; single positive for Annexin V and double positive for Annexin V and PI cells were interpreted as signs of early and late phases of apoptosis respectively. Percent of apoptotic cells was obtained from the sum of early and late apoptosis.
- Cell cycle analysis
After treatment RPMI-8226 cells were fixed in cold ethanol 70% for 1 h, then labeled with PI (Sigma Aldrich, St. Louis, MO, USA)/RNase A (EuroClone S.p.a., Pero, MI, Italy) staining solution for 30 min. Samples were acquired by NAVIOS flow cytometer and analyzed by Kaluza software (Beckman Coulter). 10,000 events were acquired for each sample.
3.2.4. Statistical Analysis
Statistical significance was determined using a paired t test by GraphPad Prism. All error bars represent the standard deviation (SD) of the average.
4. Conclusions
This study adds new knowledge about the antiproliferative activity and the possible role of smenamides, chlorinated peptide/polyketide substances originally isolated from the Caribbean sponge Smenospongiaaurea, as lead compounds in anticancer drug research. Our results have shown that the configuration at C-16 slightly affects the activity, since the 16-epi-analogue 7 was still active at nanomolar concentrations. Interestingly, it has been found that the truncated compound 8, containing the pyrrolinone terminus, was not active while compound 13, composed of the intact C12–C27 portion, retained the activity, even though its EC50 value was 1000 times smaller compared with the parent 16-epi-smenamide 7. In addition, compound 13 was able to block the cell cycle at the G0/G1 phase. It is worth noting that smenothiazoles [17], biogenetically related but structurally different from smenamides, possess the same activity. This study provides the basic knowledge needed to design simplified and synthetically easily accessible analogues that could target MM cells.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/16/6/206/s1, Figure S1: 1H NMR spectrum of compound 16 (CDCl3, 400 MHz), Figure S2: 13C NMR spectrum of compound 16 (CDCl3, 100 MHz), Figure S3: 1H NMR spectrum of compound 8 (CDCl3, 400 MHz), Figure S4: 13C NMR spectrum of compound 8 (CDCl3, 100 MHz), Figure S5: 1H NMR spectrum of compound 10 (CDCl3, 400 MHz), Figure S6: 13C NMR spectrum of compound 10 (CDCl3, 100 MHz), Figure S7: 1H NMR spectrum of compound 11 (CDCl3, 400 MHz), Figure S8: 13C NMR spectrum of compound 11 (CDCl3, 100 MHz), Figure S9: 1H NMR spectrum of compound 12 (CDCl3, 400 MHz), Figure S10: 13C NMR spectrum of compound 12 (CDCl3, 100 MHz), Figure S11: 1H NMR spectrum of compound 22 (CDCl3, 400 MHz), Figure S12: 13C NMR spectrum of compound 22 (CDCl3, 100 MHz), Figure S13: 1H NMR spectrum of compound 14 (CDCl3, 400 MHz), Figure S14: 13C NMR spectrum of compound 14 (CDCl3, 100 MHz), Figure S15: 1H NMR spectrum of compound 15 (CDCl3, 400 MHz), Figure S16: 13C NMR spectrum of compound 15 (CDCl3, 100 MHz).
Author Contributions
Conceptualization, V.P. and V.C.; Data curation, D.L. and S.T.; Funding acquisition, V.C.; Investigation, A.C., I.L. and G.E.; Supervision, V.P. and V.C.; Writing—original draft, A.C. and I.L.; Writing—review & editing, V.P. and V.C.
Funding
This research was funded by the European Union 7th Framework Programme (BlueGenics, FP7-KBBE-2012-6) under grant agreement No. 311848 and of the Università degli Studi di Napoli Federico II under the STAR project named SeaLEADS.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of the data; in the writing of the manuscript, and in the decision to publish the results.
