

VR combined with cloud computing enables surgical manipulation of real-time molecular simulations, accelerating 3D research tasks.
Abstract
We describe a framework for interactive molecular dynamics in a multiuser virtual reality (VR) environment, combining rigorous cloud-mounted atomistic physics simulations with commodity VR hardware, which we have made accessible to readers (see isci.itch.io/nsb-imd). It allows users to visualize and sample, with atomic-level precision, the structures and dynamics of complex molecular structures “on the fly” and to interact with other users in the same virtual environment. A series of controlled studies, in which participants were tasked with a range of molecular manipulation goals (threading methane through a nanotube, changing helical screw sense, and tying a protein knot), quantitatively demonstrate that users within the interactive VR environment can complete sophisticated molecular modeling tasks more quickly than they can using conventional interfaces, especially for molecular pathways and structural transitions whose conformational choreographies are intrinsically three-dimensional. This framework should accelerate progress in nanoscale molecular engineering areas including conformational mapping, drug development, synthetic biology, and catalyst design. More broadly, our findings highlight the potential of VR in scientific domains where three-dimensional dynamics matter, spanning research and education.
INTRODUCTION
It is a fundamental human instinct to build both conceptual and tangible models that help to organize and make sense of our perceptions and experience in the natural world. The practice of building and manipulating models has a rich history and profound impacts across a range of domains, including the physical sciences, the social sciences, engineering, medicine, design, and architecture. Here, our primary focus is on modeling at the nanoscale. From a modeling perspective, the nanoscale represents an interesting domain, because the objects of study (for example, molecules) are invisible to the naked eye, and their behavior is governed by physical forces and interactions significantly different from those forces and interactions that we encounter during our day-to-day phenomenological experience. In domains like this, which are imperceptible to the naked eye, effective models are vital to provide the insight required to make research progress.
In his visionary essay The Ultimate Display, Sutherland (1) highlighted the potential of immersive digital platforms to furnish an intuitive understanding of scientific and mathematical domains for which we otherwise lack intuition: “We live in a physical world whose properties we have come to know well through long familiarity. We sense an involvement with this physical world which gives us the ability to predict its properties well. For example, we can predict where objects will fall, how well-known shapes look from other angles, and how much force is required to push objects against friction… We lack corresponding familiarity with the forces on charged particles, forces in non-uniform fields, the effects of non-projective geometric transformations, and high-inertia, low friction motion…”
The fundamental forces that govern the physical mechanics of nanoscale molecular objects are relatively well characterized, owing to decades of experimental, theoretical, and computational study. The nanoscale represents a domain that is full of the sorts of “nonintuitive” physics highlighted by Sutherland: Forces acting on charged particles in nonuniform fields are the norm, and high-inertia, low-friction regimes are common in a range of applications. Moreover, molecular systems typically have thousands of degrees of freedom. As a result, their motion is characterized by a complicated, highly correlated, and elegant many-body dynamical choreography, which is nonintuitive compared to the more familiar mechanics of objects that we encounter in the everyday physical world. Their combined complexity, unfamiliarity, and importance make molecules particularly interesting candidates for investigating the potential of new digital modeling paradigms.
Tangible physical models have an important place in the history of chemistry and biochemistry. Three-dimensional (3D) molecular models have been used as conceptual and educational tools dating back to at least von Hofmann in the 1860s. More recent examples include Dorothy Hodgkin’s crystallographic model of penicillin’s structure (2) demonstrating the presence of a β-lactam ring, Pauling et al.’s (3) use of (originally paper!) models to identify the structure of α helices, and Crick and Watson’s (4) famous DNA model. Large room-sized models, made from, for example, wire, plastic, brass, balsawood, and plasticine, were used to refine and represent protein crystal structures by pioneers such as Kendrew et al. (5) and Perutz et al. (6). For example, Levitt (7) (in the 2013 Nobel lecture) recounted building a so-called Kendrew model of hen egg white lysozyme: “…slow work but at the end you really know the molecule.”
Physical models like these provide structural insight but cannot represent the often nonintuitive mechanics that determine how molecules move and flex. Going back to pioneers like Bernal (8), whose attempts to understand liquid structure involved mechanical simulation of assemblies of macroscopic spheres, molecular scientists have since made heroic efforts to include the fourth dimension (time) into their models. In the 1970s, pioneers like Levitt and Warshel (9) and McCammon, Gelin, and Karplus (10) were able to simulate the motion of complex molecules such as proteins. The year 1979 saw the first movie of a time-dependent molecular dynamics (MD) simulation (bovine pancreatic trypsin inhibitor), one of several developments that heralded the eventual replacement of tangible mechanical models by screen-based graphics. These rose to prominence in the 1980s and are now ubiquitous for the purpose of understanding and teaching molecular structure and dynamics.
Advances in computer power, and decreasing cost, have transformed many domains of computational science, including molecular visualization and simulation. For example, graphics processing units (GPUs), designed to facilitate fast rendering in video games, not only have benefitted scientific visualization but also have been adapted to accelerate a range of molecular physics algorithms (11), now allowing routine simulation of systems with hundreds of thousands of atoms at time scales ranging from hundreds of nanoseconds to microseconds, with applications to areas including protein folding (12), enzymology (13), and drug discovery (14). Beyond GPUs, application-specific integrated circuits (ASICs) (15), cloud computing platforms (16), distributed computing networks (17), and new architectures enable simulation of large systems at millisecond time scales (15, 16).
Nevertheless, computational simulation of chemical transformation in molecular systems is characterized by considerable complexity, partly because it arises from spontaneous rare events that occur during the course of a molecule’s hyperdimensional dynamical choreography. There is good evidence that mapping such transformations belongs to a class of “NP-hard” problems, for which no obviously optimal computational solution exists (18). For the foreseeable future, many important molecular-level transformations occur on time scales that will remain beyond even the most sophisticated simulation architectures. “Chemical intuition” (an oft-invoked concept that broadly refers to the chemist’s ability to efficiently navigate hyperdimensional molecular space) is therefore likely to play an important role in (bio)chemistry and synthetic biology for a long time to come. Tools accelerating the rate at which chemical intuition can be brought to bear on structurally and physically detailed models will facilitate creative teams making progress on challenging problems.
