

Summary
Interpretation of cardiac gain- and loss- of function experiments are prone to secondary, indirect effects that confound and obscure data interpretation. We advocate the use of a genetic mosaic strategy to circumvent these issues and increase precision in cardiovascular research.
Keywords: Genetic model, Cardiomyocyte maturation, Cardiac hypertrophy, Cas9 somatic mutagenesis
Honing in on direct effects using a genetic mosaic strategy
Gain- and loss- of function experiments have become mainstays to dissect gene functions. Meaningful interpretation of these experiments requires distinguishing the primary, direct consequences of genic manipulation from secondary, indirect effects. Direct effects of a genetic manipulation can lead indirect responses that alter, obscure, or even reverse the direct effects (Figure, A). Indirect responses may be further modified by the primary genetic manipulation, adding complexity to data interpretation. As a result, confounding secondary effects can be misinterpreted as the direct consequence of the genetic manipulation. To solve this problem, the genetic mosaic approach has been widely adopted in invertebrate model systems1. However, this approach is infrequently used in mammalian models.
Figure.
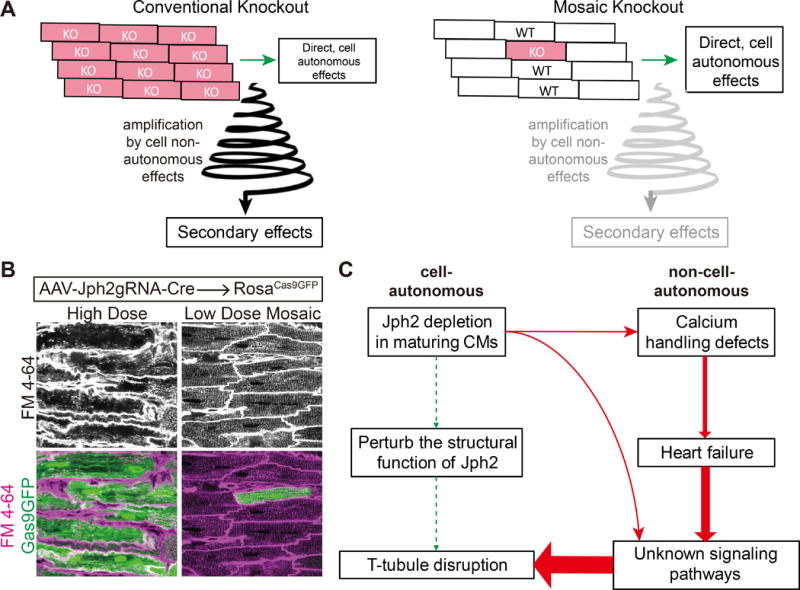
Defining cell autonomous effects using genetic mosaics. A. Comparison of whole organ versus mosaic gene manipulation. Gene in knocked out in most cardiomyocytes often results in organ dysfunction, leading to cell non-autonomous effects that amplify and obscure the direct effect. In the mosaic knockout, the unaffected cardiomyocytes limit the non-autonomous effects and thereby allow the investigator to identify the cell autonomous effects. B. Example comparing Cas9-mediated inactivation of Jph2 using high or low dose delivery of guide RNA using adenoassociated virus (AAV). Voltage-sensitive membrane dye FM4-64 staining within cardiomyocytes indicates T-tubulues. Note disruption of T-tubules in high dose, which causes heart failure, but not in low dose, which does not affect heart function. C. Interpretation of the different effects Jph2 inactivation in a high or low percentage of cardiomyocytes. Jph2 has little cell autonomous, direct effect on T-tubule formation, but its depletion in most cardiomyocytes causes heart failure, which indirectly destabilizes T-tubules.
In a genetic mosaic, a gene is perturbed in a low percentage of cells within a tissue, leaving the remaining cells unperturbed (Figure, A-B). The unperturbed cells provide a normal milieu and buffer against non-cell-autonomous secondary effect of organ dysfunction. Thus, genetic mosaics allow the investigator to focus on cell-autonomous effects that are proximal to the genetic manipulation. This approach also provides the opportunity to study otherwise lethal genetic manipulations, and to compare genetically modified and unmodified cells in the same tissue and genetic background.