References
- 1.Costantino V., Fattorusso E., Imperatore C., Mangoni A. Glycolipids from sponges. Part 17.1 Clathrosides and isoclathrosides, unique glycolipids from the Caribbean sponge Agelas clathrodes. J. Nat. Prod. 2006;69:73–78. doi: 10.1021/np050331v. [DOI] [PubMed] [Google Scholar]
- 2.Costantino V., D’Esposito M., Fattorusso E., Mangoni A., Basilico N., Parapini S., Taramelli D. Damicoside from Axinella damicornis: The Influence of a Glycosylated Galactose 4-OH Group on the Immunostimulatory Activity of α-Galactoglycosphingolipids. J. Med. Chem. 2005;48:7411–7417. doi: 10.1021/jm050506y. [DOI] [PubMed] [Google Scholar]
- 3.Costantino V., Fattorusso E., Mangoni A., Perinu C., Teta R., Panza E., Ianaro A. Tedarenes A and B: Structural and stereochemical analysis of two new strained cyclic diarylheptanoids from the marine sponge Tedania ignis. J. Org. Chem. 2012;77:6377–6383. doi: 10.1021/jo300295j. [DOI] [PubMed] [Google Scholar]
- 4.Laurenzana I., Caivano A., La Rocca F., Trino S., De Luca L., D’Alessio F., Schenone S., Falco G., Botta M., Del Vecchio L., et al. A pyrazolo[3,4-d]pyrimidine compound reduces cell viability and induces apoptosis in different hematological malignancies. Front. Pharmacol. 2016;7:416. doi: 10.3389/fphar.2016.00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teta R., Irollo E., Della Sala G., Pirozzi G., Mangoni A., Costantino V. Smenamides A and B, chlorinated peptide/polyketide hybrids containing a dolapyrrolidinone unit from the Caribbean sponge Smenospongia aurea. Evaluation of their role as leads in antitumor drug research. Mar. Drugs. 2013;11:4451–4463. doi: 10.3390/md11114451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai R., Friedman S.J., Pettit G.R., Hamel E. Dolastatin 15, a potent antimitotic depsipeptide derived from Dolabella auricularia: Interaction with tubulin and effects on cellular microtubules. Biochem. Pharmacol. 1992;43:2637–2645. doi: 10.1016/0006-2952(92)90153-A. [DOI] [PubMed] [Google Scholar]
- 7.Edwards D.J., Marquez B.L., Nogle L.M., McPhail K., Goeger D.E., Roberts M.A., Gerwick W.H. Structure and Biosynthesis of the Jamaicamides, New Mixed Polyketide-Peptide Neurotoxins from the Marine Cyanobacterium Lyngbya majuscule. Chem. Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 8.Caso A., Mangoni A., Piccialli G., Costantino V., Piccialli V. Studies toward the Synthesis of Smenamide A, an Antiproliferative Metabolite from Smenospongia aurea: Total Synthesis of ent-Smenamide A and 16-epi-Smenamide A. ACS Omega. 2017;2:1477–1488. doi: 10.1021/acsomega.7b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.La Rocca F., Airoldi I., Di Carlo E., Marotta P., Falco G., Simeon V., Laurenzana I., Trino S., De Luca L., Todoerti K., et al. EphA3 targeting reduces in vitro adhesion and invasion and in vivo growth and angiogenesis of multiple myeloma cells. Cell. Oncol. 2017;40:483–496. doi: 10.1007/s13402-017-0338-4. [DOI] [PubMed] [Google Scholar]
- 10.Caivano A., La Rocca F., Laurenzana I., Annese T., Tamma R., Famigliari U., Simeon V., Trino S., De Luca L., Villani O., et al. Epha3 acts as proangiogenic factor in multiple myeloma. Oncotarget. 2017;8:34298–34309. doi: 10.18632/oncotarget.16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esposito G., Bourguet-Kondracki M.-L., Mai L.H., Longeon A., Teta R., Meijer L., Van Soest R., Mangoni A., Costantino V. Chloromethylhalicyclamine B, a Marine-Derived Protein Kinase CK1δ/ε Inhibitor. J. Nat. Prod. 2016;79:2953–2960. doi: 10.1021/acs.jnatprod.6b00939. [DOI] [PubMed] [Google Scholar]
- 12.Esposito G., Della Sala G., Teta R., Caso A., Bourguet-Kondracki M.-L., Pawlik J.R., Mangoni A., Costantino V. Chlorinated thiazole containing polyketide-peptides from the Caribbean sponge Smenospongia conulosa: Structure elucidation on microgram scale. EJOC. 2016;16:2871–2875. doi: 10.1002/ejoc.201600370. [DOI] [Google Scholar]
- 13.Ley S.V., Norman J., Griffith W.P., Marsden S.P. Tetrapropylammoniumperruthenate, Pr4N+RuO4−, TPAP: A catalytic oxidant for organic synthesis. Synthesis. 1994:639–666. doi: 10.1055/s-1994-25538.. [DOI] [Google Scholar]
- 14.Piccialli V. Ruthenium tetroxide and perruthenate chemistry. Recent advances and related transformations mediated by other transition metal oxospecies. Molecules. 2014;19:6534–6582. doi: 10.3390/molecules19056534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zerk T.J., Moore P.W., Harbort J.S., Chow S., Byrne L., Koutsantonis G.A., Harmer J.R., Martínez M., Williams C.M., Bernhardt P.V. Elucidating the mechanism of the Ley–Griffith (TPAP) alcohol oxidation. Chem. Sci. 2017;8:8435–8442. doi: 10.1039/C7SC04260D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laurenzana I., Caivano A., Trino S., De Luca L., La Rocca F., Simeon V., Tintori C., D’Alessio F., Teramo A., Zambello R., et al. A Pyrazolo[3,4-d]pyrimidine compound inhibits Fyn phosphorylation and induces apoptosis in natural killer cell leukemia. Oncotarget. 2016;7:65171–65184. doi: 10.18632/oncotarget.11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esposito G., Teta R., Miceli R., Ceccarelli L.S., Della Sala G., Camerlingo R., Irollo E., Mangoni A., Pirozzi G., Costantino V. Isolation and assessment of the in vitro anti-tumor activity of smenothiazole A and B, chlorinated thiazole-containing peptide/polyketides from the Caribbean sponge, Smenospongiaaurea. Mar. Drugs. 2015;13:444–459. doi: 10.3390/md13010444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.