Like many domains of scientific computing, the basic workflow for molecular simulation has remained largely unchanged for the past 30 to 40 years, that is, iterative cycles of job submission to high-performance computing (HPC) resources, followed by visualization on a 2D display. Pioneers with interests spanning molecular simulation and human-computer interaction, led by Fred Brooks (19) and Kent Wilson (20), were among the first to imagine improvements to this workflow using interactive computational technologies: Following on from the ideas outlined by Sutherland (1), they speculated that interactive molecular simulation (iMS) frameworks would lead to models that would be as intuitive to manipulate as the old tangible models, but which followed rigorous physical laws, and could be used to tackle hard rare event sampling problems. Brooks and co-workers (19, 21) designed an immersive six–degree of freedom force-feedback haptic system, which users could manipulate to carry out molecular docking tasks. Inspired by this work, Schulten and co-workers subsequently miniaturized Brooks’ setup: By manipulating a desktop-mounted haptic pointer, users could steer the real-time dynamics of molecules rendered on a stereographic screen (22), a setup that has since been extended by others to interactively steer real-time molecular mechanics (23) and quantum chemistry simulations (24, 25).
These interactive setups face a well-known limitation in their ability to achieve 3D “co-location,” that is, aligning the interaction sites in 3D physical space and 3D virtual space (26). Touchscreens solve the problem of 2D co-location because the interaction site in physical space is identical to the virtual interaction site. For iMS, 3D co-location is an important design problem, given that molecules are 3D objects that move in 3D. Despite these difficulties, the utility of the iMS idea has been demonstrated by tools like Foldit (27). Using a keyboard-mouse interface, Foldit enables users to apply their intuition to explore protein conformational space and predict protein structures. Cooper et al. (27) highlighted cases where Foldit users made better predictions (that is, located deeper energetic minima, which correspond to more stable structures) than automated algorithms, owing to users’ willingness to explore high-energy (“high risk”) pathways that automated strategies avoided. Khatib et al. (28) analyzed user search strategies to construct new algorithms.
Driven mostly by the consumer gaming market, recent advances in virtual reality (VR) hardware provide commodity-priced solutions to the problem of 3D co-location. Combining infrared optical tracking, inertial movement units, and ASICs, high-end commodity VR technology such as the HTC Vive tracks a user’s real-time 3D position with errors of less than a centimeter, allowing users to reach out and touch simulated objects within the virtual world, as shown in Fig. 1 and movie S1. VR pioneers like Lanier (29) have emphasized the fact that several so-called VR frameworks do not allow this type of interaction, enabling little more than “just looking around in a spherical video.” Lanier (29) has made a point to distinguish those technologies that do afford this sort of interaction: “If you can’t reach out and touch the virtual world and do something to it, you are a second class citizen within it… a subordinate ghost that cannot even haunt” (29).
Fig. 1. Technical schematic of the HTC Vive VR setup that we designed to carry out the studies outlined here.
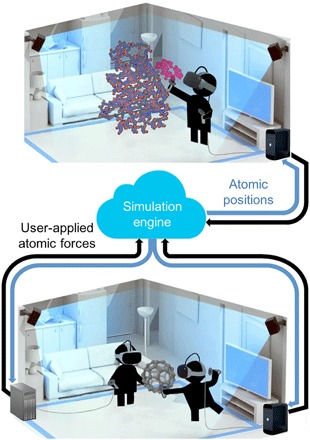
Bottom: Two users within the multiperson VR framework passing a simulated C60 molecule back and forth. Each user’s position is determined using a real-time optical tracking system composed of synchronized infrared light sources. Each user’s VR HMD is rendered locally on a computer fitted with a suitable GPU; MD calculations and maintenance of global user position data take place on a separate server, which can be cloud-mounted. As long as the network connecting the client and server enables sufficiently fast data transfer, system latency is imperceptible to the human senses. Top: Single-person setup, where the user is chaperoning a real-time GPU-accelerated MD simulation to generate an association pathway that docks a benzylpenicillin ligand (magenta) into a binding pose on the TEM-1 β-lactamase enzyme.
In the medical field, VR technologies that enable surgeons to “reach out and touch the virtual world” have an established track record: Several studies have shown that VR-trained surgeons complete surgical procedures faster, with significantly lower error rates (30). However, the use of VR in surgical contexts, where it is intended to simulate a surgeon’s experience of manipulating human tissue, is rather distinct from the use of VR to manipulate molecular structure and dynamics. Whereas surgical applications have a well-defined and measurable design reference (that is, how does the VR simulation “feel” compared to an experience involving human tissue?), molecular applications have no similarly well-defined design reference (that is, what does a molecular system feel like?). This lack of reference is what makes developing a real-time molecular simulation and manipulation framework such a fascinating challenge, which must necessarily consider aesthetics, design, and user psychology to be effective. The lack of design reference also highlights the arbitrariness of tangible (for example, plastic or metal) molecular models.
RESULTS
Building on our previous work using optical tracking technologies to interactively steer real-time molecular simulations (31), Fig. 1 (bottom) shows the framework that we have developed to interface the HTC Vive with rigorous real-time molecular simulation algorithms, which we henceforth refer to as the “iMD VR app.” Two optically tracked researchers are shown [each wearing a VR head-mounted display (HMD) and holding two small wireless controllers that function as atomic “tweezers”] manipulating the real-time MD of a C60 molecule. As shown in movie S1, the researchers can easily “grab” individual C60 atoms and manipulate their real-time dynamics to pass the C60 back and forth between each other. This is possible and immediately intuitive because the real-time C60 simulation and its associated ball-and-stick visual representation are perfectly co-located—that is, the interaction site in 3D physical space is exactly the interaction site in 3D simulation space. The cloud architecture provides further benefits: Because each VR client has access to global position data of all other users, any user can see through his/her headset a co-located visual representation of all other users at the same time. To date, our available resources have allowed us to simultaneously co-locate six users in the same room within the same simulation. The interaction shown in movie S1, in which multiple users in the same room are able to easily pass a simulated molecule between themselves (or, for example, collaboratively tie a knot in a protein) as if it were a tangible object, represents a class of simulated virtual experience which is not possible within the large-scale immersive stereoscopic CAVE environments that have become popular within academic and industrial research institutions around the world (32). The cloud-mounted framework shown in Fig. 1 also makes it straightforward to enable remotely located workers to occupy the same virtual space.
