

Abstract
Inflammasomes are innate immune signaling platforms that are required for the successful control of many pathogenic organisms, but also promote inflammatory and autoinflammatory diseases. Inflammasomes are activated by cytosolic pattern recognition receptors, including members of the NOD-like receptor (NLR) family. These receptors oligomerize upon the detection of microbial or damage-associated stimuli. Subsequent recruitment of the adaptor protein ASC forms a microscopically visible inflammasome complex, which activates caspase-1 through proximity-induced auto-activation. Following the activation, caspase-1 cleaves pro-IL-1β and pro-IL-18, leading to the activation and secretion of these pro-inflammatory cytokines. Caspase-1 also mediates the inflammatory form of cell death termed pyroptosis, which features the loss of membrane integrity and cell lysis. Caspase-1 cleaves gasdermin D, releasing the N-terminal fragment which forms plasma membrane pores, leading to osmotic lysis.
In vitro, the activation of caspase-1 can be determined by labeling bone marrow-derived macrophages with the caspase-1 activity probe FAM-YVAD-FMK and by labeling the cells with antibodies against the adaptor protein ASC. This technique allows the identification of inflammasome formation and caspase-1 activation in individual cells using fluorescence microscopy. Pyroptotic cell death can be detected by measuring the release of cytosolic lactate dehydrogenase into the medium. This procedure is simple, cost effective and performed in a 96-well plate format, allowing adaptation for screening. In this manuscript, we show that activation of the NLRP3 inflammasome by nigericin leads to the co-localization of the adaptor protein ASC and active caspase-1, leading to pyroptosis.
Keywords: This Month in JoVE, Issue 135, Immunology, innate immunity, inflammasome, caspase-1, ASC, NLRP3, pyroptosis, nigericin, murine bone marrow-derived macrophages
Introduction
Inflammasome-mediated inflammation is a critical component of the defense against pathogenic organisms1, but also underlies the etiology of many diseases2. The inflammatory response to a wide range of infections is triggered by cytosolic detection of pathogen associated molecular patterns (PAMPs) or damage associated molecular patterns (DAMPs). Pattern recognition receptors (PRR), including members of the NOD-like receptor (NLR) family, oligomerize upon the detection of these PAMPs and DAMPs. This triggers the formation of a multi-protein complex termed the inflammasome, which contains the PRR, the adaptor protein Apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain (CARD) (ASC), and the pro-form of caspase-13,4. This complex allows proximity-induced auto-activation of caspase-1. Active caspase-1 then leads to a set of events that are characteristic of the inflammatory cell death pathway pyroptosis. These events include: the cleavage and release of the inflammatory cytokines IL-1β and IL-18, lysosome exocytosis with release of lysosomal contents into the extracellular space, nuclear condensation, and gasdermin D cleavage. The released N-terminal domain of gasdermin D inserts into the plasma membrane, forming pores that cause plasma membrane rupture and release of inflammatory contents, in addition to taking away a protective niche for pathogen replication5,6,7.
Here, we focus on the well-studied NLRP3 inflammasome. The activation of the NLRP3 inflammasome occurs through a two-step process8. The first "priming" step occurs through the recognition by Toll-like receptors (TLR) of a microbial product. This is replicated in the laboratory setting by using LPS to stimulate TLR4. This stimulation upregulates NLRP3 and pro-IL-1β through NF-kB signaling. Priming additionally licenses NLRP3 through non-transcriptional mechanisms by inducing its deubiquitination9,10 and phosphorylation or dephosphorylation of specific residues11,12.
The second signal for NLRP3 activation is thought to involve mitochondrial factors, reactive oxygen species, potassium efflux and calcium signaling, although a unifying mechanism for NLRP3 activation remains elusive8. Activated NLRP3 oligomerizes through interactions between its NACHT domains, and recruits ASC via binding of the pyrin (PYD) domains13. In each cell, these macromolecular complexes form a single microscopically visible focus. ASC was originally identified as a 22 KDa protein that forms a "speck" during the apoptosis of human leukemia cells, and named apoptosis-associated speck-like protein containing a CARD14. It was later determined that ASC recruits pro-caspase-1 through the interaction of the CARD of ASC with the CARD of pro-caspase-1, forming the inflammasome15.
Not all NLRs require the presence of ASC to induce caspase-1 activation. Unlike NLRP3, NLRC4 and NLRP1b have CARD domains and can directly recruit pro-caspase-1 via CARD-CARD interactions to induce caspase-1 activation. In the absence of ASC, active caspase-1 remains diffuse throughout the cytosol and does not form a single focus. This diffuse active caspase-1 is sufficient to induce pyroptotic cell death, but is unable to process pro-IL-1β13,16.
