

Abstract
CD4+ T lymphocytes are the principal target of HIV infection, but infected macrophages also contribute to viral pathogenesis. Killing of infected cells by CD8+ cytotoxic T lymphocytes (CTLs) leads to control of viral replication. Here we demonstrate that CTL killing of macrophages is impaired compared to killing of CD4+ T cells, resulting in inefficient HIV suppression. Macrophage killing depends on caspase-3 and granzyme B, whereas rapid killing of CD4+ T cells is caspase-independent and does not require granzyme B. Moreover, impaired killing of macrophages is associated with prolonged effector-target contact time and greater CTL interferon-γ expression, inducing macrophage production of pro-inflammatory chemokines that recruit monocytes and T cells. Similar results were observed when macrophages presented other viral antigens, suggesting a general mechanism for macrophage persistence as antigen-presenting cells that enhance inflammation and adaptive immunity. Inefficient CTL killing of macrophages may contribute to chronic inflammation, a hallmark of chronic HIV disease.
Accumulating evidence suggests that infected macrophages contribute to HIV persistence and pathogenesis. Whereas HIV-infected CD4+ T cells die within a few days of infection, in vitro studies suggest that macrophages are resistant to the cytopathic effects of HIV replication resulting in continuous viral propagation1. Moreover, infected macrophages efficiently disseminate virus to CD4+ T cells via neutralization-evading cell-to-cell spread2, 3, 4. Animal models of HIV infection further support in vivo infection and persistence of macrophages5, 6, 7, 8, even during combination antiretroviral therapy (cART)6, 8, and suggest macrophages contribute to pathogenesis9. In addition, infected myeloid cells and macrophages have been observed in the lung, gut and lymph tissues of HIV-infected patients (reviewed in10), including the brain, which contributes to the development of HIV-1 associated dementia and HIV-associated neurocognitive disorder (reviewed in11). Finally, macrophage-associated diseases, such as atherosclerosis, metabolic diseases and cancer, have been described in HIV+ subjects (reviewed in12), with chronic inflammation contributing to these comorbidities, which afflict cART-treated individuals13.
CD8+ cytotoxic T lymphocytes (CTL) control virus levels during acute and chronic stages of HIV infection and reduce HIV disease progression14, 15. Most studies have focused on CTL control of infected CD4+ T cells with less focus on infected macrophages. Previous work shows that HIV-specific CTL can eliminate HIV-infected macrophages in vitro16, 17, 18, 19. However, the relative efficiency of CTL-mediated killing of HIV-infected CD4+ T cells versus macrophages is poorly characterized. Studies suggest that SIV-infected macrophages are relatively resistant to CTL killing, but the mechanism behind their differential susceptibility is unknown20, 21. In fact, CTL killing of infected macrophages, unlike CD4+ T cells, appears to be relatively unaffected by Nef-mediated MHC-I downregulation16, 20. An improved understanding of CTL responses to HIV-infected macrophages will inform strategies to eliminate this population and combat HIV-associated inflammation.
Here, we characterize and compare the interactions of ex vivo HIV-specific CTLs with HIV-infected CD4+ T cell and macrophage targets. We show that macrophages are less susceptible to CTL-mediated killing than CD4+ T cells, and that this is an intrinsic characteristic of macrophages that is independent of HIV infection. Although CTL cytotoxic granules mediate killing of both cell types, CD4+ T cells undergo rapid caspase-independent cell death, while macrophages undergo a slower granzyme B- and caspase-3-dependent death. Inefficient CTL-mediated killing of macrophages drives prolonged synapse formation between effectors and targets, greater CTL secretion of IFN-γ (a major macrophage-activating cytokine) and induction of macrophage pro-inflammatory chemokines that recruit monocytes and T cells. Furthermore, similar results were observed for cytomegalovirus (CMV), Epstein-Barr Virus (EBV) and influenza virus (Flu) responses, indicating that delayed killing of macrophages by CTLs may be a general mechanism whereby antigen-presenting cells promote inflammation.
RESULTS
HIV-infected macrophages are inefficiently killed by CTLs
We developed an in vitro system to simultaneously study interactions of freshly isolated (“ex vivo”) CTLs with HIV-infected CD4+ T cells and macrophages (Supplementary Fig. 1). Because HIV controllers, who spontaneously control plasma viremia below 50 RNA copies/ml (elite controllers) or between 50-2000 RNA copies/ml (viremic controllers), exhibit potent ex vivo CTL responses to infected CD4+ T cells (reviewed in22) and macrophages18, 19, we used elite and viremic controller samples for this study. Monocyte–derived macrophages (MDM – differentiated using the growth factors GM-CSF and M-CSF) and activated CD4+ T cells were infected with HIV and co-cultured with autologous ex vivo CTL (isolated using negative enrichment kits that deplete NK cells). Elimination of HIV-infected Gag p24+ target cells was assessed by flow cytometry after four hours of co-culture (Fig. 1a, b, and Supplementary Fig. 2). Infected CD4+ T cells were more efficiently eliminated by autologous ex vivo CTL (57.0 ± 5.5%, mean ± SEM, residual Gag+ targets at an effector: target ratio of 4:1) than infected macrophages (94.3 ± 1.8% residual Gag+ targets, p < 0.0001). Killing was HIV-specific, as evidenced by the lack of killing using ex vivo CTL from uninfected healthy donors (Fig. 1b – dotted lines). Similar results were observed with MDM differentiated using human serum (Supplementary Fig. 3a, p < 0.0001), suggesting that this resistance to CTL-mediated killing is a general feature of macrophages, not only GM-CSF, M-CSF, or fetal bovine serum (FBS)-derived macrophages. To evaluate macrophage susceptibility to CTL-mediated killing further, we employed HIV peptide-expanded CTL effectors, which have been shown to kill macrophages within 4-24 hours16, 18, 20. These peptide-expanded cells killed HIV-infected macrophages within four hours of co-culture (65.0 ± 7.8% residual Gag+ targets) (Fig. 1c). However, infected macrophages still survived significantly more than infected CD4+ T cells (39.8 ± 5.3% residual Gag+ targets, p = 0.0121). Together, these data indicate that HIV-infected macrophages are more resistant to CTL-mediated killing than infected CD4+ T cells.
Fig. 1. HIV-infected macrophages are less susceptible to CTL-mediated killing compared to HIV-infected CD4+ T cells.
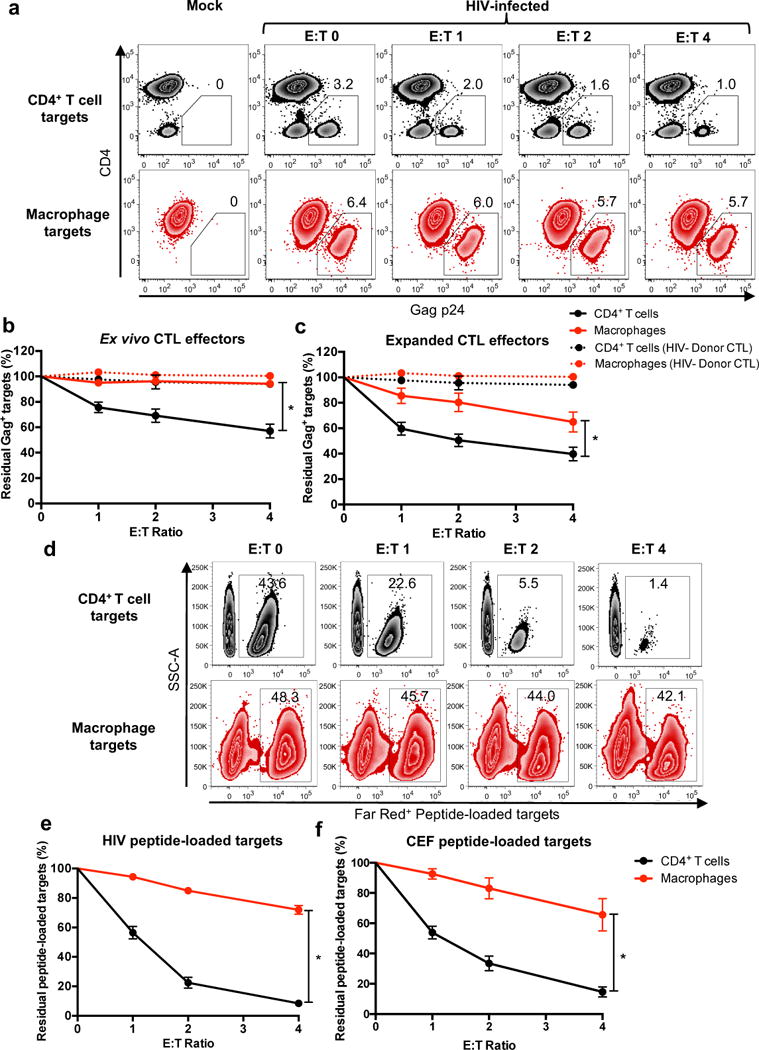
(a) HIV-infected target elimination assay. HIV-infected CD4+ T cells and macrophages were co-cultured with CTL effectors for four hours, followed by quantitation of infected cells by flow cytometry. Live infected cells are depicted in the gate outlining positive Gag-p24 intracellular staining with down-modulation of surface CD4, representing one of four independent experiments. See also Supplementary Figs. 1 and 2. (b) Summary of elimination assays using ex vivo CTL (n=16 distinct samples from four independent experiments) and (c) HIV peptide-expanded CTL from HIV-infected donors (n=16 distinct samples from four independent experiments) and CTL from HIV− healthy donors (n=3 distinct samples from two independent experiments) as a negative control. Shown are the means +/- SEM. Statistical analysis: two-sided unpaired t-test, *p<0.0001 for (b) and *p=0.0121 for (c). (d) Elimination assay using HIV peptide-loaded, uninfected targets. Activated CD4+ T cells and macrophages, each 50% loaded with HIV peptides and stained with CellTrace Far Red, were co-cultured with autologous HIV peptide-expanded CTL from an HIV+ donor for four hours followed by flow cytometry analysis. Shown is one of two independent experiments. (e) Summary of HIV peptide target elimination assays using CTL from HIV+ donors (n=8 distinct samples from two independent experiments). Shown are means +/- SEM. Statistical analysis: two-sided unpaired t-test, *p<0.0001. See also Supplementary Fig. 3. (f) Summary of CMV, EBV and Flu (CEF) peptide-loaded target cell elimination assays using CEF-specific CTL from HIV− donors (n=6 distinct samples from three independent experiments). Shown are the means +/- SEM. Statistical analysis: two-sided unpaired t-test, *p=0.0011.