While a real-time MD simulation of C60 is relatively cheap, the cloud architecture enables access to more powerful computational servers as needed. For example, Fig. 1 (top) and movie S2 show a researcher taking hold of a fully solvated benzylpenicillin ligand and interactively guiding it to dock it within the active site of the TEM-1 β-lactamase enzyme (with both molecules fully flexible and dynamic) and generate the correct binding mode (33), a process that is important to our understanding of antimicrobial resistance. The β-lactamase example [which benefits from a plug-in that communicates with the GPU-accelerated molecular simulation package OpenMM (34) via PLUMED (35)] illustrates that our framework is sufficiently intuitive and easy to control to enable a researcher to quickly formulate and evaluate dynamical hypotheses in large molecular systems. Generating a benzylpenicillin docking pathway involves a nonlinear sequence of complex molecular manipulations that would be difficult to formulate algorithmically. Thus, the framework has potential as an effective tool for understanding molecular docking and binding kinetics (standard tasks in structure-based drug design) and may prove particularly effective as a complement to enhanced sampling techniques for challenges such as identifying allosteric and cryptic binding sites that are not present in crystal structures (36).
An essential question is whether co-located interaction of the sort enabled by VR affords any real advantage in accelerating complex 3D manipulation tasks compared to more conventional interaction technologies. The application of interest, in this case is molecular modeling. To tackle this question, we exploited the fact that the Fig. 1 framework runs on a wide range of different client interaction hardware, including mice (connected to personal computers/laptops) and tablets. Both are extremely familiar interfaces: the former offering non–co-located interaction, and the latter offering co-located interaction in 2D. We designed three specific tasks (Fig. 2 and movie S1) for users to carry out and compared the rates of task accomplishment across different interaction hardware. Each task involves an increasingly complicated choreography: (i) guiding a methane molecule (CH4) through a carbon nanotube, (ii) changing the screw sense of an organic helicene molecule, and (iii) tying a knot in a small polypeptide [17-alanine (17-ALA)]. Each task represents a class of dynamics that is important across natural and engineered nanosystems. For example, nanopores are ubiquitous across bio/materials chemistry (37), induced changes in molecular helicity offer a strategy for synthetic biologists to transmit chemical messages (38), and protein knots are associated with neurodegenerative diseases (39). To control for variations in the graphics and computational capabilities of different interaction hardware, each interaction platform used the same renderer and the same back-end MD simulation engine. The Supplementary Materials include details on the simulation setups, along with instructions enabling readers to use the executables at isci.itch.io/nsb-imd to initialize a cloud-based interactive simulation instance so that they can attempt the Fig. 2 tasks on any of the three interaction platforms.
Fig. 2. Experimental iMS tasks.
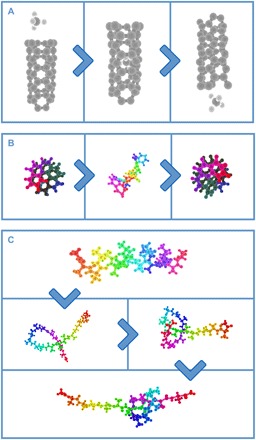
Experimental iMS user studies featured the following tasks: (i) threading CH4 through a nanotube, (ii) changing the screw sense of a helicene molecule, and (iii) tying a knot in a polypeptide (17-ALA). Colors selected in this figure are chosen for the sake of clarity.
Figure 3 shows the rates at which cohorts of 32 users (who reported little or no previous VR experience) accomplished each of the three tasks in Fig. 2 on different hardware platforms. Before their participation, study candidates were warned of the potential for VR sickness and instructed to inform the demonstrators if they wanted to exit at any point. We recorded zero such instances, suggesting that our design produces little user discomfort. Reported errors in the measured rates were obtained using Poisson statistics, a common strategy in chemical kinetics. Rare event (or “task accomplishment”) rates are said to be statistically distinguishable if their corresponding Poisson error bars do not overlap.
Fig. 3. User study results.
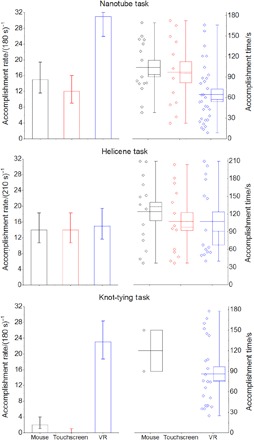
Left-hand panel shows user accomplishment rates for the tasks outlined in Fig. 2 (n = 32 for all tasks), with Poisson error estimates. Right-hand panel shows the corresponding distribution of task accomplishment times, along with box-and-whisker plots. Whiskers indicate the data range, and box limits indicate the standard error of the distribution. The mean is shown as a solid line, and the median is shown as a dashed line.
DISCUSSION
A significant fraction of study participants were able to use the real-time VR simulation framework to accomplish (in a relatively short time) the molecular rearrangement tasks that we set for them (shown in Fig. 2). Specifically, 97% were able to thread methane through the nanotube, 47% were able to change the helicene screw sense, and 72% were able to tie a protein knot. This indicates that our preset simulation parameters (outlined in further detail below) enabled most of the participants to accomplish the Fig. 2 tasks. For tasks A and C, Fig. 3 indicates that VR provides a clear acceleration benefit compared to the other platforms. The more inherently 3D the task, the greater the benefit. Thus the knot-tying results (Fig. 3C) are the most dramatic, showing that this task is very difficult to accomplish using a mouse or a touchscreen. To ascertain whether the knot-tying results were reproducible, we carried out another set of tests with a different cohort; reassuringly, the results were the same within Poisson error limits (see the Supplementary Materials). For the nanotube task (Figs. 2A and 3A), the accomplishment rates, mean time, and median time in VR are approximately a factor of 2 faster than on other platforms.