The highly integrated structure and function of the heart makes cardiac research especially vulnerable to erroneous interpretation of data accurately recorded from whole heart gene manipulations. Recent advances have made the genetic mosaic approach feasible for mammalian cardiovascular biologists2–5. Here, we advocate for adoption of the genetic mosaic strategy to increase the precision of conclusions made from gain- and loss- of function experiments. First, we cover technical aspects of a genetic mosaic experiment. Next, we review examples of genetic mosaic experiments to highlight how this strategy improves precision in data interpretation.
Techniques to generate and study genetic mosaics
Genetic mosaics can be using Cre-loxP technology, in which Cre is expressed in a small fraction of cells within a tissue. In these cells, Cre catalyzes the inactivation of a floxed target gene. Neighboring cells lacking Cre remain unaffected. The most convenient method to deliver Cre to a low percentage of cardiomyocytes is to systemically administer AAV9-Tnnt2-Cre at a dose titrated to induce recombination in a low percentage (e.g. < 15%) of cardiomyocytes3,5. This treatment selectively targets cardiomyocytes in the heart, because AAV9 is cardiotropic, and Cre expression is driven from the cardiomyocyte-selective troponin T (Tnnt2) promoter. A ligand-dependent Cre, such as CreERT2 or MerCreMer, can also generate genetic mosaics, by administering a limiting dose of the ligand2,5. An alternative method that does not require a floxed allele is CRISPR/CAS9/AAV-mediated somatic mutagenesis (CASAAV)4,6. AAV9 that expresses both a guide RNA (gRNA) and Cre is delivered to Rosa26fsCas9 mice. Cre recombination activates Cas9 expression in cardiomyocytes, where it cleaves the gRNA-targeted gene, leading to frameshift mutations. CASAAV allows many genes to be inactivated in parallel without acquiring and mating the corresponding floxed alleles. However, CASAAV mutagenesis generates variable mutations through non-homologous end joining, and typical knockout efficiency in AAV-transduced cells is 50-70%, compared to 85-95% with Cre-loxP.
It is important to distinguish genetic mosaics from the more classical use of Cre-loxP in lineage-specific genetic manipulation. The latter method is effective to define gene functions within a lineage, but it is vulnerable to confounding secondary effects due to widespread gene manipulation within the the targeted lineage. Although lineage-specific Cre-loxP experiments are commonly described as revealing “cell-autonomous” gene functions, this is inaccurate since they define the function of a gene at the resolution of a lineage, rather than a single cell.
Identifying the mutant cells in a genetic mosaic is pivotal to data interpretation. When a suitable antibody is available, this can be achieved by immunofluorescent staining. A Cre-activated fluorescent reporter can be a useful surrogate and enables flow cytometry-based purification of Cre-exposed cells. Defining the extent to the reporter corresponds to gene ablation is crucial to carefully evaluate experimental results. With low dose AAV-Cre, generally over 85% of reporter-positive cells have undergone target gene ablation. This concordance may be lower using low dose tamoxifen, because this method intentionally limits Cre activity to a level that does not reliably recombine all floxed targets in a cell. With CASAAV, we typically observe concordance rates of 50-70%, because some alleles might not be mutagenized by Cas9 and others will acquire in-frame mutations that permit continued protein synthesis.
Applications of genetic mosaics: cardiomyocyte maturation and beyond
Our lab has used the genetic mosaic strategy to study postnatal cardiomyocyte maturation. While cardiomyocyte lineage specification has been studied extensively, relatively less is known about the fetal-to-adult transition of cardiomyocytes7. One barrier that has contributed to this knowledge gap is the lack of tools to interrogate this process in vivo. A second barrier is that heart dysfunction itself strong affects cardiomyocyte maturation. CASAAV and the genetic mosaic strategy offer avenues to surmount these barriers.