Resistance to CTL-mediated killing is an intrinsic characteristic of macrophages
HIV infection enhances macrophage survival through multiple proposed mechanisms1, which might mediate resistance to CTL killing. To determine whether infection-associated differences in macrophage survival might explain the differences in killing between infected CD4+ T lymphocyte and macrophage targets, or whether this was an intrinsic property of macrophages, we next performed killing assays using uninfected, peptide-loaded macrophages and activated CD4+ T cells (Fig. 1d, e). HIV peptide-loaded macrophages were significantly more resistant to expanded CTL-mediated killing (72.0 ± 3.0% mean ± SEM, residual peptide-loaded targets) than CD4+ T lymphocytes (8.4 ± 1.8% residual peptide-loaded targets, p < 0.0001). Similar results were observed with ex vivo CTL (Supplementary Fig. 3b). Furthermore, there was no difference in CTL killing of activated CD4+ T cells or resting ex vivo CD4+ T cells (Supplementary Fig. 3c), suggesting that enhanced CD4+ T cell sensitivity to CTL-mediated killing is not an effect of activation. To determine whether these results extend beyond HIV, targets were loaded simultaneously with CMV, EBV and Flu (CEF) peptides and co-cultured with CEF peptide-expanded CTLs from HIV-negative donors. Similar to HIV peptide-loaded cells, CEF peptide-loaded macrophages exhibited relative resistance to CTL-mediated killing (65.6 ± 10.7% residual peptide-loaded targets) compared to CD4+ T cells (14.6 ± 3.4% residual peptide-loaded targets, p = 0.0011) (Fig. 1f). Together, these results indicate that resistance to CTL-mediated killing is an intrinsic feature of macrophages that does not depend on mode of differentiation or HIV infection.
Macrophages, unlike CD4+ T cells, die by slow caspase-3-dependent apoptosis, resulting in inefficient viral suppression
The above elimination assay measured the impact of HIV-specific CTLs on infected cell survival following a short incubation. To determine whether macrophages might still be killed, but more slowly, we co-cultured ex vivo CTL with peptide-loaded CD4+ T cells or macrophages for up to 24 hours and monitored target cell survival over time. In contrast to killing of peptide-loaded CD4+ T cells, which was detected within 4 hours, killing of macrophages was delayed until 12 hours (Fig. 2a). Furthermore, at the 24-hour time point, significantly more CD4+ T cells were eliminated (50.0% ± 5.5% residual peptide-loaded targets) compared to macrophages (72.3% ± 2.0% residual peptide-loaded targets, p = 0.0156). Similar trends were observed using expanded CTL effectors (Supplementary Fig. 4). Thus, for the macrophages that succumb to CTL-mediated killing, the kinetics of cell death are slower compared to activated CD4+ T cells.
Fig. 2. CTL induce delayed, caspase-3 dependent apoptosis of macrophages resulting in less efficient control of HIV infection.
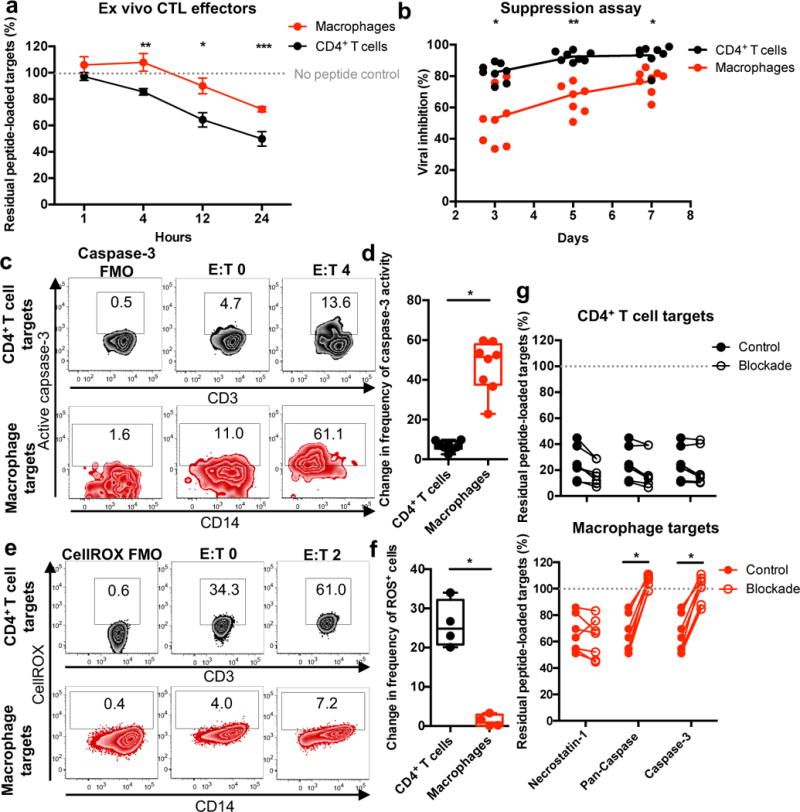
(a) Target killing time course. Peptide-loaded targets were incubated with ex vivo CTL from HIV+ donors (n=4 distinct samples from two independent experiments) for the indicated times, followed by analysis for live FarRed staining via flow cytometry. Shown are the means +/- SEM. Statistical analysis: two-sided unpaired t-test, *p=0.0201, **p=0.0165, ***p=0.0156. See also Supplementary Fig. 4. (b) Viral inhibition assay. Ex vivo CTL from HIV+ donors (n=8 distinct samples from three independent experiments) were co-cultured with HIV-infected CD4+ T cells or macrophages for seven days, followed by measurement of culture supernatant Gag-p24 antigen. Statistical analysis: two-sided unpaired t-test, * p=0.0005, **p<0.0001. (c) Caspase-3 activity was assessed in live target cells described in Fig. 1a. FMO: Fluorescence minus one (staining control). Shown is one representation of two independent experiments. (d) Caspase-3 activity in live targets. Summary of the absolute change in caspase-3 activity in live target cells for assays using expanded CTL from HIV+ patients (n=8 distinct samples from two independent experiments). Box elements, center line, limits and whiskers are the median, 25th-75th percentiles and min-max, respectively. Statistical analysis: two-sided unpaired t-test, *p<0.0001. (e) Detection of oxidative stress. Levels of reactive oxygen species (ROS) were measured in live target cells described in Fig. 1d. Shown is a representation of one of four samples from one experiment. (f) Detection of oxidative stress in live cells. Summary of the absolute change in frequency of ROS+ live target cells for assays using expanded CTL from HIV+ patients (n=4 distinct samples from one experiment). Box elements, center line, limits and whiskers are the median, 25th-75th percentiles and min-max, respectively. Statistical analysis: two-sided unpaired t-test, *p=0.0002. (g) Inhibiting target cell elimination. Peptide-loaded targets were pre-treated with inhibitors, followed by co-culture with expanded CTL for four hours and analyzed for live cell FarRed staining via flow cytometry. Shown are summaries from HIV+ patients (n=7 distinct samples from two independent experiments). Statistical analysis: two-sided paired t-test, *p<0.0001.
We next examined the ability of CTL to mediate viral suppression in macrophages and CD4+ T cells. We monitored Gag p24 antigen in culture supernatants of HIV-infected CD4+ T cells or macrophages, cultured for 7 days, with or without ex vivo CTL (Fig. 2b). CD4+ T cell infection was robustly suppressed on day 3 of co-culture (82.3 ± 2.0%, mean ± SEM, reduction in p24 compared to cultures without CTLs) and plateaued on day 5 of co-culture (92.6 ± 1.1% reduction in p24). In contrast, ex vivo CTLs were less efficient at suppressing macrophage infection after 3 days of co-culture (53.1 ± 6.2% reduced p24, p = 0.0005). Inhibition of macrophage infection remained significantly less than inhibition of CD4+ T cell infection even after 7 days of culture (p = 0.0005). These data indicate that slow macrophage killing results in less efficient suppression of HIV infection in macrophages than in CD4+ T cells.
Based on these results, we hypothesized that differential mechanisms of cell death might explain the relative delay in macrophage killing. CTLs trigger both caspase-dependent and independent cell death pathways. We assessed activation of caspase-3 (the primary executioner caspase23) in live HIV-infected target cells after four hours of co-culture with effectors (Fig. 2c and d). HIV-infected macrophages exhibited significantly more caspase-3 activity than infected CD4+ T cell targets following co-culture with expanded CTLs, as measured by a fixable caspase-3 activity indicator (p < 0.0001). In contrast, live CD4+ T cells exhibited significantly increased levels of reactive oxygen species (ROS) (resulting from disruption of the mitochondria, which occurs in both caspase-dependent and caspase-independent killer cell-mediated programmed cell death24) following co-culture with CTL (Fig. 2e and f; p = 0.0002). Furthermore, co-culture of expanded CTLs with peptide-loaded target cells treated with either pan-caspase or caspase-3 inhibitors did not inhibit CD4+ T cell killing, but dramatically blocked macrophage killing (from 65.7 ± 5.3%, mean ± SEM, residual peptide-loaded targets for the control to 105.1 ± 2.0% for the pan-caspase inhibitor, p < 0.0001, and 100.5 ± 3.9% for the caspase-3 inhibitor, p < 0.0001), Fig. 2g). As a control, necrostatin-1, which blocks necroptosis (an alternative programmed cell death pathway25), did not inhibit target cell killing. These data suggest that macrophages die by caspase-3-dependent apoptosis, whereas CD4+ T cells die by a caspase-independent process.
CTL killing of macrophages, but not CD4+ T cells, requires granzyme B
We next sought to further examine the mechanism by which CTLs initiate caspase-3-dependent macrophage death. CTLs kill their target cells either by releasing their cytotoxic granules or by engaging death receptors, such as FAS or TRAIL, which trigger caspase-mediated apoptosis26. Granule-mediated killing can be either caspase-independent or dependent, depending on which granzymes are involved and whether the target cell is resistant to caspase-mediated cell death27. To determine the mechanism of CTL-mediated killing, HIV-infected target and effector cells were co-cultured in the presence of an MHC-I blocking antibody to prevent T cell receptor (TCR) engagement; concanamycin A (CMA) to indirectly inhibit perforin28; a granzyme B-specific inhibitor (Ac-IETD-CHO); a FAS neutralizing antibody; or recombinant TRAIL-R1-Fc protein to block TRAIL engagement (Fig. 3a and b). Killing of both HIV-infected CD4+ T cells and macrophages was blocked by the MHC-I blocking antibody and CMA (p < 0.0001 and p < 0.0001, respectively, for CD4+ T cells, and p = 0.0097 and p = 0.0036, respectively, for macrophages), whereas blockade of FAS and TRAIL had no effect, suggesting that CTL killing of target cells is mediated by class I restricted TCR recognition triggering granule exocytosis and granule-mediated death. Specific blockade of granzyme B inhibited killing of HIV-infected or peptide-loaded macrophages (Fig. 3b; p = 0.0153 and Fig. 3c; p < 0.0001) but had no effect on killing of HIV-infected or peptide-loaded CD4+ T cells (Fig.s 3a and 3c, respectively). Together with the observations from Fig. 2, these data suggest that CTL killing of macrophages is mediated by granzyme B and subsequent caspase-3 activation (Fig. 2c, d, and g), while killing of CD4+ T cells may be mediated by alternative granzymes (other than B) that disrupt the mitochondria (as evidenced by increased ROS), to induce caspase-independent programmed cell death (Fig. 2e, f and g). Both CD4+ T cells and macrophages express high levels of SERPINB9, a natural granzyme B inhibitor27 (Fig. 3d), suggesting both targets might be resistant to granzyme B. Given that macrophage killing by CTL requires granzyme B, SERPINB9 expression likely delays initiation of granzyme B-mediated cell death (similar to Bcl-2 overexpression29), which could explain the slow timing of macrophage apoptosis compared to CD4+ T cells.