At first glance, the helicene task (Fig. 3B) is a case in which VR appears to provide little significant rate enhancement compared to other platforms. Observation suggests that this is because changes in helicene screw sense are most efficiently accomplished using a simple 2D circular motion, as shown in movie S1. Essentially, the 2D limitations of the mouse and touchscreen constrain the user to carrying out a motion that is well suited to inducing changes in molecular screw sense. However, closer inspection of the helicene time distributions suggests that VR does afford some advantage: The median time required to change molecular screw sense in VR is 30 to 40% less than the median time required on a touchscreen. Significantly, every task considered in Figs. 2 and 3 shows a median VR time that (i) is always faster than the mean and (ii) lies outside the standard error limits. This distribution of waiting times is clearly not Gaussian; rather, it resembles the waiting time distribution that characterizes first-order kinetics, suggesting a cluster of VR users who accomplish tasks quickly with a longer tail of slow users.
Figure 3 shows that our VR platform provides a clear advantage over conventional visualization/interaction platforms, and this advantage appears to increase as the complexity of the problem increases. To better understand the results, we carried out further evaluation in the form of participant questionnaires and interviews (detailed in the Supplementary Materials). Despite the fact that very few of the study participants had previous VR experience, participants indicated an overwhelming preference for VR in carrying out the Fig. 3 tasks (compared to a mouse or touchscreen) for three reasons: (i) quick perception of depth, (ii) the ability to inspect the molecule by walking around it, and (iii) the use of both hands to accomplish the tasks.
The results presented here represent a first attempt to quantify the acceleration that can be achieved by carrying out 3D molecular sampling, modeling, and manipulation tasks in VR. Our exclusive focus in this article has been on interactive dynamics with molecular mechanics force fields, rather than quantum mechanical force fields. There are two main reasons for this: Quantum mechanical (electronic structure) methods are only feasible for small molecules because they require greater processing resources, and quantum mechanical methods are generally less stable than molecular mechanics force fields, owing to difficulties that arise as one attempts to numerically converge quantum mechanical self-consistent field equations on-the-fly. Because molecular mechanics force fields are more robust than their quantum mechanical equivalents, they afforded better control during user studies reported here. However, we have recently undertaken work to interface our VR framework with the quantum mechanical force field engine described by Mühlbach et al. (40). This enables us to perform real-time simulations of chemical reactivity in a fully co-located multiperson VR environmental and to carry out a range of studies that we will report on in the near future.
For biomolecular conformational problems like dynamical path sampling or drug docking, where computational search spaces are too large for brute force approaches, our results suggest that VR frameworks, like those that we have outlined here, may offer a powerful tool enabling research experts (for example, structural biologists, molecular modelers, and medicinal chemists) to occupy the same virtual environment and efficiently express their 3D biomolecular intuition to test hypotheses for collaboratively tackling important microscopic questions linked to molecular mechanism and design. For example, using VR technologies to generate seeds for subsequent automated searches could represent a powerful strategy for obtaining molecular insight, which is likely to save on valuable computational clock cycles. It may even be possible to imagine a scenario where machines “learn” efficient strategies for navigating hyperdimensional biomolecular search spaces by accumulating observational data on how human experts navigate hyperdimensional biomolecular spaces in VR.
Recent advances in HPC, the cloud, data science, robotics, and machine learning have led many workers across academia and industry to begin speculating about the future of scientific practice, asking important questions as to the sort of scientific future that we should be consciously working to design over the next few decades (29, 41). In an increasingly automated future that is reliant on machines, it is extremely important to think carefully about and discuss the role that human creative expression will play. Many scientists and engineers express a philosophical sentiment that automation is often desirable because it eliminates mistakes and unpredictability; however, these are staple features that are inevitably entangled with creative expression. So long as human creativity continues to play an important role in the process of scientific discovery and design, then we believe that VR frameworks like that outlined here may play a crucial role in our emerging scientific future. In our view, advanced visualization and interaction frameworks are complementary to research activities aimed at increasing the automation of research tasks and scientific discovery, because they provide an efficient means for humans to express high-level creative scientific and design insight, leaving automated frameworks to subsequently sort out the computational and mechanistic details. Another practical role for VR frameworks in an era of increasing automation pertains to quality assurance: As robotic automation becomes more prevalent (for example, in molecular simulation workflows), we are going to need efficient new visualization tools that enable us to continually review and evaluate robot performance to understand the actions and decisions they are taking, determine whether those actions and decisions make sense, and decide whether the robots need additional training.
Moving forward, there are a range of related domains that we intend to explore. For example, we aim to better understand how changes in simulation parameters (for example, force strength, MD timestep, simulation temperature, holonomic constraints, and rendering options) affect the ability of users to intuitively accomplish molecular tasks and furnish meaningful microscopic insight. We are interested in designing analytical methods that can be combined within interactive sampling strategies so that mechanistic observables (for example, free energies, barrier heights, and conformational probabilities) can be extracted from VR simulations. We are also exploring the extent to which other sensory modalities (for example, sound) might be engaged to provide additional information within a real-time immersive simulation environment (42). As we explore the potential of VR across both molecular mechanics–based and quantum mechanics–based simulations and study its ability to enable specialists and nonspecialists alike to better understand and more efficiently manipulate complex data, we look forward to understanding how it may be used to advance molecular science in a range of research, communication, and educational contexts.
MATERIALS AND METHODS
MD simulation setup
The broad strategies that we have previously used to carry out interactive MD (iMD) were outlined by Glowacki et al. (31) and briefly summarized here for the sake of completeness. In classical mechanics, the time-dependent dynamics of molecular systems are solved by numerically integrating Newton’s equations of motion. The vector of forces acting on a set of atoms F(t) can be written in terms of the system’s potential energy V, that is
| (1) |
where q is a vector containing the position of each atom in the ensemble. Our system effectively allows users to interactively chaperone a real-time MD simulation by splitting V into two different components
| (2) |
where Vint corresponds to the system’s internal potential energy, and Vext corresponds to the additional potential energy added when a user exerts a force on a specific atom (or group of atoms) when he/she grabs it using the handheld wireless controller shown in Fig. 1. Substituting Eq. 2 into Eq. 3 then gives
| (3) |
The external forces were implemented by projecting a spherical Gaussian field into the system at the point specified by the user and applying the field to the nearest atom j through the following formula
| (4) |
where mj is the atomic mass of the atom, c is a scale factor that tunes the strength of the interaction, qj is the position of atom j, gi is the position of the interaction site, and σ controls the width of the interactive fields. For all tasks on all the platforms, c was set to 2000 (kJ mol−1)/(atomic mass unit), a value that achieves responsive interaction while preserving dynamical stability, and σ was set to the default value of 1 nm. While an interaction is active, it is always applied to the same atom (or group of atoms), which means that a user can dynamically adjust the course and strength of the interaction by repositioning their field with respect to the atoms with which they are interacting, until he/she decides to “let go.” As discussed in the main text, the position of the interaction field in VR is co-located to precisely where the user’s controller is; for the mouse and touchscreen interfaces, the field is attached to the nearest atom (measured in the 2D plane of the screen). Movements of the field are only possible in 2D, so 3D motions must be built up from successive 2D motions followed by repositioning of the camera.