As an example, we review our study4 of genes required for development of transverse tubules (T-tubules), the membrane invaginations that facilitate calcium handling in adult cardiomyocytes and that are hallmarks of cardiomyocyte maturation. Junctophilin-2 (JPH2) tethers T-tubules to the sarcoplasmic reticulum. Broad cardiomyocyte-specific knockdown of Jph2 caused dramatic T-tubule loss, which led to the conclusion that Jph2 is required for T-tubule maturation8,9. However, this manipulation was also associated with severe cardiac dysfunction, which itself causes T-tubule abnormalities10. Using CASAAV at different AAV doses, we depleted Jph2 in either a low or high percentage of cardiomyocytes. High percentage depletion caused heart failure and T-tubule disarray, consistent with prior studies. However, mosaic depletion in a low percentage of cardiomyocytes did not disturb heart function and was associated with minimal disorganization of T-tubules4 (Figure, B). This study demonstrated that Jph2 has a minimal cell autonomous essential role in T-tubule formation. This study highlighted how mosaic analysis can overcome confounding secondary effects of organ-wide, cardiomyocyte-specific gene depletion to improve precision in data interpretation.
We also used the mosaic inactivation strategy to assess whether the neonatal shift to oxidative phosphorylation is required for overall cardiomyocyte maturation5. We inactivated Tfam, a nuclear gene that is required for mitochondrial DNA replication and transcription5, to broadly impair mitochondrial function. Previous studies showed that fetal or perinatal Tfam inactivation in cardiomyocytes caused death by 3-4 weeks of life11, but the effects on cardiomyocyte maturation were not explored. We studied mosaic knockout of Tfam in fetal cardiomyocytes and showed that it is cell autonomously required for cardiomyocyte proliferation. Elevated reactive oxygen species in Tfam-ablated cardiomyocytes activated DNA damage response pathways. Surprisingly, mosaic Tfam ablation in perinatal cardiomyocytes was compatible with normal structural maturation, including maturational hypertrophy, myofibril expansion, and T-tubulation5. These findings suggest that the switch to oxidative phosphorylation is not essential for attaining other hallmarks of cardiomyocyte maturation.
We also used the genetic mosaic strategy to study transcriptional regulation of cardiomyocyte maturation. Previous work showed that Gata4 and Gata6, key cardiac transcription factors, are required to maintain heart function and pathological cardiomyocyte hypertrophy12,13. Using low dose AAV-TNT-Cre, we achieved mosaic ablation of both genes while preserving heart function, which revealed an essential, cell autonomous requirement for Gata4 and Gata6 for normal maturational cardiomyocyte growth3.
Although we have focused on the application of the genetic mosaic strategy to the dissection of cardiomyocyte maturation, this approach is likely to improve precision in many areas of cardiovascular research, especially in postnatal stages that are most amenable to stage-specific, mosaic gene inactivation using AAV and either Cre/loxP or CASAAV. For instance, a major focal point of cardiovascular research has been pathological cardiac hypertrophy, an independent risk factor for adverse cardiac outcomes14. Under pathological conditions, intracellular signaling pathways and transcriptional programs result in cell-autonomous cardiomyocyte hypertrophy, which predisposes to cell death or dysfunction. In traditional lineage-specific overexpression or knockout models, perturbation of these pathways frequently causes heart dysfunction and dilation, which then complicates interpreting the direct effects of the manipulation. In vitro culture and time course experiments have been used to disentangle these interlocking variables, but a genetic mosaic approach that perturbs these pathways without global disruption of organ function or chamber size would more clearly delineate cell-autonomous relationships and better link perturbations to molecular mechanisms in vivo. Similar arguments could be made regarding cardiac metabolism, gene transcription, fibrosis, and apoptosis, and their links to heart failure. Past research in these areas has been built on cardiac gene manipulations that affect most cells within a lineage. It will be worthwhile revising these studies using adult-specific, genetic mosaic approaches to dissect cell-autonomous direct effects from indirect, confounding secondary effects.
We also anticipate that genetic mosaic studies will yield another bounty: in vivo, forward genetic screens in cardiomyocytes. While murine forward genetic screens have been performed15, they are time and resource intensive. The major driver of cost is the need to breed mice and study many mice per gene. Using CASAAV-based mosaic mutagenesis, the unit of study becomes the individual cardiomyocytes, and there are about 2 million cardiomyocytes per mouse. Therefore, using pooled libraries of guide RNAs, it should be possible to study many genes per mouse. We anticipate that forward genetics will take us in new, unexpected directions when applied to cardiac problems such as maturation and hypertrophy.