Fig. 3. Killing of target cells is MHC-I and perforin-dependent, but granzyme B is dispensable for CD4+ T cell killing.
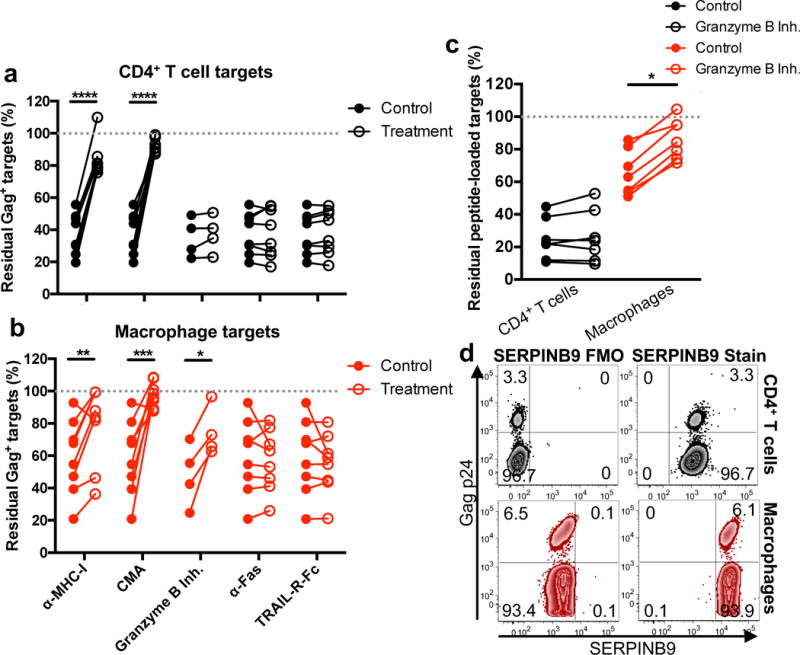
(a and b) Inhibiting HIV-infected target cell elimination. HIV-infected CD4+ T cells (a) or macrophages (b) were incubated with HIV-peptide expanded CTL in the presence of an MHC-I blocking antibody, concanamycin A (CMA – an indirect perforin inhibitor), a granzyme B inhibitor, a FAS neutralizing antibody, or recombinant human TRAIL-R1-Fc for four hours. Target cells were analyzed for live cell Gag-p24 intracellular staining via flow cytometry. Shown is the elimination assay summary using HIV+ patients (n = 8 distinct samples from four independent experiments). Statistical analysis: two-sided paired t-test, *p=0.0153, **p=0.0097, *** p=0.0036, ****p<0.0001. (c) Inhibition of granzyme B. HIV-peptide expanded CTLs were pre-incubated with the granzyme B inhibitor for one hour prior to co-culture with peptide-loaded, activated CD4+ T cells or macrophages. Shown are the results of the elimination assay using HIV+ patients (n=7 distinct samples from two independent experiments). Statistical analysis: two-sided unpaired t-test, *p<0.0001. (d) Granzyme B inhibitor, SERPINB9, expression in CD4+ T cells and macrophages. Shown is a representative flow cytometry plot of intracellular SERPINB9 staining on HIV-infected CD4+ T cell and macrophages from three independent experiments.
HIV-specific CTLs exhibit low co-expression of perforin and granzyme B
Ex vivo CTL-mediated killing of HIV-infected macrophages is significantly reduced compared to killing of HIV-infected CD4+ T cells (Fig. 1b and 2a). However, killing of macrophages is improved when expanded CTL are engaged as effectors (Fig. 1c and Supplementary Fig. 4). To assess the cytolytic potential of both effector cell populations, ex vivo CTL were first stained with a pool of HIV tetramers matched to each subject’s HLA alleles, and assessed for perforin and granzyme expression, with naive T cell staining used as a negative control (Supplementary Fig. 5a and b). Although the majority of HIV-specific cells expressed granzymes (mean ~26%, 60%, 54%, 67%, and 79% expression of granzymes A, B, H, K, and M, respectively), the frequencies of cells co-expressing perforin and granzyme (representing the cytolytic population) was lower (mean ~9%, 21%, 20%, 7% and 30% perforin co-expression with granzymes A, B, H, K and M, respectively) (Figs. 4a – c). This was not an effect of poor ex vivo staining of CTL as ex vivo CMV-specific CTL exhibited high co-expression levels of perforin and granzymes (Supplementary Fig. 5c). Similar results were observed by phenotyping ex vivo CTL that degranulated in response to HIV-infected targets (CD107a+ - Fig. 4d and e; staining controls are shown in Supplementary Fig. 5d). However, in vitro expanded HIV-specific CTL effectors exhibited significantly higher frequencies of perforin+granzyme+ cells, except for granzyme M, compared to ex vivo CTL (Fig. 4d and e, p = 0.0156, p < 0.0001, p = 0.0115, p = 0.005 for granzymes A, B, H, and K, respectively), supporting the enhanced cytolytic function of expanded CTL compared to ex vivo CTL. Comparisons of perforin and granzyme co-expression in response to CD4+ T cells and macrophages revealed no differences (Supplementary Fig. 5e), suggesting that CTL engaged by either target cell exhibit similar cytolytic potential.
Fig. 4. Ex vivo HIV-specific CTL exhibit low perforin+granzyme+ expression compared to expanded CTL.
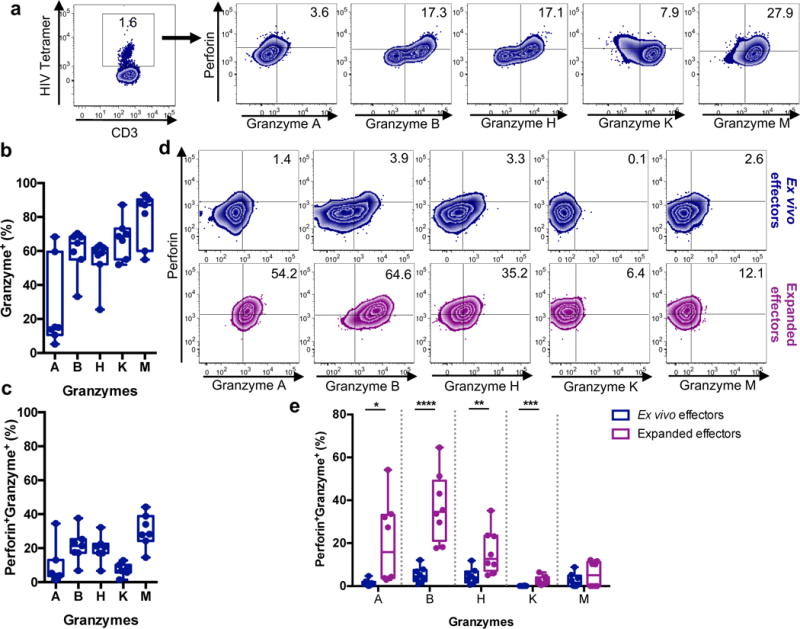
(a) Perforin and granzyme staining of ex vivo HIV tetramer+ CD8+ T cells. Shown is a representative plot of A02-SL9 (HIV) tetramer staining from two independent experiments. Tetramer+ cells were phenotyped for perforin, and granzyme A, B, H, K, and M staining. Staining controls are shown in Supplementary Fig. 5a and b. Also see Supplementary Fig. 5c for CMV-specific CTL perforin and granzyme expression as a comparison. Shown is a representation of one experiment from two independent experiments. (b) Summary of total granzyme expression on HIV+ controller ex vivo HIV-specific CTL (n=7 distinct samples from two independent experiments). Box elements, center line, limits and whiskers are the median, 25th-75th percentiles and min-max, respectively. (c) Summary of dual perforin and granzyme expression on ex vivo HIV-specific CD8+ T cells (n=7 distinct samples from two independent experiments). Box elements, center line, limits and whiskers are the median, 25th-75th percentiles and min-max, respectively. (d) Cytolytic capacity of HIV-specific ex vivo and expanded CTL. Ex vivo or expanded CTLs were incubated with HIV-infected CD4+ T cells for six hours followed by flow cytometry analysis of degranulation (surface CD107a expression), perforin and granzyme expression. Shown is a representative analysis of CTL perforin and granzyme phenotyping of degranulated cells from two independent experiments. Supplementary Fig. 5d shows staining controls. (e) Summary of the perforin and granzyme co-expression of degranulated ex vivo and expanded CTL for HIV+ patients (n=8 distinct samples from two independent experiments). Box elements, center line, limits and whiskers are the median, 25th-75th percentiles and min-max, respectively. Statistical analysis: two-tailed unpaired t-test, *p=0.0156, **p=0.0115, ***p=0.005, ****p<0.0001.
CTL efficiently recognize and produce more IFN-γ in response to HIV-infected macrophages
Variations in surface MHC-I density could alter CTL recognition of target cells and contribute to differences in elimination. To determine the relative MHC-I surface densities of CD4+ T cells and macrophages, cells were analyzed using imaging flow cytometry to calculate their surface areas and mean fluorescence intensity of MHC-I (Supplementary Fig. 6a and b). There were no differences in the relative MHC-I surface densities of CD4+ T cells and macrophages (p = 0.2209), suggesting that the amount of antigen presented on macrophages, which have much greater surface area than T cells, could not explain the difference in killing (Fig. 5a). To examine whether HIV-infected CD4+ T cells and macrophages were similarly recognized, we co-cultured infected cells with autologous ex vivo or expanded CTL and measured the frequency of degranulation by surface CD107a staining (Fig. 5b). Ex vivo CTL degranulated similarly in response to infected CD4+ T cells and macrophages (Fig. 5c) and degranulation was HIV-specific, since CD8+ T cells from HIV− donors did not degranulate (Supplementary Fig. 6c). Peptide-expanded CTL degranulated significantly more in response to HIV-infected macrophages than CD4+ T cells (p = 0.0036; Fig. 5c). Similar results were observed for CTLs responding to CEF peptide-loaded targets (p<0.0001; Fig. 5c) suggesting that enhanced recognition of macrophages compared to CD4+ T cells is not an HIV-specific phenomenon but is broadly applicable. As described above, the CD107a+ CTLs responding to infected CD4+ T cells and macrophages had comparable cytolytic potential as shown by expression of perforin and granzymes A, B, H, K and M (Supplementary Fig. 5e). Thus, reduced macrophage killing is not due to impaired recognition, degranulation, or cytolytic potential of the HIV-specific CTLs.