Simulating the dynamics of a particular molecular system requires specifying an engine to calculate the internal forces. Here, we benefit from the fact that our framework has been designed to flexibly communicate with a wide range of force engines via a defined application programming interface. For the hydrocarbon systems in the nanotube and helicene task, we used our own “in-house” implementation of the MM3 force field (43). The 17-ALA protein knot task used the GPU-accelerated Amber99SB-ILDN force field provided within the OpenMM MD package (34). For the simulation of larger biomolecular systems such as β-lactamase, we used a custom build of PLUMED (35) to communicate with simulations running within OpenMM on a workstation consisting of four NVIDIA 1080 Ti GPUs. This workstation enables us to simulate proteins using the Amber99SB-ILDN force field and optionally include either implicit (for example, continuum) or explicit (for example, TIP3P water) solvent models. In cases where we modeled an explicit solvent, we did not typically visualize the solvent molecules to maintain clarity and high-quality rendering. To integrate the forces, all tasks used a Velocity Verlet integrator, with a Berendsen thermostat (44) set to a target temperature of 200 K. For the nanotube and helicene tasks, a time step of 1 fs was used, while the protein knot task used a time step of 2 fs along with the RATTLE holonomic constraint, owing to the fact that the conformational change occurs over a longer time scale.
To carry out the user studies (detailed further in what follows), the simulations were hosted on separate machines within our own local area network. We used one machine for each task to avoid any latency that might arise from excessive computational load on a single machine. The three machines that we used as simulation servers during user studies included the following: (i) a mid-range gaming desktop with a 3.5 GHz quad-core Intel i5-6600K processor and an NVIDIA GTX 970 graphics card, (ii) a high-end Alienware13 R3 gaming laptop with a 2.6 GHz quad-core Intel Core i7-6700HQ processor and an NVIDIA 1060 dedicated VR graphics card, and (iii) a high-end Alienware15 R3 gaming laptop with a 2.6 GHz quad-core Intel Core i7-6700HQ and an NVIDIA 1070 GTX dedicated VR graphics card. The user tests involving mice (described in the “User study one” section) were run at two different sites: (i) our laboratory at the University of Bristol (UoB), using the mid-range gaming desktop along with an external USB mouse and a 21-inch external monitor and (ii) the CCPBioSim (Collaborative Computational Project for Biomolecular Simulation) 2017 conference using the screen on the Alienware15 along with an external USB mouse. During all user studies, we throttled the performance on all platforms to guarantee that (despite differences in each machine’s specifications) they were capable of running at 30 frames per second. This ensured that the latency across all test platforms gave an equally fluid user experience. For the touchscreen version of the tasks, we used a Samsung Galaxy S3 tablet, connected to the simulation over an 802.11ac Dual Band Gigabit WiFi connection.
Controlled user studies
User study one
A total of 32 participants were recruited for this study. To mitigate any learning or fatigue effect, the platform on which any given participant started was randomly selected. Twelve participants began with the mouse platform, 9 participants started with the touchscreen platform, and 11 participants started with the VR platform. Participants were rotated using a Latin square in the order of VR, mouse, and touchscreen.
Before starting on a platform, participants were given the buckminsterfullerene task (see the “Buckminsterfullerene task” section for details) to familiarize themselves with the feel of the molecular interaction on a given platform. Once participants indicated to the study facilitators that they had a sufficient level of familiarity, they were moved onto the nanotube/methane task (see the “Nanotube/methane task” section for details). Study facilitators moved a participant onto the helicene task if they either managed to accomplish the nanotube task or time expired (see the “Helicene task” section for details). Once the user had attempted both tasks, the process was repeated on the next platform until the participant had tried the task on all three platforms. Once participants had attempted both tasks on all three platforms, they were given a short questionnaire to fill out, details of which are discussed in further detail below.
In total, 32 articipants were recruited through email to staff and students at the UoB and offered a £10 Amazon gift voucher for their time. Seventeen (53.125%) of the participants were ages 18 to 24, 10 (31.25%) were ages 25 to 34, 4 (12.5%) were ages 35 to 44, and 1 (3.125%) was ages 45 to 54. Twenty-two (68.75%) of the participants were male, and 10 (31.25%) were female. Participants reported a range of education levels. Eleven (34.375%) of the participants were undergraduate students, 16 (50%) were postgraduate students, 3 (9.375%) were postdoctoral researchers, and 2 (6.25%) were researchers.
Participants were given a Likert scale question to complete to indicate their familiarity with using VR and tablets, where 1 represents having no experience and 5 represents being very experienced. A breakdown of responses can be found in table S1. Altogether, self-reported VR experience was found to be low, where tablet use was more prevalent. Given the education level of the group, and the fact that they were drawn from a university chemistry department, we assumed that mouse familiarity was high.
User study two
Here, 12 people were recruited and interviewed afterward. We used a smaller sample size, because our emphasis during these studies was on gaining qualitative user feedback on attitudes to the three platforms, achieved via interview. Task accomplishment rates for this study are presented in fig. S2. In addition to administering questionnaires used during the first user study, qualitative analysis of participants’ subjective feedback (45) was performed on the recorded interview transcripts, a summary of which is presented below.
Participants were recruited in group sizes varying between one and three. To mitigate any learning or fatigue effect, the platform on which participants started was randomized. Specifically, four people started with the mouse platform, four started with touchscreen platform, and four started with the VR platform. Participants were rotated using a Latin square in the order of VR, mouse, and touchscreen. Again, participants were first given the buckminsterfullerene task so that they could familiarize themselves with the interactive feel. Before starting on a platform, participants were first shown a short, instructional video of the specific trefoil knot that they were being asked to tie. Once they grasped what they were being asked to do, participants were moved onto the knot-tying task. A more detailed description of what both tasks entailed can be found below. Once the task was completed (or once time had elapsed), the process was repeated on the next platform until the participant had tried the task on all three platforms.