In this manuscript, we will discuss two ways to assess inflammasome activation. The first uses the fluorescent activity probe, FAM-YVAD-FMK, which binds the caspase-1 family of proteases. This family includes caspase-1, but also mouse caspase-11 and human caspase-4 and caspase-5. By using macrophages from caspase-1/11 deficient mice in all experiments, the specificity of this probe will be addressed. This method can be combined with antibody labeling of inflammasome components, which we will also describe. Microscopic visualization allows for the identification of individual cells containing active caspase-1 and oligomerized ASC. Using this method, researchers will be able to determine where in the inflammasome formation cascade their manipulations of the host cell or the pathogenic organism under study have an effect. For example, one can distinguish whether a given intervention prevents the recruitment of ASC to the NLRP3 complex or targets the subsequent recruitment and activation of caspase-117. Examining outcomes of caspase-1 activation such as IL-1β secretion would not be able to distinguish between these two possibilities. Also, the secretion of IL-1β could be altered without altering the cells ability to activate caspase-1 and undergo pyroptotic cell death16.
The second method measures the release of lactate dehydrogenase (LDH) into the cell supernatant, which occurs during pyroptotic macrophage lysis following caspase-1 activation as described above. This second method is a population-based approach as the release of LDH in the entire well is measured. This simple approach allows for the rapid analysis (30 min of incubation time) of samples to determine if there is caspase-1-mediated cell death and can be performed in a 96-well plate format.
These methods are complementary, each with different advantages, and both are easily amenable to modifications. For example, macrophage treatment with targeted small molecular weight compounds prior to inflammasome stimulation can be used to investigate the role certain proteins may have on controlling inflammatory responses.
Protocol
All animal procedures were approved and conducted under University of Washington Institutional Animal Care and Use Committee guidelines.
1. Harvest of Bone Marrow
Aseptically remove the femur and tibia from a mouse and clean the bones using sterile instruments. Place the cleaned bones in Dulbecco Modified Eagle Medium (DMEM) + 5 mM HEPES + 0.2 mg/mL L-glutamine + 0.05 mM 2-mercaptoethanol + 50 mg/mL gentamicin sulfate + 10,000 U/mL penicillin/streptomycin + 10% fetal bovine serum (FBS, DMEM-10 complete) and incubate the tube on ice for 15 min.
Remove the proximal and distal ball joints using scissors. Insert a 25 G needle attached to a 10 mL syringe filled with medium into the hollow of the bones. Flush out the bone marrow using DMEM-10 complete. Resuspend any clumps by pipetting the medium up and down gently.
Centrifuge the cells for 10 min at 300 x g and count the number of cells using a hemocytometer. At this point, either freeze the cells using 90% FBS + 10% dimethyl sulfoxide (DMSO) or plate the cells in a 15 cm Petri dish to differentiate macrophages using L929 conditioned medium (step 2.1).
2. Differentiation of Bone Marrow-derived Macrophages
Using pre-warmed differentiation medium (21 mL of DMEM-10 complete and 9 mL of L929 cell conditioned medium as described by Swanson et al.18), put bone marrow cells in a 15 cm non-tissue culture treated Petri dish (1.2-1.5 x 107 cells). Incubate the plate at 37 °C and 5% CO2 for 7 days. Add an additional 30 mL of differentiation medium to the plate at day 3 or 4. NOTE: It is important not to use tissue culture treated plates, as macrophages will be difficult to detach and harvest.
To collect macrophages, wash the plate with phosphate-buffered saline (PBS) and add 20 mL of cold PBS + 1 mM ethylenediaminetetraacetic acid (EDTA). Incubate the plate at 4 °C for 10 min. Harvest the cells by pipetting the PBS + EDTA over the cells and wash the plate with 10 mL of PBS. Combine both washes into two 15 mL conical tubes.
Centrifuge the cells at 300 x g for 10 min and resuspend the cells in 10 mL of phenol-red free DMEM + 5 mM HEPES + 0.2 mg/mL L-glutamine + 0.05 mM 2-mercaptoethanol + 5% FBS (DMEM-5) and count the cells using either a hemocytometer or a Coulter counter. Keep the cells on ice during the counting.