Fig. 5. HIV-infected macrophages induce stronger CTL cytokine responses than infected CD4+ T cells.
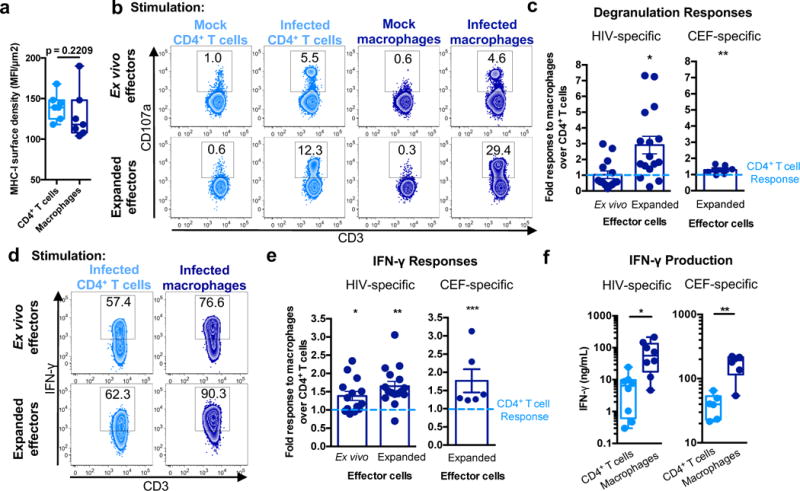
(a) MHC-I surface density. Imaging flow cytometry was used to calculate relative surface densities of MHC-I on CD4+ T cells and macrophages (see Supplementary Fig. 6a and b) (n=7 distinct samples from three independent experiments). Box elements, median and 25th-75th percentiles. Statistical analysis, two-sided Mann-Whitney test. p=0.2209. (b) CTL recognition assay. Ex vivo and expanded CTL were incubated with mock/uninfected or HIV-infected CD4+ T cells or macrophages for six hours followed by analysis of degranulation (CD107a expression). See also Supplementary Fig. 6c. Shown is one of five independent experiments. (c) Comparison of CTL responses to CD4+ T cells and macrophages. Data shown are the ratios of CTL degranulation in response to macrophages over CD4+ T cells for ex vivo (n=14 distinct samples from five independent experiments) and expanded CTL (n=16 distinct samples from five independent experiments). Responses against CEF peptide-loaded targets were also assessed (n=6 distinct samples from three independent experiments). Shown are the means +/- SEM. Statistical analysis: two-sided one sample t-test, *p=0.0036, and two-sided Wilcoxon signed rank test, **p<0.0001. (d) Representative analysis of IFN-γ expression for CD107a+ cells, showing one of five independent experiments. (e) Summary of IFN-γ expression for CTLs. Shown is the ratio of responses to macrophage targets over CD4+ T cell targets using ex vivo (n=14 distinct samples from five independent experiments) and expanded CTL (n=16 distinct samples from five independent experiments). In addition, responses against CEF peptide-loaded targets were assessed (n=6 distinct samples from three independent experiments). Shown are the means +/- SEM. Statistical analysis: two-sided one sample t-test, *p=0.008, **p=0.0002, and two-sided Wilcoxon signed rank, ***p<0.0001. (f) CTL IFN-γ production. Expanded CTLs were co-cultured with HIV-infected or CEF peptide-loaded targets for 18 hours followed by ELISA-based detection of IFN-γ in the culture supernatants. Shown are data from HIV+ patients (n=8 distinct samples from two independent experiments) and HIV− donors (n=6 distinct samples from three independent experiments). Box elements, center line, limits and whiskers are the median, 25th-75th percentiles and min-max, respectively. Statistical analysis, two-tailed Mann-Whitney test; *p=0.0047, **p=0.0043.
We next examined whether HIV-infected CD4+ T cells and macrophages differentially triggered other CTL effector functions. Effector cells responding to HIV-infected macrophages produced more IFN-γ, assessed by intracellular cytokine staining, compared to those responding to infected CD4+ T cells (Fig. 5d and e). This was significant for ex vivo CTL (p = 0.008) and expanded CTL (p=0.0002). Similar differential responses were observed for CTLs with CEF peptide-loaded target cells (p < 0.0001; Fig. 5e). The difference in intracellular cytokine staining was also associated with a significant increase in secreted IFN-γ after overnight co-culture with infected macrophages compared to CD4+ T cell targets, as assessed by ELISA analysis of culture supernatants (p = 0.0047; Fig. 5f). Approximately 10-fold more IFN-γ was released after exposure to infected macrophages compared to CD4+ T cell targets. Again, similar results were observed for CEF-specific CTLs with equivalent peptide-loaded target cells (p = 0.0043; Fig. 5f). Approximately 4.2-fold more IFN-γ was released after exposure to CEF peptide-loaded macrophages than CD4+ T cell targets. Together, these data suggest that CTLs efficiently recognize and produce more cytokine in response to cognate antigen expressed on macrophages compared to CD4+ T cell targets.
Inefficient macrophage killing drives enhanced cytokine responses through prolonged effector-target interactions
CTLs detach from their specific target cells when the target cell is killed. CTLs that are unable to kill have a prolonged synapse time with the target cell during which many cytokines, including IFN-γ, are hypersecreted30. In addition, children with profound genetic defects in granule-mediated cytotoxicity develop an often-fatal syndrome, familial hemophagocytic lymphohistiocytosis, caused by elevated levels of IFN-γ and uncontrolled macrophage activation31. Since CTL inefficiently kill macrophages (Fig. 1) and produce significantly more IFN-γ after co-culture with macrophages versus CD4+ T cells (Fig. 5e and f), we hypothesized that poor killing leads to prolonged synapses between the CTL and macrophage, driving excessive secretion of IFN-γ. To test this hypothesis, we used imaging flow cytometry to examine the duration of target cell synapse formation of violet-stained, expanded CTL effectors with peptide-loaded, Far Red-stained, activated CD4+ T cell or macrophage targets over 1 hour (Fig. 6a-c). Target-effector conjugates were distinguished by first gating on total peptide-loaded targets (Fig. 6a, red box), followed by gating on dually stained target-effector conjugates (Fig. 6a, blue box). Immunological synapse formation of conjugates was confirmed by assessing actin concentration at the cell-cell interface (Fig. 6b)32. The frequencies of effector-target synapses were low for CD4+ T cells, likely reflecting short synapse time, whereas macrophages formed significantly higher frequencies of synapses at all time points (Fig. 6c; p = 0.0001, p = 0.001, p < 0.0001 for 10, 30, and 60-minute time points, respectively). Although synapse frequency waned after 30 minutes for CD4+ T cells, they continued to increase for macrophages, suggesting that macrophage-CTL synapses were longer-lived and accumulated over time. Thus, CTLs stay in contact with target macrophages for much longer than with CD4+ T cells.
Fig. 6. Antigen-loaded macrophages accumulate more immunological synapses with effector cells compared to antigen-loaded CD4+ T cells.
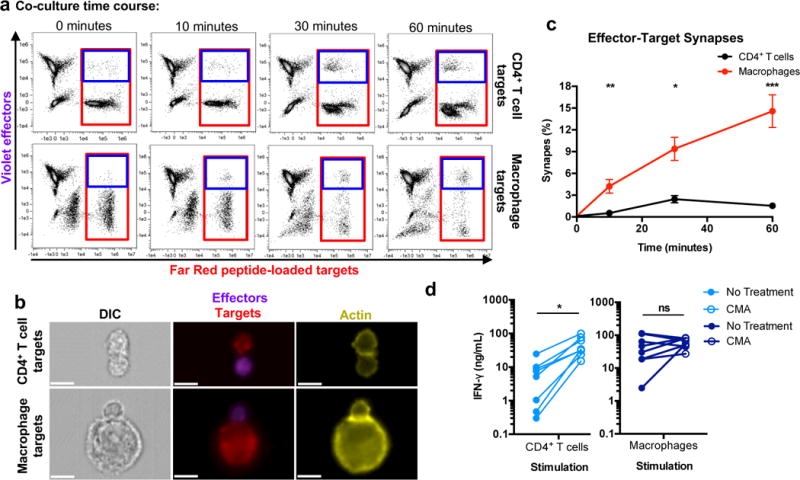
(a) Assessment of target-effector doublets. Peptide-loaded, FarRed-labeled targets were co-cultured for the indicated times with Violet-labeled expanded CTLs followed by fixation, actin staining, and analysis via imaging flow cytometry. Shown are representative plots (one of two independent experiments) of effector-target co-culture samples and the gating strategy used to quantitate total peptide-loaded targets (red box) and target-effector doublets (blue box). (b) Immunological synapses formed between target-effector pairs. Data points within the doublet gate were assessed for concentrated actin (yellow) at the interface between the effectors and targets32. As in (a), shown is a representation of one of two independent experiments. White scale bars denote 10μm. (c) Summary of immunological synapse accumulation over 60 minutes (n=6 distinct samples from two independent experiments). Frequencies of immunological synapses were calculated as described in the Methods. Shown are means +/- SEM. Statistical analysis: two-sided unpaired t-test, *p=0.001, **p=0.0001, ***p<0.0001. (d) Inhibition of target cell killing in CD4+ T cells results in more robust cytokine production. Prior to co-culture with targets, HIV-peptide expanded CTLs were pre-incubated with concanamycin A (CMA – an indirect perforin inhibitor) for one hour. HIV-infected CD4+ T cells (left) and macrophages (right) were co-cultured for 18 hours with effector cells, followed by assessment of culture supernatant IFN-γ via ELISA (n=8 distinct samples from two independent experiments). Statistical analysis: two-sided paired t-test, *p=0.0021.
To confirm whether poor killing and prolonged contact time were responsible for greater CTL cytokine production, we assessed whether inhibiting CD4+ T cell killing would enhance IFN-γ secretion by CTL exposed to HIV-infected CD4+ T cells. HIV peptide-expanded CTLs, pre-treated or not with CMA to inhibit perforin-mediated lysis, were cultured overnight with HIV-infected CD4+ T cells or macrophages, followed by measurement of IFN-γ in the culture supernatants. Inhibition of CD4+ T cell killing significantly increased the amount of IFN-γ in the supernatants from 7.3 ± 2.8 ng/mL to 46.6 ± 10.4 ng/mL (mean ± SEM, p = 0.0021, which was comparable to the IFN-γ levels in co-cultures of infected macrophages and untreated CTLs (50.6 ± 15.0 ng/mL) (Fig. 6d). These data suggest that macrophage resistance to CTL-mediated killing drives IFN-γ hypersecretion.