After each group had attempted the knot-tying task on each of the three platforms, they were interviewed about their experience using each of the four tasks. During this interview stage, the following points were covered: (i) How had the participants found the task in general? (ii) Was there a preferred platform (or platforms) for completing the task? (iii) Why did participants prefer a given platform over others? (iv) Was there a least preferred platform (or platforms) for completing the task? (v) Are there any suggestions for how the platforms can be improved? (vi) Any further points?
Once again, the participants for this study were recruited by email to staff and students at the UoB and offered a £10 Amazon gift voucher for their time. Six (50%) of the participants were ages 18 to 24, five (41.7%) were ages 25 to 34, and one (8.3%) was age 35 to 44. Seven (58.3%) participants were male, and five were female (41.7%). Participants reported a range of education levels. Four (33.3%) reported themselves as being undergraduate students, three (25%) reported themselves as being postgraduate students, four (33.3%) reported themselves as being postdoctoral researchers, and one (8.3%) reported themselves as being a research technician.
Participants were given a Likert scale question to complete to clarify their familiarity with using VR and tablets, where 1 represents having no experience and 5 represents being very experienced. A breakdown of responses can be found in table S1. Together, self-reported VR experience was found to be low, where tablet use was more prevalent among the cohort. Given the education level of the group, mouse familiarity was again assumed to be high.
User study three
We decided to repeat the methodology from the second user study with a larger sample size of 32 participants, identical to the sample sizes selected for the nanotube and helicene talks in the first user study. The primary aim of this leg was to obtain better statistics on knot task completion; therefore, no questionnaire or interview was given afterward.
Participants were recruited in group sizes varying between one and three. To mitigate any learning or fatigue effect, the platform on which participants started was randomly selected. Specifically, 10 participants started with the mouse platform, 11 participants started with the touchscreen platform, and 11 participants started with the VR platform. Participants were rotated using a Latin square in the order of VR, mouse, and touchscreen.
Participants were recruited during the fifth annual UK CCPBioSim conference (13 to 14 September 2017), held at the University of Southampton (www.ccpbiosim.ac.uk). The chance to participate in the user study was advertised by email, flyers, and word of mouth. Six (18.75%) of the participants were ages 18 to 24, 20 (62.5%) were ages 25 to 34, four (12.5%) were ages 35 to 44, one (3.125%) was ages 45 to 54, and one (3.125%) was over the age of 65. Twenty-four (75%) of the participants were male, and 7 (22%) were female. One (3%) participant chose not to state their gender. Participants reported a range of education levels. Nineteen (59.375%) of the participants were postgraduate students, 11 (34.375%) were postdoctoral researchers, and 2 (6.25%) reported themselves as researchers.
Participants were given a Likert scale question to complete to ascertain their familiarity with using VR and tablets, where 1 represents having no experience and 5 represents being very experienced. A breakdown of responses can be found in table S1. Self-reported VR experience was low; tablet use was far more prevalent. Given the education level of the group, mouse familiarity was again assumed to be high.
Molecular simulation tasks
Buckminsterfullerene task
Two buckminsterfullerene molecules were loaded into the iMD VR app. Users were then instructed to grab the molecule and experiment with moving it around within the virtual space, and also to familiarize themselves with resizing and rotating the virtual space, thus giving them an understanding of the platform controls and the feel of the molecular interaction. There was no time limit to this task, nor did it have any specific end goal. The simulation details are included in section S1.
Nanotube/methane task
One C60 nanotube and one methane molecule were loaded into the iMD VR app. Users were instructed to grab the methane molecule and lead it through the center of the nanotube, from one end to the other. The task had a time limit of 180 s, and completion was marked as the point that the user had successfully pulled the methane molecule through the entire nanotube, that is, leaving the opposite side from which it entered. The simulation details are included in section S1.
Helicene task
One 12-helicene molecule was loaded into the iMD VR app. Users were instructed to manipulate the helicene molecule so that the screw sense of the helix was reversed. The task had a time limit of 210 s, and completion was marked as the point at which the screw sense of the helicene molecule had gone from its clockwise initial conditions to being completely anticlockwise. The simulation details are included in section S1.
Protein knot-tying task
One 17-ALA molecule was loaded into the iMD VR app. Before starting the task on each of the platforms, users were shown a brief instructional video on the task. Users were then instructed to tie a 31 or trefoil knot in 17-ALA. The task had a time limit of 180 s, and completion was marked as the point at which a stable trefoil knot was formed in 17-ALA. The simulation details are specified above. The simulation details are included in section S1.