Seed the macrophages in tissue culture coated plates at 2 x 105 cells/mL. For microscopy experiments, seed 1 mL of cells on coverslips in 24 well plates. For LDH release assays, seed 100 µL of cells in a 96 well plate. For the LDH assay, seed 9 wells (3 wells untreated, 3 wells with 100% lysis controls and 3 wells for the experimental stimulus).
Centrifuge the 96 well plate for 5 min at 300 x g to make certain the cells are equally distributed across the well.
Incubate the plate overnight at 37 °C + 5% CO2 before starting the priming process.
3. Priming
Replace the medium with fresh medium (DMEM-5) containing 100 ng/mL LPS (500 μL for the 24 well plate or 50 μL for the 96 well plate). NOTE: There are a variety of structural variations in LPS, which affect the ability to stimulate TLR4-mediated priming19. LPS from Salmonella minnesota R595 (Re) is available from multiple vendors and is recommended.
Incubate the plate for 3 h at 37 °C and 5% CO2.
4. Activation of the NLRP3 Inflammasome with Nigericin and Labeling with FAM-YVAD-FMK
Remove the medium and replace it with 290 μL of DMEM-5 containing 5 μM nigericin and 5 mM glycine. Glycine is added to reduce the amount of cell lysis that occurs during caspase-1 activation5. Incubate for 60 min at 37 °C and 5% CO2. Use both wild type and caspase-1 deficient macrophages to ensure that the observed labeling is specific for caspase-1.
During the last 45 min, add 10 μL of 30x FAM-YVAD-FMK, prepared according to the manufacturer's instructions.
After 60 min, remove the medium and wash the cells three times for 5 min each with 1 mL of cold PBS.
Add 250 μL of 2% paraformaldehyde to each well and incubate on ice covered with aluminum foil for 30 min.
During the last 5 min, add 4 μL of 0.2 mM far-red fluorescent nucleic acid stain to label the nuclei. 4',6-diamidino-2-phenylindole (DAPI) or other dyes can also be used to label DNA.
Wash the cells three times with 1 mL of cold PBS and mount the coverslips onto microscope slides using 7 μL anti-fade mounting medium. Let the mounting medium harden over night before sealing the coverslip to the microscope slide using nail polish.
Image the macrophages by confocal microscopy. Ex/Em for FAM-YVAD-FMK is 492/520nm and 642/661 for far-red fluorescent nucleic acid stain. Make sure to set up the microscope such that untreated cells do not show any background staining in the 488 channel. Otherwise, background FAM-YVAD-FMK staining will be detected and not actual active caspase-1. NOTE: Microscope set-up is described in section 6.
5. Antibody Staining
NOTE: To label the cells with antibodies to detect ASC, seed, prime and expose the cells to nigericin identical to the previous section. The processing afterwards is as follows:
Wash the cells three times for 5 min each with 1 mL of cold PBS. Add 250 μL of fixation and permeabilization solution. Incubate the cells on ice, and covered for 30 min.
Wash the macrophages three times for 5 min each with 1 mL of wash buffer.
Add 250 μL of primary antibody against ASC diluted 1:500 in wash buffer and incubate for 1 h on ice. Include one coverslip that does not receive any primary antibody, but will only receive the secondary. This coverslip will be used during the microscope setup to set the correct offset.
Wash the cells three times for 5 min each with 1 mL wash buffer.
Add 250 μL of secondary antibody (fluorescent dye conjugated goat-anti-mouse) diluted 1:500 in wash buffer and incubate for 1 h on ice. Add 4 μL of 0.2 mM far-red fluorescent nucleic acid stain for the last 5 min of this incubation to label nuclei.
Wash the cells 3x with 1 mL of cold wash buffer and mount the coverslips onto microscope slides using 7 μL anti-fade mounting medium. Let the mounting medium harden over night before sealing the coverslip to the microscope slide using clear nail polish.
Image macrophages by confocal microscopy. Ex/Em for the secondary antibody is 555/580 nm and 642/661 for far-red fluorescent nucleic acid stain (as described below). NOTE: Labeling with FAM-YVAD-FMK and antibody labeling can be combined to show co-localization of active caspase-1 and ASC. For this, follow the conditions for FAM-YVAD-FMK labeling and then switch to the antibody labeling protocol for further processing.
6. Imaging of Labeled Macrophages by Confocal Microscopy
NOTE: The stained cells can be viewed after the coverslips have been allowed to cure overnight and have been sealed to the microscope slide with nail polish. Here, imaging using a confocal microscope is described. Cells can also be viewed by standard fluorescence microscopy.
Place the slide with the untreated control cells on the microscope stage and focus the microscope on the cells.