CTLs induce macrophage production of pro-inflammatory chemokines
IFN-γ induces macrophage production of pro-inflammatory chemokines that recruit monocytes and T cells33, 34, 35. To examine whether interaction of CTL with macrophages induces pro-inflammatory chemokine secretion, we measured the levels of macrophage-derived chemokines, CXCL9, CXCL10, CXCL11, MIP-1α (CCL3), MIP-1β (CCL4), RANTES (CCL5), and CCL2, in cell culture supernatants after 24 hours of CTL and macrophage co-culture. Co-culture resulted in significant increases in each of the 7 chemokines assessed (Fig. 7a). Fold induction of each of these chemokines in co-cultures compared to single cultures was significant (Fig. 7b; p = 0.0118, p = 0.009, p = 0.0211, p = 0.0002, p = 0.0006, p=0.0286, p = 0.0023 for CXCL9, CXCL10, CXCL11, MIP-1α, MIP-1β, RANTES, and CCL2, respectively). In addition, recombinant chemokines added at concentrations measured in these co-cultures (Table 1) mediated trans-well chemotaxis of T cells (CXCL9, CXCL10, MIP-1β, and CCL2) and monocytes (MIP-1α and CCL2) (Fig. 7c), consistent with the chemokine receptor profile of each cell type (Supplementary Fig. 7). Together, these in vitro results indicate that inefficient killing of macrophages by CTL may promote inflammatory chemotaxis of other immune cells.
Fig. 7. CTL interaction with macrophages induces pro-inflammatory chemokine production from macrophages.
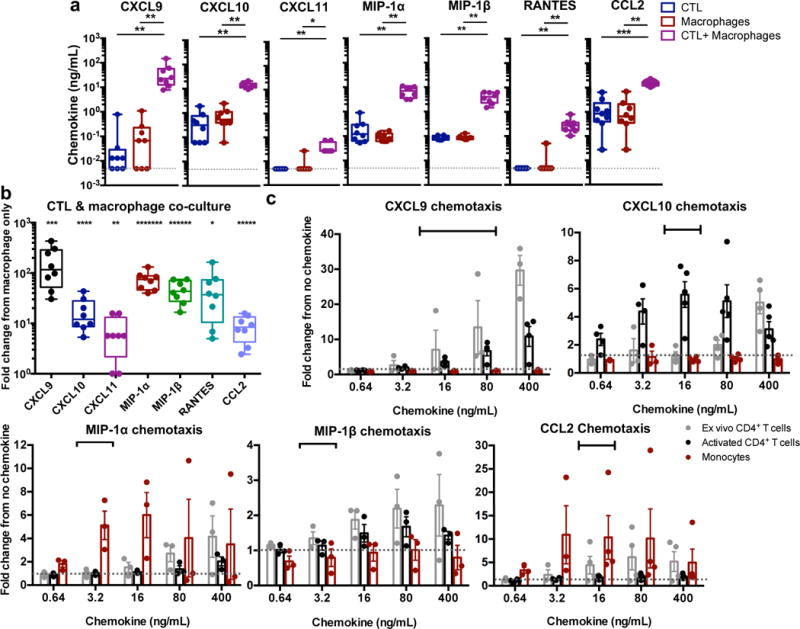
(a) Detection of pro-inflammatory chemokines. CTLs were co-cultured with peptide-loaded macrophages for 24 hours, followed by assessment of chemokines in the culture supernatant by cytokine bead array. The dotted gray line indicates the limit of detection for the assay. Shown are results from HIV+ patients (n=8 distinct samples from two independent experiments). Box elements, center line, limits and whiskers are the median, 25th-75th percentiles and min-max, respectively. Statistical analysis: two-sided Mann-Whitney test, *p=0.0011, **p=0.0002, ***p<0.0001. (b) Summary of the fold change response in chemokine production in the CTL + macrophage co-cultures versus the macrophage only cultures. Box elements, center line, limits and whiskers are the median, 25th-75th percentiles and min-max, respectively. Statistical analysis: two-sided one sample t-test, *p=0.0286, **p=0.0211, ***p=0.0118, ****p=0.009, *****p=0.0023, ******p=0.0006, *******p=0.0002. (c) Chemotaxis assays. Ex vivo CD4+ T cells, activated CD4+ T cells, and ex vivo monocytes were subjected to a transwell chemotaxis assay with titered CXCL9, CXCL10, MIP-1α, MIP-1β, and CCL2. Responses shown are the fold number of migrated cells from no chemokine conditions. Shown are results from three independent experiments (n=3 distinct samples for MIP-1α and MIP-1β) and four independent experiments (n=4 distinct samples for CXCL9, CXCL10 and CCL2), with means +/- SEM. Black bars indicate the range of chemokine observed in co-culture conditions as described in Table 1. See also Supplementary Fig. 7.
Table 1. Chemokine concentrations in CTL and macrophage co-cultures.
Shown are the means ± SEM of the chemokines measured CTL and macrophage co-cultures (n = 8 distinct samples from two independent experiments).
| Chemokine | Concentration (ng/mL) |
|---|---|
| CXCL9 | 42.1 ± 16.5 |
| CXCL10 | 13.7 ± 1.33 |
| CXCL11 | 0.03 ± 0.07 |
| MIP-1α | 7.04 ± 1.28 |
| MIP-1β | 4.07 ± 0.67 |
| RANTES | 0.32 ± 0.07 |
| CCL2 | 15.5 ± 1.33 |
DISCUSSION
Our results demonstrate that macrophages are inherently more resistant to CTL-mediated killing than CD4+ T cells. Resistance is associated with less efficient CTL inhibition of HIV replication, prolonged CTL synapse formation, and increased secretion of pro-inflammatory cytokines and chemokines. Killing of HIV-infected CD4+ T cells and macrophages is mediated by CTL death-inducing perforin and granzymes. However, in contrast to CD4+ T cell killing, which is rapid, more effective at suppressing HIV, and caspase- and granzyme B-independent, macrophage killing is slow, caspase-dependent and requires granzyme B. While our initial observations of differences in suppression of HIV-infected CD4+ T cells and macrophages disagree with one previous report18, potentially due to differences in viral strains, infection protocols, source of CTL or effector-to-target ratios, both studies consistently demonstrate that although killing is delayed, HIV-infected macrophages can be eliminated by CTL16, 18, 19. Our results agree with and extend results previously described for SIV-infected CD4+ T cell and macrophage targets20, 21 by demonstrating a mechanism whereby poor target cell killing induces hyperinflammatory responses30, which could contribute to chronic inflammation, a hallmark of HIV infection.
Human CTL can express any combination of 5 granzymes, each of which activates an independent cell death pathway. While granzymes B and M induce caspase-dependent apoptosis, granzymes A, H and K are caspase-independent24, 26, 36, 37. Our results suggest that activated CD4+ T cells may be susceptible to killing by at least one granzyme that induces caspase-independent apoptosis. However, due to lack of granzyme-specific inhibitors, it is unclear which granzyme is responsible for this killing. T cell differentiation and activation state have also been shown to contribute to susceptibility to rapid CTL killing38. While we observed no differences between killing of resting and activated CD4+ T cell targets in our assay, the precise mechanism of CD4+ T cell killing remains to be identified. In contrast, macrophages require granzyme B to induce apoptosis through caspase-3-dependent mechanisms, and appear to be resistant to more rapid, caspase-independent killing. SERPINB9, a natural granzyme B inhibitor27, is expressed by both target cell types. Resistance to granzyme B-mediated apoptosis via SERPINB9 expression in CD4+ T cells is likely overcome by susceptibility to an alternative granzyme that induces apoptosis through a caspase-independent mechanism. In contrast, given that macrophages require granzyme B for CTL-mediated killing, SERPINB9 expression may slow the initiation of or protect the cell from apoptosis, like Bcl-229. Enhanced resistance of macrophages to CTL-mediated killing may represent a mechanism to preserve antigen presentation and induce more potent adaptive immune responses, by increasing the survival of antigen-presenting cells from killing by sentinel CTLs. To this end, it has been reported that mature dendritic cells are more resistant to CTL lysis than immature dendritic cells38, 39. We show that macrophages are also resistant to killing by CMV, EBV and Flu-specific CTLs, suggesting this resistance and subsequent pro-inflammatory consequences broadly apply. Although SERPINB9 represents a mechanism to resist granzyme B, there are currently no known mechanisms of resistance to the other granzymes. Therefore, understanding the mechanism behind macrophage resistance to rapid killing by a caspase-independent mechanism will require future study.
In this study, ex vivo CTL and in vitro expanded CTL were examined for their cytolytic capacity to eliminate HIV-infected CD4+ T cells and macrophages. Despite killing of CD4+ T cells, killing of macrophages by ex vivo CTL was barely detectable, likely due to a combination of macrophage resistance to granzymes and low frequencies of perforin+granzyme+ CTL. Expanded CTL, which expressed higher levels of perforin and granzymes, exhibited greater macrophage killing; however, similar to ex vivo CTL, this was relatively inefficient compared to killing of CD4+ T cells. As HIV controller CTL maintain their proliferative capacity and express more perforin upon restimulation than non-controllers40, the in vitro expanded CTL used in this study likely represent a more physiologically relevant scenario for elite controllers. In contrast, HIV+ chronic progressor CTL appear exhausted, exhibiting deficiencies in proliferative ability and low cytolytic potential, but maintenance of IFN-γ production41, 42, 43, 44, 45. Indeed, CTL from chronic progressors exhibit poorer killing of both CD4+ T cell46 and macrophage18 targets, and function is not fully restored by cART47. Inefficient killing in these contexts may contribute to infected macrophage persistence and potentially to the chronic inflammatory state observed in all HIV+ patients.
The timing of programmed cell death has consequences. Slow killing by killer lymphocytes in patients bearing hypomorphic Prf1 mutations results in prolonged CTL interaction with their targets and enhanced production of IFN-γ and other pro-inflammatory cytokines30. High levels of IFN-γ have been postulated to lie at the root of the often-fatal symptoms of familial hemophagocytic lymphohistiocytosis, as neutralization of this cytokine induced recovery in two animal models of this disease48. HIV-infected individuals also experience chronic inflammation13. Our results suggest that non-cytolytic interaction of T cells with target cells could potentially contribute to elevated IFN-γ production and enhanced chronic inflammation in HIV+ patients. For ART-suppressed HIV+ individuals, waning peripheral blood mononuclear cells (PBMC) and tissue T cell responses may prove beneficial in the context of inflammation49, as lower IFN-γ would yield less macrophage activation. Indeed, markers of chronic inflammation, including soluble CD163, a monocyte/macrophage activation marker, are lower in ART-treated individuals compared to elite controllers50. IFN-γ-induced macrophage activation and recruitment of immune cells to sites of infection could contribute to inflammatory macrophage-associated diseases, including atherosclerosis and neurological disease, described in HIV+ subjects11, 12, 13.