Supplementary Material
Acknowledgments
We thank T. Mitchell, J. Hyde, I. Cressy, B. de Kosnik, G. Anderson, and R. Arbon for their contribution during the Barbican Open Lab residency, where the aesthetics of the multiperson VR environment took shape; R. Male (UFI Charitable Trust), A. Wilson (Barbican), S. Khajuria (Barbican), C. Sharp (Barbican), J. Upton (Royal Society), S. McIntosh-Smith (UoB), and D. Woolfson (UoB) for encouragement and support throughout; J. A. Glowacki (UoB) for assisting H.M.D. with data collection; G. Viedma Nunez and J. Tsai-Smith (Oracle) for guidance in enabling the cloud-mounted simulation engine; J. Sandilands and L. Cullingford (Interactive Scientific) for respective cloud-mounted interaction design contributions and project management; and the 2017 CCPBioSim annual conference and its organizers (M. van der Kamp, J. Essex, E. Rosta, and D. Huggins) for enabling us to run user studies as part of the conference program (www.ccpbiosim.ac.uk). Funding: M.O.’s studentship (supervised by D.R.G.) is supported through an industrial CASE, funded by the Engineering and Physical Sciences Research Council (EPSRC) and Interactive Scientific. H.M.D.’s PhD studentship (supervised by D.R.G., A.J.M., A.R., and O.M.) is funded by the EPSRC. D.R.G. acknowledges additional funding from Oracle Corporation (University Partnership Cloud Award), the Royal Society (UF120381), the EPSRC (Impact Acceleration Award, Institutional Sponsorship Award, and EP/P021123/1), the Leverhulme Trust (Philip Leverhulme Prize), and the London Barbican (Open Lab Funding). R.S. and P.T. acknowledge support from the UFI Charitable Trust. A.R. acknowledges support from the Leverhulme Trust and the EPSRC (EP/P004342/1). A.J.M. thanks the EPSRC for funding (EP/M022609/1) and also the CCPBioSim, supported by the EPSRC. O.M. acknowledges EPSRC Fellowship EP/N00616X/2. Author contributions: M.O., P.T., M.W., and R.S. designed and implemented the cross-platform, real-time, cloud-mounted multiperson iMD framework. H.M.D. and E.D. carried out user studies and performed data analysis. H.M.D., O.M., and A.R. designed the user studies. M.S. constructed the video figures. B.R.G. and D.R.G. designed the molecular tasks in Fig. 2. A.J.M. and D.R.G. conceived the TEM-1 β-lactamase/benzylpenicillin application, which was developed by H.M.D. under their supervision. P.B. provided crucial support implementing the cloud-mounted simulation infrastructure. D.R.G. designed the overall project concept, organized execution of the work strands, analyzed the data, and wrote the initial paper draft along with H.M.D. and M.O., with subsequent input from A.J.M., L.M.T., B.R.G., O.M., and A.R. Competing interests: M.O.’s studentship (supervised by D.R.G.) is supported through an industrial CASE award, partially funded by Interactive Scientific, for whom D.R.G. serves as a nonexecutive scientific consultant. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaat2731/DC1
section S1. Launching a cloud-hosted iMD session
section S2. User study data
section S3. Platform design
section S4. Qualitative analysis of participants’ subjective feedback
fig. S1. Distribution of round-trip latencies, measured from Bristol to each of the cloud data centers.
fig. S2. User study results.
fig. S3. Screenshots of a user’s view from within VR carrying out the nanotube task.
fig. S4. User’s view of the molecular manipulation application when using either a mouse or touchscreen.
fig. S5. Number of participants (y axis) self-reporting their attitudes on the importance of depth perception, navigating the virtual space, and controlling molecules with two hands.
table S1. Self-reported familiarity with the VR and tablet platforms on a Likert scale.
movie S1. Sampling molecular conformational dynamics in VR (www.vimeo.com/244670465).
movie S2. Interactively sampling dynamical pathways of benzylpenicillin binding to β-lactamase (www.vimeo.com/235894288).
REFERENCES AND NOTES
- 1.Sutherland I. E., The ultimate display. Proc. IFIP Cong. 2, 506–508 (1965). [Google Scholar]
- 2.D. Crowfoot, C. W. Bunn, B. W. Rogers-Low, A. Turner-Jones, X-ray Crystallographic Investigation of the Structure of Penicillin (Princeton Univ. Press, 1949), pp. 310–367. [Google Scholar]
- 3.Pauling L., Corey R. B., Branson H. R., The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. U.S.A. 37, 205–211 (1951). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crick F. H. C., Watson J. D., The complementary structure of deoxyribonucleic acid. Proc. R. Soc. A 223, 80–96 (1954). [Google Scholar]
- 5.Kendrew J. C., Bodo G., Dintzis H. M., Parrish R. G., Wyckoff H., Phillips D. C., A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature 181, 662–666 (1958). [DOI] [PubMed] [Google Scholar]
- 6.Perutz M. F., Rossmann M. G., Cullis A. F., Muirhead H., Will G., North A. C. T., Structure of haemoglobin: A three-dimensional Fourier synthesis at 5.5-Å. Resolution, obtained by x-ray analysis. Nature 185, 416–422 (1960). [DOI] [PubMed] [Google Scholar]
- 7.Levitt M., Birth and future of multiscale modeling for macromolecular systems (Nobel Lecture). Angew. Chem. Int. Ed. 53, 10006–10018 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Bernal J. D., The Bakerian Lecture, 1962. The structure of liquids. Proc. R. Soc. Lond. A Math. Phys. Sci. 280, 299–322 (1964). [Google Scholar]
- 9.Levitt M., Warshel A., Computer simulation of protein folding. Nature 253, 694–698 (1975). [DOI] [PubMed] [Google Scholar]
- 10.McCammon J. A., Gelin B. R., Karplus M., Dynamics of folded proteins. Nature 267, 585–590 (1977). [DOI] [PubMed] [Google Scholar]
- 11.Stone J. E., Hardy D. J., Ufimtsev I. S., Schulten K., GPU-accelerated molecular modeling coming of age. J. Mol. Graph. Model. 29, 116–125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dill K. A., MacCallum J. L., The protein-folding problem, 50 years on. Science 338, 1042–1046 (2012). [DOI] [PubMed] [Google Scholar]
- 13.van der Kamp M. W., Mulholland A. J., Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 52, 2708–2728 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Ganesan A., Coote M. L., Barakat K., Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 22, 249–269 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Lindorff-Larsen K., Piana S., Dror R. O., Shaw D. E., How fast-folding proteins fold. Science 334, 517–520 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Kohlhoff K. J., Shukla D., Lawrenz M., Bowman G. R., Konerding D. E., Belov D., Altman R. B., Pande V. S., Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 6, 15–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.A. L. Beberg, D. L. Ensign, G. Jayachandran, S. Khaliq, V. S. Pande, Proceedings of the 2009 IEEE International Symposium on Parallel & Distributed Processing, Rome, Italy, 23 to 29 May 2009 (IEEE, 2009). [Google Scholar]
- 18.Hart W. E., Istrail S., Robust proofs of NP-hardness for protein folding: General lattices and energy potentials. J. Comput. Biol. 4, 1–22 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Brooks F. P. Jr, Ouh-Young M., Batter J. J., Kilpatrick P. J., Project GROPEHaptic displays for scientific visualization. ACM SIGGraph Comput. Graph. 24, 177–185 (1990). [Google Scholar]
- 20.Atkinson W. D., Bond K. E., Tribble G. L. III, Wilson K. R., Computing with feeling. Comput. Graph. 2, 97–103 (1977). [Google Scholar]
- 21.Surles M. C., Richardson J. S., Richardson D. C., Brooks F. P. Jr, Sculpting proteins interactively: Continual energy minimization embedded in a graphical modeling system. Protein Sci. 3, 198–210 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.L. Pollack, VMD: Twenty years of history and innovation (2012); www.ks.uiuc.edu/History/VMD/.