Using the microscope Look Up Table (LUT) settings, adjust the offset such that there is no positive FAM-YVAD-FMK staining in the untreated cells. This process can also be performed using caspase-1 deficient macrophages. NOTE: The offset has to be adjusted for each channel used in the experiment, but setting the correct offset is most critical for the FAM-YVAD-FMK channel and fluorescent antibody channels. The offset for the DNA channel is less critical. Once the offset has been set, do not adjust this setting for the rest of the experiment.
Switch the slide to the nigericin treated sample. Find a field that contains both cells that are positive and negative for FAM-YVAD-FMK staining. Adjust the imaging plane of the FAM-YVAD-FMK channel to the plane that has the highest intensity of positive staining. This will normally be the plane where the center of the inflammasome focus is imaged.
Adjust the gain such that foci are visible in cells that have condensed nuclei. After setting both gain and offset, leave these settings for the rest of the imaging session. NOTE: One easy way to determine if a cell is pyroptotic is by looking at nuclear morphology. Cells with active caspase-1 have rounded and condensed nuclei, while nucleoli are visible in cells without caspase-1 activation and the nuclear morphology is similar to those of untreated cells.
Collect images of 5 randomly selected fields at 100X total magnification. This will normally result in about 40 cells per image. Repeat this process for each sample.
Count the number of cells in each image using image analysis software. Then count the number of cells that have positive FAM-YVAD-FMK staining. Represent the level of caspase-1 activation as the percentage of FAM-YVAD-FMK positive cells.
7. LDH Release Assay
NOTE: For the LDH release assay, seed and prime the cells as described in the previous section, except use a 96 well plate. Do not remove the medium after priming.
Add 50 μL of DMEM-5 containing 10 μM nigericin to the experimental wells and 50 μL DMEM-5 to the spontaneous and 100% lysis control wells. Do not add glycine in this experiment since the goal is to measure cell lysis. Incubate for 60 min at 37 °C and 5% CO2.
After 30 min, add 10 μL of 10x lysis buffer to the 100% lysis control wells and add 10 μL of DMEM-5 to the other wells. During this incubation, remove 1 vial of substrate mix (1.5 mL of freeze-dried substrate) and 1 aliquot of assay buffer (~12 mL) from the freezer and let them thaw protected from light to room temperature. NOTE: The substrate mix contains a tetrazolium salt (iodonitro-tetrazolium violet), which is converted to a red formazan product.
After the 60 min total incubation, centrifuge the plate for 5 min at 500 x g at 10 °C. Transfer 50 μL of the supernatant to a clear flat-bottom plate. During this time, add 12 mL of assay buffer to the vial of substrate mix and incubate at room temperature for 5 min before using.
Add 50 μL of substrate to each well and incubate for 30 min in the dark. Include medium only wells for blank values. Check the plate after 15 min to make sure that the signal will not exceed the detection limit of the plate reader.
After 30 min, add 50 μL of stop solution (1 M acetic acid) and measure OD490 using a plate reader.
Calculate the percentage of cell lysis using the following formula: % Cytoxicity = ((Experimental - Spontaneous)/(Maximum - Spontaneous))×100 Experimental: nigericin treated cells; spontaneous: untreated cells; maximum: cells exposed to lysis buffer
Representative Results
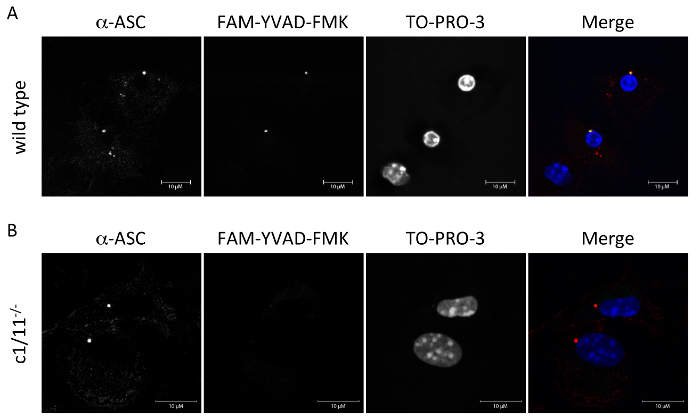
To detect caspase-1 activation following the exposure to nigericin, the cells were processed as described in the protocol section. We combined both FAM-YVAD-FMK and ASC antibody labeling in order to show that ASC and active caspase-1 co-localize following NLRP3-mediated inflammasome activation. Figure 1A shows that wild type macrophages exposed to 5 μM nigericin have formation of an ASC focus in the perinuclear region. These foci also contain active caspase-1 as seen by the co-localization of ASC with FAM-YVAD-FMK. The cells that are positive for active caspase-1 have nuclear condensation showing that these cells are undergoing pyroptosis. Figure 1B shows that while macrophages of caspase-1/11 deficient mice do have the formation of an ASC focus, they do not have any active caspase-1 associated with this focus as shown by the absence of FAM-YVAD-FMK staining. The nuclei of these cells are not condensed, indicating that there is no pyroptotic cell death occurring. Images were taken using a scanning confocal microscope with a 63X oil immersion objective. Images were saved as TIFF files without any further processing.