This study suggests that non-cytolytic CTL interactions with HIV-infected macrophages may contribute to ongoing inflammatory conditions. Small animal models of HIV infection are currently inadequate to address the underlying mechanism due to high levels of background inflammation (graft-versus-host disease) and poor myeloid cell reconstitution. Future studies with improved small animal models of HIV infection could help to confirm a causal link between poor CTL killing and chronic inflammation. Finally, an enhanced understanding of the precise mechanisms underlying the observed resistance to target cell killing and resulting hypersecretion of pro-inflammatory cytokines and chemokines will be necessary to develop approaches capable of efficiently eliminating infected macrophages and suppressing excessive inflammation. The inflammatory consequences of poor macrophage killing may also have implications for infections with other pathogens that infect macrophages, such as tuberculosis, chikungunya, and adenovirus.
ONLINE METHODS
Human subjects
HIV+ subjects were recruited from outpatient clinics at local Boston area clinics and from outside Boston. The institutional review board of Massachusetts General Hospital approved the studies of cells derived from human blood samples. All human subjects gave written, informed consent. Peripheral blood mononuclear cells (PBMCs) from HIV− healthy donors and HIV+ controllers were collected by Ficoll gradient separations from leukapheresis samples, cryopreserved and stored at −150°C for future use. Controller status was classified as previously described51.
Cell isolations and culture
For preparation of target cells, frozen PBMCs were thawed and subjected to CD14 positive isolation (Stemcell Technologies) per the manufacturer’s instructions to isolate monocytes. CD4+ T cells were isolated via negative enrichment (Stemcell Technologies) using the leftover CD14-depleted PBMCs. Macrophages were obtained via seven-day maturation of monocytes with 50 ng/mL recombinant human GM-CSF (BioLegend) and 50 ng/mL recombinant human M-CSF (BioLegend) in R10 media (RPMI 1640, 10% FBS, 1 U/ml penicillin, 100 mg/ml streptomycin, 2 mM glutamine, 10 mM HEPES; Sigma) in low attachment 24-well plates (400,000 cells/well; Corning). In addition, different lots of “Certified FBS” (Invitrogen) were screened for the best induction of macrophage maturation that yielded efficient levels of HIV-infection. Half of the maturation media was exchanged for fresh media containing GM-CSF and M-CSF on Day 4. Alternative methods to obtain macrophages included maturation using 10% Human Serum AB (Gemini Bio-Products – Supplementary Fig. 3a only). Successful maturation was assessed via spreading of the cells onto the surface of the plate4. CD4+ T cells were rested overnight in R10 with 30 U/mL IL-2 (R10/30; NIH AIDS Reagent Program), then activated using plate-bound anti-CD3 (clone OKT3; BioLegend) and soluble anti-CD28 (clone 28.8; BioLegend) in R10/30 to permit HIV infection. After three days of activation, cells were removed from the plate and rested in R10/30 for an additional three days before infection. For experiments using resting ex vivo CD4+ T cells (Supplementary Fig. 3), CD4+ T cells were isolated from frozen PBMCs immediately before co-culture with CTL effectors.
For preparation of ex vivo effector cells, autologous PBMCs were thawed and rested in R10 media with 5 U/mL recombinant IL-2 (R10/5) for 8-10 hours before co-culture with target cells. CD8+ T cells were isolated via negative enrichment (Stemcell Technologies), followed by CD62L-depletion (Miltenyi) per the manufacturer’s instructions. This negative enrichment kit contains antibodies for CD56 and CD16 to deplete NK cells. For preparation of HIV peptide-expanded and CMV, EBV, and Flu (CEF) peptide-expanded effector cells, autologous PBMCs were thawed three days prior to co-culture and stimulated in R10 with 50 U/mL IL-2 (R10/50) and 200 ng/mL/peptide of a pool of HIV or CEF peptides representing optimal MHC class I restricted epitopes matched for each subject’s HLA type. PBMC from HIV− healthy donors were stimulated with 0.5 μg/mL of anti-CD3/anti-CD28 for negative controls in elimination assays. Seven days following activation, effector CTLs were isolated as described above and rested in R10/50 for an additional five days before use in co-culture assays.
Preparation of HIV Stocks
HEK293T17 cells (obtained from ATCC – cells were not tested for mycoplasma) were transfected with 89.6 or JR-CSF proviral plasmids (NIH AIDS Reagent Program) using 25kDa polyethylenimine (PEI; Polysciences). 40-48 hours post-transfection, culture supernatants were collected, centrifuged at 3000 xg for 10 minutes to remove cell debris, filtered through a 0.45 μm membrane to remove smaller aggregates and concentrated with PEG-it Virus Precipitation Solution (System Biosciences) as per manufacturer’s instructions and frozen at −80°C. Viral stocks were titered via p24 ELISA (Frederick National Laboratory for Cancer Research).
Preparation of HIV-infected target cells
HIV89.6, an R5X4 dual-tropic strain, was used to infect GM-CSF/M-CSF-derived macrophages and CD4+ T cells for co-culture assays. HIVJRCSF, an R5 strain, was used for assays containing human serum-derived macrophages and CD4+ T cells. For HIVJRCSF experiments, macrophages were pre-transduced with SIV Vpx for 24 hours to enhance HIV infection (Supplementary Fig. 3a). Macrophages and CD4+ T cells were infected as previously described4. Macrophages were incubated for six hours at 37°C with 100 ng p24 of HIV89.6 in each well of a 24-well plate, followed by removal of the virus. In parallel, activated CD4+ T cells were infected in 96-well flat bottom plates at 1 million cells/well with 40 ng p24 of HIV89.6 and spinoculated at 800 xg for one hour, incubated at 37˚C for three hours, followed by removal of the virus. Two days following initial infection with HIV, levels of infection were assessed for each target cell via flow cytometry before setting up of co-culture assays. Macrophages and CD4+ T cells were surface stained with anti-CD14-APC/Cy7 (clone M5E2; BioLegend) and anti-CD3-APC/Cy7 (clone HIT3a; BioLegend), respectively, anti-CD4-APC (clone OKT4; BioLegend), and LIVE/DEAD Blue (ThermoFisher), followed by intracellular staining with anti-Gag p24-RD1 (clone KC-57; Beckman Coulter). Intracellular staining for SERPINB9 was performed using anti-SERPINB9-AF488 (PI-9, clone 7D8; BioRad, Hercules, CA). Flow cytometric data were acquired using a FACSCanto instrument with FACSDiva software (BD Biosciences). Uninfected cells were added to normalize infection levels between the two target cell types. Target cell infection ranged from 2.0% to 16.6% in elimination, suppression, and recognition assays.
Preparation of peptide-loaded target cells
For assays using peptide-loaded targets, uninfected, activated CD4+ T cells or macrophages were incubated at 37°C with 1 μg/mL/peptide of the HIV or CEF optimal peptides used to make their autologous expanded CTLs for 30 minutes. Where indicated, resting CD4+ T cells (unstimulated, total ex vivo CD4+ T cells) were used in place of activated CD4+ T cells. During the last 5 minutes of incubation, CellTrace FarRed (ThermoFisher) was added to the culture. Cells were washed twice in R10, and then mixed 1:1 with unloaded, unlabeled target cells, producing a population of 50% peptide-loaded target cells. These targets were used for elimination, recognition, and conjugate/synapse formation assays.
Tetramer staining and perforin/granzyme phenotyping of ex vivo CD8+ T cells
Rested ex vivo CD8+ T cells were assessed for CMV and HIV specificity using HLA-matched tetramers. Tetramers were made via biotinylated monomer conjugation to BV510 streptavidin (BioLegend). Monomers were obtained from Dr. Soren Buus (University of Copenhagen). The A02-NV9 tetramer was used to stain for CMV-specificity while pools of A02-SL9, B27-KK10, B53-QW9, B53-YY9, B57-IW9(p24), B57-IW9(RT), B57-KF11, B57-QW9, and B57-TW10 tetramers were used to stain for HIV-specificity. Subsequent surface stains included anti-CD62L-PE-Cy5 (clone DREG-56; BioLegend), anti-CD3-AF700 (clone HIT3a; BioLegend), anti-CD8-FITC (clone SK1; BioLegend), anti-CD45RA-BV605 (clone HI100; BioLegend), and LIVE/DEAD Blue. Following permeabilization, the cells were intracellular stained using anti-perforin PE/Cy7 (clone B-D48; BioLegend), anti-granzyme A-PerCp/Cy5.5 (clone CB9; BioLegend), anti-granzyme B-Pacific Blue (clone GB11; BioLegend), anti-granzyme H (biotinylated and secondary stained with BV650 streptavidin) (polyclonal antibody, R&D), anti-granzyme K-PE (clone GM26E7; BioLegend), and anti-granzyme M-eFluor660 (clone 4B2G4; eBioscience). The cells were analyzed by flow cytometry. Naïve CD8+ T cells within the mixed population of CD8+ T cells were used as internal gating controls for perforin and each granzyme as these cells do not express these effector molecules.
CTL elimination assays
Infected or peptide-loaded CD4+ T cell and macrophage target cells and effector CTLs (ex vivo and peptide-expanded) were prepared as described above. Effector cells were stained with CellTrace Violet (ThermoFisher) to distinguish target cells and effector cells via flow cytometry. For assays assessing inhibition of killing, effector cells were pre-incubated with either 100 ng/mL of concanamycin A28, 52 (Tocris), 100 μM granzyme B inhibitor II53 (Millipore), 2 μg/mL anti-FAS antibody52 (clone ZB4; Millipore), or 1 μg/mL TRAIL-R1-Fc54 (R&D), while target cells were pre-incubated with 50 μM necrostatin-1 (Millipore), 100 μM caspase inhibitor I (pan-caspase inhibitor; Millipore), 100 μM caspase-3 inhibitor (R&D), or 80 μg/mL of anti-MHC-I antibody (clone W6/32; BioLegend). Isotype and vehicle controls were included for comparison. Effector cells were co-cultured with mock, peptide-loaded, or infected target cells at E:T of 0, 1, 2, and 4 for 4 hours (or 15 minutes, 1 hour, 4 hours, 12 hours, and 24 hours for the time course) at 37°C. During the last hour of incubation, a fixable fluorescent inhibitor of caspase-3 (FAM-DEVD-FMD; ThermoFisher) or ROS detection reagent, CellROX (ThermoFisher) was added as indicated. Cells were harvested and surface stained with anti-CD14-APC/Cy7 (for macrophages) (clone M5E2; BioLegend) or anti-CD3-APC/Cy7 (for CD4+ T cells) (clone HIT3a; BioLegend), in addition to anti-CD4-APC (for infected target cells only) (clone OKT4; BioLegend) and LIVE/DEAD Blue (ThermoFisher), followed by intracellular staining with anti-Gag p24-RD1 (for infected targets only) (clone KC-57; Beckman Coulter) and flow cytometric analysis.