- 23.Dreher M., Piuzzi M., Turki A., Chavent M., Baaden M., Férey N., Limet S., Raffin B., Robert S., Interactive molecular dynamics: Scaling up to large systems. Proc. Comp. Sci. 18, 20–29 (2013). [Google Scholar]
- 24.Haag M. P., Vaucher A. C., Bosson M., Redon S., Reiher M., Interactive chemical reactivity exploration. ChemPhysChem 15, 3301–3319 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Luehr N., Jin A. G. B., Martínez T. J., Ab initio interactive molecular dynamics on graphical processing units (GPUs). J. Chem. Theory Comput. 11, 4536–4544 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Swapp D., Pawar V., Loscos C., Interaction with co-located haptic feedback in virtual reality. Virtual Real. 10, 24–30 (2006). [Google Scholar]
- 27.Cooper S., Khatib F., Treuille A., Barbero J., Lee J., Beenen M., Leaver-Fay A., Baker D., Popović Z. Foldit Players , Predicting protein structures with a multiplayer online game. Nature 466, 756–760 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khatib F., Cooper S., Tyka M. D., Xu K., Makedon I., Popović Z., Baker D. Foldit Players , Algorithm discovery by protein folding game players. Proc. Natl. Acad. Sci. U.S.A. 108, 18949–18953 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.J. Lanier, Dawn of the New Everything: A Journey Through Virtual Reality (Penguin Random House, 2017). [Google Scholar]
- 30.Seymour N. E., Gallagher A. G., Roman S. A., O’Brien M. K., Bansal V. K., Andersen D. K.; Satava R. M., Virtual reality training improves operating room performance: Results of a randomized, double-blinded study. Ann. Surg. 236, 458–464 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glowacki D. R., O’Connor M., Calabró G., Price J., Tew P., Mitchell T., Hyde J., Tew D. P., Coughtrie D. J., McIntosh-Smith S., A GPU-accelerated immersive audio-visual framework for interaction with molecular dynamics using consumer depth sensors. Faraday Discuss. 169, 63–87 (2014). [DOI] [PubMed] [Google Scholar]
- 32.C. Cruz-Neira, D. J. Sandin, T. A. DeFanti, Surround-screen projection-based virtual reality: The design and implementation of the CAVE, Proceedings of the 20th Annual Conference on Computer Graphics and Interactive Techniques (SIGGRAPH 93), Anaheim, CA, 1 to 6 August 1993. [Google Scholar]
- 33.Chudyk E. I., Limb M. A. L., Jones C., Spencer J., van der Kampa M. W., Mulhollanda A. J., QM/MM simulations as an assay for carbapenemase activity in class A β-lactamases. Chem. Commun. 50, 14736–14739 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Eastman P., Friedrichs M. S., Chodera J. D., Radmer R. J., Bruns C. M., Ku J. P., Beauchamp K. A., Lane T. J., Wang L.-P., Shukla D., Tye T., Houston M., Stich T., Klein C., Shirts M. R., Pande V. S., OpenMM 4: A reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theory Comput. 9, 461–469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonomi M., Branduardi D., Bussi G., Camilloni C., Provasi D., Raiteri P., Donadio D., Marinelli F., Pietrucci F., Broglia R. A., Parrinello M., PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 180, 1961–1972 (2009). [Google Scholar]
- 36.Oleinikovas V., Saladino G., Cossins B. P., Gervasio F. L., Understanding cryptic pocket formation in protein targets by enhanced sampling simulations. J. Am. Chem. Soc. 138, 14257–14263 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Kalra A., Hummer G., Garde S., Methane partitioning and transport in hydrated carbon nanotubes. J. Phys. Chem. B 108, 544–549 (2004). [Google Scholar]
- 38.De Poli M., Zawodny W., Quinonero O., Lorch M., Webb S. J., Clayden J., Conformational photoswitching of a synthetic peptide foldamer bound within a phospholipid bilayer. Science 352, 575–580 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Ziegler F., Lim N. C. H., Mandal S. S., Pelz B., Ng W.-P., Schlierf M., Jackson S. E., Riefa M., Knotting and unknotting of a protein in single molecule experiments. Proc. Natl. Acad. Sci. U.S.A. 113, 7533–7538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mühlbach A. H., Vaucher A. C., Reiher M., Accelerating wave function convergence in interactive quantum chemical reactivity studies. J. Chem. Theory Comput. 12, 1228–1235 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Aspuru-Guzik A., Lindh R., Reiher M., The matter simulation (r)evolution. ACS Cent. Sci. 4, 144–152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.R. E. Arbon, A. J. Jones, L. A. Bratholm, T. Mitchell, D. R. Glowacki, Sonifying stochastic walks on biomolecular energy landscapes. https://arxiv.org/abs/1803.05805 (2018).
- 43.Allinger N. L., Yuh Y. H., Lii J. H., Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 111, 8551–8566 (1989). [Google Scholar]
- 44.Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., Haak J. R., Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984). [Google Scholar]
- 45.Braun V., Clarke V., Using thematic analysis in psychology. Qual. Res. Psychol. 3, 77–101 (2006). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaat2731/DC1
section S1. Launching a cloud-hosted iMD session
section S2. User study data
section S3. Platform design
section S4. Qualitative analysis of participants’ subjective feedback
fig. S1. Distribution of round-trip latencies, measured from Bristol to each of the cloud data centers.
fig. S2. User study results.
fig. S3. Screenshots of a user’s view from within VR carrying out the nanotube task.
fig. S4. User’s view of the molecular manipulation application when using either a mouse or touchscreen.
fig. S5. Number of participants (y axis) self-reporting their attitudes on the importance of depth perception, navigating the virtual space, and controlling molecules with two hands.
table S1. Self-reported familiarity with the VR and tablet platforms on a Likert scale.
movie S1. Sampling molecular conformational dynamics in VR (www.vimeo.com/244670465).
movie S2. Interactively sampling dynamical pathways of benzylpenicillin binding to β-lactamase (www.vimeo.com/235894288).