In order to determine the level of cell death that is associated with exposure to nigericin, we performed an LDH release assay on the culture supernatant. Figure 2 shows that wild type macrophages undergo cell death after the exposure to nigericin. In contrast, macrophages deficient in caspase-1 do not release LDH into the cell supernatant, indicating that the cells are intact.
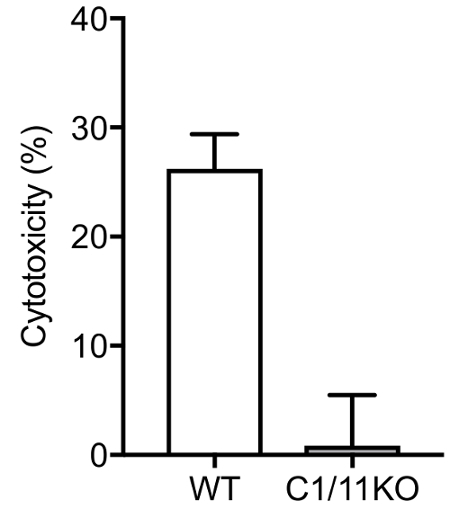
Figure 1: FAM-YVAD-FMK and anti-ASC labeling. (A) Wild type (WT) and (B) caspase-1/11 deficient (c1/11-/-) macrophages were exposed to 5 μM nigericin for 1 h and analyzed for active caspase-1 by confocal microscopy using FAM-YVAD-FMK to detect active caspase-1, and anti-ASC primary and goat-anti-mouse secondary antibodies to detect ASC focus formation. Please click here to view a larger version of this figure.
Figure 2: LDH release assay results. Wild type and caspase-1/11 deficient macrophages were exposed to 5 μM nigericin for 1 h and cell supernatant was analyzed for the presence of LDH. Data are means ± SD of three replicates. Please click here to view a larger version of this figure.
Discussion
In this manuscript, we have presented two techniques to examine inflammasome activation and the consequence of caspase-1 activation following NLRP3 stimulation in murine bone marrow-derived macrophages. The first technique allows the researcher to determine the level of caspase-1 activation on a cellular basis using the fluorescent reporter FAM-YVAD-FMK. This reporter is highly specific for binding the caspase-1 family of enzymes, as seen by the absence of staining in caspase-1/11 deficient macrophages. Specificity was also shown by LaRock et al. in their manuscript describing the role of the Yersinia protein YopM on caspase-1 activation. In their manuscript, they show that FAM-YVAD-FMK foci overlaps with foci that were labeled with antibodies directed against caspase-117.
As is the case with any experiment, there are some critical steps that need to be followed in order to eliminate false positive results. First of all, it is imperative to use both wild type and caspase-1/11 deficient macrophages. By using these two cell types, conditions that lead to non-specific staining can be excluded from further analysis. Second, as with any antibody or activity probe labeling assay, it is important to thoroughly wash the cells after staining with FAM-YVAD-FMK. By using three 5 min washes, the level of background signal is greatly reduced. A third critical step occurs during the setup of the confocal microscope. Here, it is important that untreated cells labeled with FAM-YVAD-FMK are used to set the offset of the confocal microscope in such a way that no positive cytoplasmic staining is observed in the untreated cells. When these steps are followed, this technique is a useful tool to assess activation of caspase-1 on a cellular basis.
This protocol can be modified to suit the researchers needs. For example, the cells can be treated with small molecular weight compounds at several steps during the entire process. This would allow for the identification of pathways that modulate the oligomerization of the NLR, the recruitment of either ASC or pro-caspase-1, or the activation of caspase-1 itself. The modifications to this protocol have to be such that they do not activate caspase-1 by itself. If this occurs, a dose response curve of the drug in question is required. Even though we focused on the activation of the NLRP3 inflammasome, this protocol can also be used to examine inflammasomes triggered by other PRRs. Another modification we have included in this manuscript is the combination of FAM-YVAD-FMK staining with antibody labeling of inflammasome components. We show that there is co-localization of the inflammasome adaptor ASC with active caspase-1. A limitation of this method as described, is that it requires access to a confocal microscope. However, a related strategy to detect ASC focus formation by flow cytometry has been reported20,21.