Calculations for elimination assay analysis
Elimination of infected target cells was assessed via quantitation of live (LIVE/DEAD Blue negative), CD4−p24+ target cells, while early apoptotic LIVE/DEAD negative, CD4−p24+ target cells were detected via induction of active caspase-3 or ROS activity. Percent residual Gag+ targets was calculated by dividing the %CD4−p24+ at E:T 1, 2, or 4 by the %CD4−p24+ at E:T 0 and multiplying by 100. Casapse-3 activity was calculated by subtracting %active caspase-3 at E:T 0 from %active caspase-3 at E:T 4. ROS activity was calculated by subtracting %ROS at E:T 0 from %ROS at E:T 2. Elimination of peptide-loaded target cells was assessed via the loss of LIVE/DEAD Blue negative, Far Red+ target cells. Percent residual peptide-loaded targets was calculated by dividing %Far Red+ targets at E:T 1, 2, or 4 by %Far Red+ targets at E:T 0 and multiplying by 100.
Viral inhibition assays
Infected CD4+ T cells, macrophages, and ex vivo CTLs were prepared as described above. Co-cultures were performed in 96-well plates in 200 μl of R10/50 at an E:T of 2. On days three, five, and seven of co-culture, 100 μl of culture supernatant was harvested and replaced with 100 μl of new R10/50. Quantitation of virus in the culture supernatants was performed using p24 ELISA (NCI Frederick). Percent viral suppression was calculated by subtracting the p24 concentration at E:T 2 from the paired E:T 0, dividing by the p24 concentration at E:T 0 and multiplying by 100.
Recognition assays
For flow cytometry-based recognition assays, infected or peptide-loaded CD4+ T cells and macrophages were prepared as described above followed by co-culture with ex vivo or peptide-expanded CTLs at an E:T of 2 in the presence of GolgiPlug/GolgiStop (BD Biosciences) and anti-CD107a-AF488 (clone H4A3; BioLegend). Following a 6 hour incubation at 37°C, cells were harvested and surface stained with anti-CD62L-PE-Cy5 (clone DREG-56; BioLegend), anti-CD3-APC/Cy7 (clone HIT3a; BioLegend), anti-CD8-AF700 (clone SK1; BioLegend), anti-CD45RA-BV650 (clone HI100; BioLegend), and LIVE/DEAD Blue. Intracellular staining was performed using anti-IFN-γ-BV510 (clone 4S.B3; BioLegend) and cells were analyzed by flow cytometry. For perforin/granzyme phenotyping of degranulated cells, the previously described perforin/granzyme-staining panel was used with anti-IFN-γ-BV510 replacing the tetramer-BV510 stain, anti-CD3-BUV395 (clone HIT3a; BD Biosciences) replacing anti-CD3-AF700, and anti-CD8-AF700 (clone SK1; BioLegend) replacing anti-CD8-FITC.
For ELISA-based recognition assays, effector and target cell co-cultures were set up as described above without GolgiPlug/GolgiStop or CD107a-AF488 for an overnight co-culture to allow for accumulation of IFN-γ and chemokines. For inhibition of target cell killing, effector cells were pre-incubated with DMSO or 100 ng/mL CMA for one hour. Eighteen hours following co-culture, 100 μl of culture supernatant was collected and IFN-γ levels were assessed via ELISA (BioLegend). For quantitation of chemokine, peptide-loaded macrophages and expanded effector CTLs were cultured individually or co-cultures at an E:T of 2 (150,000 targets/well or 300,000 CTLs per well) in 24-well in R10/50 for 24 hours at 37°C. 100 μl of culture supernatant was collected for cytokine bead array measurement of CXCL9, CXCL10, CXCL11, MIP-1α, MIP-1β, and CCL2 levels (LEGENDPlex, BioLegend).
Calculations for the recognition assay analysis
CD107a frequencies were corrected for the background frequencies observed in mock conditions. For samples with infected target cells, CD107a frequencies were also normalized to levels of CD4+ T cell productive infection. The final analysis of degranulation was expressed as the CTL fold response to macrophages over CD4+ T cells. Only samples showing at least a 1% CD107a response over mock background for either paired CD4+ T cell or macrophages were included in the analysis. For assessment of effector response quality, frequencies IFN-γ were assessed in CD107a+ CTLs. The final analysis of IFN-γ was expressed as the CTL fold response to macrophages over CD4+ T cells. For ELISA-based recognition assays, levels of IFN-γ were normalized to levels of CD4+ T cell productive infection as described for normalization of CD107a frequencies.
Imaging flow cytometry (ImageStream) assays
For assessment of MHC-I surface density, activated CD4+ T cells and macrophages were harvested and stained with anti-MHC-I-BV510 (clone W6/32; BioLegend) and LIVE/DEAD Near-IR (Invitrogen), followed by fixing and intracellular staining for actin using Phalloidin-AF555 (Invitrogen). 5,000 – 10,000 events were collected from each sample on the ImageStream X Mark II imaging flow cytometer (Amnis) and analyzed using IDEAS Software (Millipore). Events in focus were gated followed by cells with an actin staining Aspect Ratio of 0.9-1.0, representing cells with near circular morphology. Live cells were gated followed by assessment MHC-I mean fluorescence intensity (MFI) and cell height (diameter in μm). “Corrected” MFI was calculated by subtracting the MFI of the fluorescence minus one (FMO) staining control from the MHC-I-BV510-stained sample. Surface area was calculated via the diameter of the cells using the formula: Surface Area = π*(diameter)2. Thus, relative surface density of MHC-I for both cell types was calculated by dividing the corrected MFI of MHC-I by the surface area.
For the conjugate/synapse formation assay, peptide-loaded CD4+ T cells, macrophages, and expanded effector CTLs were co-cultured at an E:T of 2 in 96-well round bottom plates at 30 million/mL in R10/50 for 0, 10, 30 or 60 minutes at 37°C before 100 μl of 4% paraformaldehyde was added directly to the cells at 4°C for 15 minutes. Cells were washed, permeabilized and stained with Phalloidin-AF555 (Invitrogen) for actin staining. 50,000 events were collected from each sample on the ImageStream X Mark II imaging flow cytometer and analyzed using IDEAS Software. Frequencies of immunological synapses were calculated by dividing the number of conjugates (effector-target pairs) with immunological synapses (concentrated actin at the cell-cell interface32) by the total number of FarRed+ peptide-loaded targets for each time point.
Chemotaxis assays
CD4+ T cells were activated as describe above for HIV infection, and rested for 3 days in R10/50. CD4+ T cells and monocytes were isolated from PBMCs as described above to obtain ex vivo cells. All three cell types were resuspended to 2.6 million/mL in R10 and 75μl was plated onto 3.0 μm pore polycarbonate membranes of a HTS transwell 96 well plate (Corning, Corning, NY). Recombinant human chemokines CXCL9, CXCL10, MIP-1α, MIP-1β, and CCL2 (BioLegend) were titered from 400ng/mL to 0.64ng/mL in R10. 235μl of each chemokine concentration was plated in the lower wells of the transwell plate. The membrane support containing the cells was then lowered onto the bottom chamber containing the chemokine media and incubated at 37°C for 1.5 hours. The membrane support was carefully removed and the media transferred to a 96 well round bottom plate and centrifuged to pellet the cells. 135μl of media was removed and 100μl of CellTiter-Glo 2.0 Assay reagent (Promega, Madison, WI) was mixed with the remaining media and bioluminescence readings were taken in black 96 well plates (Corning). In parallel, a standard curve was created for each cell type, with top cell numbers per well of 200,000 and 10-fold dilutions down to 20. Bioluminescence readings from the cell dilutions were used to create a standard curve to allow for calculation of the number of cells migrated to the lower chambers of each well. Each cell type was also phenotyped for chemokine receptor expression using antibodies anti-CD4-AF488 (clone OKT4; BioLegend), anti-CCR2-PE (clone K036C2; BioLegend), anti-CD3-APC/Cy7 (clone HIT3a; BioLegend), anti-CD14-AF700 (clone M5E2; BioLegend), anti-CXCR3-AF647 (clone G025H7; BioLegend), and anti-CCR5-Pacific Blue (clone J418F1; BioLegend), and LIVE/DEAD Blue (ThermoFischer).
Statistical analyses
The data were summarized using descriptive measures such as mean, standard deviation, median, inter quartile range (IQR), frequency and percent (%). Statistical tests such as one-sample, two-sample and paired t-tests and their non-parametric alternatives (Wilcoxon signed rank, Mann-Whitney, Wilcoxon matched-pairs signed rank) were used to compare outcome variables. Normality was assessed using diagnostic plots (e.g., histogram, box and normal probability plots) and statistical tests (e.g., skewness, kurtosis and Shapiro-Wilks). All p-values are two-sided and p < 0.05 was considered significant. Statistical analysis and graphing were performed using GraphPad Prism 6.0.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. All data generated or analyzed during this study are included in this published article (and its supplementary information files). All Fig.s have associated raw data.
Reporting Summary
The “Life Sciences Reporting Summary” and “Flow Cytometry Reporting Checklist” containing more specific experimental details are published along with this manuscript.
Supplementary Material
Acknowledgments
We thank Drs. G. Mylvaganam, G. Gaiha, T. Diefenbach and A. Balazs for their helpful comments. We thank Dr. Joseph Trapani for his helpful discussions regarding granzymes, and A. Piechocka-Trocha for experimental help. We thank the Flow Cytometry and Sample Processing Cores at the Ragon Institute for their help with instrumentation and processing of the samples. Funding support was provided by the Howard Hughes Medical Institute (D.R.C. and B.D.W.), the Ragon Institute of MGH, MIT and Harvard (B.D.W.), the Harvard University Center for AIDS Research (P30 AI060354, B.D.W., which is supported by the following NIH co-funded and participating Institutes and Centers: NIAID, NCI, NICHD, NHLBI, NIDA, NIMH, NIA, FIC, and OAR), NIH R01 AI118544 (B.D.W.), and amfAR (Grant ID 109326-59-RGRL, KLC and B.D.W.). K.L.C also acknowledges salary support from the Canadian Institute of Health Research (CIHR).
Footnotes
DATA AVAILIBILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request. All data generated or analyzed during this study are included in this published article (and its supplementary information files). All Fig.s have associated raw data.