Acknowledgments
Sources of Funding
WTP is supported by NIH (R01 HL128694 and UM1HL098166), AHA (IRG33410894), and charitable donations from the Edwin August Boger, Jr. Fund.
Footnotes
Disclosures
None
References
- 1.Xu T, Rubin GM. The effort to make mosaic analysis a household tool. Development. 2012;139:4501–4503. doi: 10.1242/dev.085183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, van Gorp PRR, Zhou B, Chen J, Seidman JG, Wang D-Z, Pu WT. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res. 2014;115:354–363. doi: 10.1161/CIRCRESAHA.115.303632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prendiville TW, Guo H, Lin Z, Zhou P, Stevens SM, He A, VanDusen N, Chen J, Zhong L, Wang D-Z, Gao G, Pu WT. Novel Roles of GATA4/6 in the Postnatal Heart Identified through Temporally Controlled, Cardiomyocyte-Specific Gene Inactivation by Adeno-Associated Virus Delivery of Cre Recombinase. PLoS One. 2015;10:e0128105. doi: 10.1371/journal.pone.0128105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo Y, VanDusen NJ, Zhang L, Gu W, Sethi I, Guatimosim S, Ma Q, Jardin BD, Ai Y, Zhang D, Chen B, Guo A, Yuan G-C, Song L-S, Pu WT. Analysis of Cardiac Myocyte Maturation Using CASAAV, A Platform for Rapid Dissection of Cardiac Myocyte Gene Function In Vivo. Circ Res [Internet] 2017 doi: 10.1161/CIRCRESAHA.116.310283. Available from: http://dx.doi.org/10.1161/CIRCRESAHA.116.310283. [DOI] [PMC free article] [PubMed]
- 5.Zhang D, Li Y, Heims-Waldron D, Bezzerides V, Guatimosim S, Guo Y, Gu F, Zhou P, Lin Z, Ma Q, Liu J, Wang D-Z, Pu WT. Mitochondrial Cardiomyopathy Caused by Elevated Reactive Oxygen Species and Impaired Cardiomyocyte Proliferation. Circ Res. 2018;122:74–87. doi: 10.1161/CIRCRESAHA.117.311349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.VanDusen NJ, Guo Y, Gu W, Pu WT. CASAAV: A CRISPR-Based Platform for Rapid Dissection of Gene Function In Vivo. Curr Protoc Mol Biol. 2017;120:31.11.1–31.11.14. doi: 10.1002/cpmb.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galdos FX, Guo Y, Paige SL, VanDusen NJ, Wu SM, Pu WT. Cardiac Regeneration: Lessons From Development. Circ Res. 2017;120:941–959. doi: 10.1161/CIRCRESAHA.116.309040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, Skapura DG, Landstrom AP, Song L-S, Ackerman MJ, Wehrens XHT. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc Res. 2013;100:44–53. doi: 10.1093/cvr/cvt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen B, Guo A, Zhang C, Chen R, Zhu Y, Hong J, Kutschke W, Zimmerman K, Weiss RM, Zingman L, Anderson ME, Wehrens XHT, Song L-S. Critical roles of junctophilin-2 in T-tubule and excitation–contraction coupling maturation during postnatal development. Cardiovasc Res. 2013;100:54–62. doi: 10.1093/cvr/cvt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo A, Zhang C, Wei S, Chen B, Song L-S. Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc Res. 2013;98:204–215. doi: 10.1093/cvr/cvt020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Brüning JC, Kahn CR, Clayton DA, Barsh GS, Thorén P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- 12.van Berlo JH, Elrod JW, van den Hoogenhof MMG, York AJ, Aronow BJ, Duncan SA, Molkentin JD. The transcription factor GATA-6 regulates pathological cardiac hypertrophy. Circ Res. 2010;107:1032–1040. doi: 10.1161/CIRCRESAHA.110.220764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 14.Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122:2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, Chang C, Yagi H, San Agustin JT, Thahir M, Anderton S, Lawhead C, Vescovi A, Pratt H, Morgan J, Haynes L, Smith CL, Eppig JT, Reinholdt L, Francis R, Leatherbury L, Ganapathiraju MK, Tobita K, Pazour GJ, Lo CW. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015;521:520–524. doi: 10.1038/nature14269. [DOI] [PMC free article] [PubMed] [Google Scholar]