The second technique we discussed allows for the rapid detection of cell lysis using an LDH release assay. It is important to note that LDH release indicates loss of plasma membrane integrity and cell lysis, which also occurs during many other forms of cell death, and is not specific to pyroptosis. However, the use of caspase-1 deficient cells can identify caspase-1-dependent cell lysis, a defining feature of pyroptosis.
A critical step in this protocol (especially when using an infectious agent) is to centrifuge the plate after seeding the cells in a 96 well plate. Centrifugation of the plate allows for equal distribution of macrophages across the entire surface of the well. With equal distribution, each macrophage will be exposed to the same number of bacteria. Without centrifugation, the meniscus effect of the fluid in the well will lead to more macrophages accumulating at the edge of the well. This would lead to lower than intended multiplicity of infection (MOI) close to the well wall and higher than intended MOI in the center of the well.
A potential disadvantage of the LDH release assay is that it measures lysis on a population level and at a single time point per assay. An alternative approach is to quantify uptake of cell impermeant dyes by microscopy, which identifies loss of membrane integrity on a single cell basis. Combined with live cell imaging platforms, this approach can also provide a temporal assessment of pyroptosis22. Small DNA-binding fluorescent dyes such as propidium iodide or ethidium bromide can pass through the gasdermin D pore, bind cellular nucleic acids, and fluoresce5,22,23. Pore formation and uptake of these small dyes temporally precedes terminal cell lysis. Lysis allows uptake of larger dyes and loss of large cellular proteins such as LDH. The use of glycine can experimentally uncouple pore formation from lysis, as glycine does not block gasdermin D-mediated pore formation, small dye uptake or IL-1β secretion, but glycine does prevent rapid lysis and LDH release5,22,23,24. We suggest including glycine in FAM-YAVD-FMK staining experiments to prevent complete loss of membrane integrity, but glycine must be omitted when measuring pyroptotic LDH release.
Disclosures
The authors have no conflicts of interest to disclose
Acknowledgments
All microscopy was done at the W. M. Keck Microscopy Center with support of Nathaniel Peters and with support of NIH award S10OD016240. S.L.F. is supported by NIH K08AI119142 and R21AI130281. We thank Dr. Richard Flavell and Dr. Brad Cookson for caspase-1/11 deficient mice, and Dr. Brad Cookson for sharing laboratory facilities and equipment.
References
- Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265(1):130–142. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8(11):1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, den Hartigh AB, Loomis WP, Cookson BT. Coordinated host responses during pyroptosis: caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J Immunol. 2011;187(5):2748–2754. doi: 10.4049/jimmunol.1100477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. 2014;1319:82–95. doi: 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliana C, et al. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287(43):36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49(2):331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Song N, et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell. 2017;68(1):185–197. doi: 10.1016/j.molcel.2017.08.017. [DOI] [PubMed] [Google Scholar]
- Stutz A, et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med. 2017;214(6):1725–1736. doi: 10.1084/jem.20160933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- Masumoto J, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274(48):33835–33838. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, et al. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor molecules. Genes Cells. 2004;9(11):1055–1067. doi: 10.1111/j.1365-2443.2004.00789.x. [DOI] [PubMed] [Google Scholar]
- Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243(1):206–214. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRock CN, Cookson BT. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe. 2012;12(6):799–805. doi: 10.1016/j.chom.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63(9):3609–3620. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3(1):36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- Sester DP, et al. A novel flow cytometric method to assess inflammasome formation. J Immunol. 2015;194(1):455–462. doi: 10.4049/jimmunol.1401110. [DOI] [PubMed] [Google Scholar]
- Sester DP, et al. Assessment of Inflammasome Formation by Flow Cytometry. Curr Protoc Immunol. 2016. [DOI] [PubMed]
- DiPeso L, Ji DX, Vance RE, Price JV. Cell death and cell lysis are separable events during pyroptosis. Cell Death Discov. 2017. [DOI] [PMC free article] [PubMed]
- Russo HM, et al. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J Immunol. 2016;197(4):1353–1367. doi: 10.4049/jimmunol.1600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold CL, et al. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity. 2017. [DOI] [PMC free article] [PubMed]