AUTHOR CONTRIBUTIONS
K.L.C designed and performed the experiments and wrote the manuscript. D.R.C and B.D.W. contributed to the design of the experiments and writing of the manuscript. J. Lengieza performed experiments and optimized macrophage infection conditions. M.G provided advice for statistical analysis. F.D contributed to the discussions of granzyme-induced cell death experiments. J. Lieberman provided guidance and design for apoptosis, granzyme and perforin experiments, and editing of the manuscript. B.D.W. provided overall supervision of the project.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Swingler S, Mann AM, Zhou J, Swingler C, Stevenson M. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS pathogens. 2007;3:1281–1290. doi: 10.1371/journal.ppat.0030134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Groot F, Welsch S, Sattentau QJ. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood. 2008;111:4660–4663. doi: 10.1182/blood-2007-12-130070. [DOI] [PubMed] [Google Scholar]
- 3.Duncan CJ, et al. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. Journal of virology. 2014;88:2025–2034. doi: 10.1128/JVI.03245-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins DR, Lubow J, Lukic Z, Mashiba M, Collins KL. Vpr Promotes Macrophage-Dependent HIV-1 Infection of CD4+ T Lymphocytes. PLoS pathogens. 2015;11:e1005054. doi: 10.1371/journal.ppat.1005054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Honeycutt JB, et al. Macrophages sustain HIV replication in vivo independently of T cells. The Journal of clinical investigation. 2016;126:1353–1366. doi: 10.1172/JCI84456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honeycutt JB, et al. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nature medicine. 2017;23:638–643. doi: 10.1038/nm.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avalos CR, et al. Quantitation of Productively Infected Monocytes and Macrophages of Simian Immunodeficiency Virus-Infected Macaques. Journal of virology. 2016;90:5643–5656. doi: 10.1128/JVI.00290-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avalos CR, et al. Brain Macrophages in Simian Immunodeficiency Virus-Infected, Antiretroviral-Suppressed Macaques: a Functional Latent Reservoir. MBio. 2017;8:e01186–01117. doi: 10.1128/mBio.01186-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nowlin BT, et al. SIV encephalitis lesions are composed of CD163(+) macrophages present in the central nervous system during early SIV infection and SIV-positive macrophages recruited terminally with AIDS. The American journal of pathology. 2015;185:1649–1665. doi: 10.1016/j.ajpath.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sattentau QJ, Stevenson M. Macrophages and HIV-1: An Unhealthy Constellation. Cell host & microbe. 2016;19:304–310. doi: 10.1016/j.chom.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiNapoli SR, Hirsch VM, Brenchley JM. Macrophages in Progressive Human Immunodeficiency Virus/Simian Immunodeficiency Virus Infections. Journal of virology. 2016;90:7596–7606. doi: 10.1128/JVI.00672-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamers SL, et al. HIV-1 Nef in macrophage-mediated disease pathogenesis. Int Rev Immunol. 2012;31:432–450. doi: 10.3109/08830185.2012.737073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu DC, Sereti I, Ananworanich J. Serious Non-AIDS events: Immunopathogenesis and interventional strategies. AIDS Res Ther. 2013;10:29. doi: 10.1186/1742-6405-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Streeck H, Nixon DF. T cell immunity in acute HIV-1 infection. The Journal of infectious diseases. 2010;202(Suppl 2):S302–308. doi: 10.1086/655652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goulder PJ, Walker BD. The great escape - AIDS viruses and immune control. Nature medicine. 1999;5:1233–1235. doi: 10.1038/15184. [DOI] [PubMed] [Google Scholar]
- 16.Fujiwara M, Takiguchi M. HIV-1-specific CTLs effectively suppress replication of HIV-1 in HIV-1-infected macrophages. Blood. 2007;109:4832–4838. doi: 10.1182/blood-2006-07-037481. [DOI] [PubMed] [Google Scholar]
- 17.Severino ME, et al. Inhibition of human immunodeficiency virus type 1 replication in primary CD4(+) T lymphocytes, monocytes, and dendritic cells by cytotoxic T lymphocytes. Journal of virology. 2000;74:6695–6699. doi: 10.1128/jvi.74.14.6695-6699.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker-Sperling VE, Buckheit RW, 3rd, Blankson JN. Comparative analysis of the capacity of elite suppressor CD4+ and CD8+ T cells to inhibit HIV-1 replication in monocyte-derived macrophages. Journal of virology. 2014;88:9789–9798. doi: 10.1128/JVI.00860-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker-Sperling VE, et al. Short Communication: HIV Controller T Cells Effectively Inhibit Viral Replication in Alveolar Macrophages. AIDS research and human retroviruses. 2016;32:1097–1099. doi: 10.1089/aid.2016.0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rainho JN, et al. Nef Is Dispensable for Resistance of Simian Immunodeficiency Virus-Infected Macrophages to CD8+ T Cell Killing. Journal of virology. 2015;89:10625–10636. doi: 10.1128/JVI.01699-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vojnov L, et al. The majority of freshly sorted simian immunodeficiency virus (SIV)-specific CD8(+) T cells cannot suppress viral replication in SIV-infected macrophages. Journal of virology. 2012;86:4682–4687. doi: 10.1128/JVI.06324-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker BD, Yu XG. Unravelling the mechanisms of durable control of HIV-1. Nature reviews Immunology. 2013;13:487–498. doi: 10.1038/nri3478. [DOI] [PubMed] [Google Scholar]
- 23.Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. The Journal of biological chemistry. 2001;276:7320–7326. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]
- 24.Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nature reviews Immunology. 2003;3:361–370. doi: 10.1038/nri1083. [DOI] [PubMed] [Google Scholar]
- 25.Belizario J, Vieira-Cordeiro L, Enns S. Necroptotic Cell Death Signaling and Execution Pathway: Lessons from Knockout Mice. Mediators Inflamm. 2015;2015:128076. doi: 10.1155/2015/128076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lieberman J. Cell-Mediated Cytotoxicity. In: Paul WE, ebrary Inc, editor. Fundamental immunology. Seventh. p. 1. online resource (1303 pages) [Google Scholar]
- 27.Kaiserman D, Bird PI. Control of granzymes by serpins. Cell death and differentiation. 2010;17:586–595. doi: 10.1038/cdd.2009.169. [DOI] [PubMed] [Google Scholar]
- 28.Kataoka T, et al. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell-mediated cytotoxicity. J Immunol. 1996;156:3678–3686. [PubMed] [Google Scholar]
- 29.Sutton VR, et al. Granzyme B triggers a prolonged pressure to die in Bcl-2 overexpressing cells, defining a window of opportunity for effective treatment with ABT-737. Cell death & disease. 2012;3:e344. doi: 10.1038/cddis.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenkins MR, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. The Journal of experimental medicine. 2015;212:307–317. doi: 10.1084/jem.20140964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stepp SE, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 32.Calabia-Linares C, et al. Endosomal clathrin drives actin accumulation at the immunological synapse. J Cell Sci. 2011;124:820–830. doi: 10.1242/jcs.078832. [DOI] [PubMed] [Google Scholar]
- 33.Corbera-Bellalta M, et al. Blocking interferon gamma reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis. 2016;75:1177–1186. doi: 10.1136/annrheumdis-2015-208371. [DOI] [PubMed] [Google Scholar]
- 34.Foley JF, et al. Roles for CXC chemokine ligands 10 and 11 in recruiting CD4+ T cells to HIV-1-infected monocyte-derived macrophages, dendritic cells, and lymph nodes. J Immunol. 2005;174:4892–4900. doi: 10.4049/jimmunol.174.8.4892. [DOI] [PubMed] [Google Scholar]
- 35.Reinhart TA, et al. Increased expression of the inflammatory chemokine CXC chemokine ligand 9/monokine induced by interferon-gamma in lymphoid tissues of rhesus macaques during simian immunodeficiency virus infection and acquired immunodeficiency syndrome. Blood. 2002;99:3119–3128. doi: 10.1182/blood.v99.9.3119. [DOI] [PubMed] [Google Scholar]
- 36.de Poot SA, et al. Granzyme M targets topoisomerase II alpha to trigger cell cycle arrest and caspase-dependent apoptosis. Cell death and differentiation. 2014;21:416–426. doi: 10.1038/cdd.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ewen CL, Kane KP, Bleackley RC. Granzyme H induces cell death primarily via a Bcl-2-sensitive mitochondrial cell death pathway that does not require direct Bid activation. Molecular immunology. 2013;54:309–318. doi: 10.1016/j.molimm.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Roederer M. Differential susceptibility of leukocyte subsets to cytotoxic T cell killing: implications for HIV immunopathogenesis. Cytometry A. 2007;71:94–104. doi: 10.1002/cyto.a.20363. [DOI] [PubMed] [Google Scholar]
- 39.Medema JP, et al. Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte-induced apoptosis: differential modulation by T helper type 1 and type 2 cells. The Journal of experimental medicine. 2001;194:657–667. doi: 10.1084/jem.194.5.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Migueles SA, et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nature immunology. 2002;3:1061–1068. doi: 10.1038/ni845. [DOI] [PubMed] [Google Scholar]
- 41.Wherry EJ. T cell exhaustion. Nature immunology. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 42.Hersperger AR, et al. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS pathogens. 2010;6:e1000917. doi: 10.1371/journal.ppat.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trimble LA, Lieberman J. Circulating CD8 T lymphocytes in human immunodeficiency virus-infected individuals have impaired function and downmodulate CD3 zeta, the signaling chain of the T-cell receptor complex. Blood. 1998;91:585–594. [PubMed] [Google Scholar]
- 44.Draenert R, et al. Persistent recognition of autologous virus by high-avidity CD8 T cells in chronic, progressive human immunodeficiency virus type 1 infection. Journal of virology. 2004;78:630–641. doi: 10.1128/JVI.78.2.630-641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Appay V, et al. HIV-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. The Journal of experimental medicine. 2000;192:63–75. doi: 10.1084/jem.192.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Migueles SA, et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity. 2008;29:1009–1021. doi: 10.1016/j.immuni.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Migueles SA, et al. Defective human immunodeficiency virus-specific CD8+ T-cell polyfunctionality, proliferation, and cytotoxicity are not restored by antiretroviral therapy. Journal of virology. 2009;83:11876–11889. doi: 10.1128/JVI.01153-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pachlopnik Schmid J, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO molecular medicine. 2009;1:112–124. doi: 10.1002/emmm.200900009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Critchfield JW, et al. Magnitude and complexity of rectal mucosa HIV-1-specific CD8+ T-cell responses during chronic infection reflect clinical status. PloS one. 2008;3:e3577. doi: 10.1371/journal.pone.0003577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pereyra F, et al. Increased coronary atherosclerosis and immune activation in HIV-1 elite controllers. AIDS. 2012;26:2409–2412. doi: 10.1097/QAD.0b013e32835a9950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pereyra F, et al. HIV Control Is Mediated in Part by CD8+ T-Cell Targeting of Specific Epitopes. Journal of virology. 2014;88:12937–12948. doi: 10.1128/JVI.01004-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salerno-Goncalves R, Fernandez-Vina M, Lewinsohn DM, Sztein MB. Identification of a human HLA-E-restricted CD8+ T cell subset in volunteers immunized with Salmonella enterica serovar Typhi strain Ty21a typhoid vaccine. J Immunol. 2004;173:5852–5862. doi: 10.4049/jimmunol.173.9.5852. [DOI] [PubMed] [Google Scholar]
- 53.Metkar SS, et al. Granzyme B activates procaspase-3 which signals a mitochondrial amplification loop for maximal apoptosis. J Cell Biol. 2003;160:875–885. doi: 10.1083/jcb.200210158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bellucci R, et al. Tyrosine kinase pathways modulate tumor susceptibility to natural killer cells. The Journal of clinical investigation. 2012;122:2369–2383. doi: 10.1172/JCI58457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. All data generated or analyzed during this study are included in this published article (and its supplementary information files). All Fig.s have associated raw data.