

Abstract
Background: Arrhythmogenic ventricular cardiomyopathy (AVC) is an inherited cardiac disorder affecting 1 in 1000 individuals worldwide. The mean diagnosed age of disease is 31 years. In this article, an Iranian family reported that they were affected by ARVC due to a novel PKP2 mutation.
Methods: Clinical evaluations, 12-lead ECG, CMR, and signal-averaged ECG were performed. After DNA extraction, genetic testing was done, and PCR-sequencing was applied to find causal mutations. Segregation analysis was also performed for the family.
Results: ARVC criteria were documented in the patients. Genetic testing revealed a novel chain termination mutation (p.Tyr168Ter) in PKP2 gene; this mutation was transmitted from the mother to her 23-year-old son, but only the son was affected with ARVC.
Conclusion: Modifier genes were indicated using interactome analysis of Plakophilin 2 protein (PKP2); they might have led to phenotypic variability through cellular mechanisms, such as nonsense-mediated mRNA decay. At least, 9 proteins were identified that might have affected Plakophilin 2 protein function, and consequently, rationalizing this intrafamilial phenotypic variability. This study highlighted the role of modifier genes involved in ARVC as well as the major role of PKP2 mutation in developing the disease in our population.
Keywords: ARVC/D, PKP2 gene, Phenotypic variability
↑ What is “already known” in this topic:
ARVC/D frequency affects 1 in 1000 individuals, with a mean diagnosed age of 31 years. Heterozygous mutation in PKP2 gene, encoding Plakophilin-2, is the common cause of ARVC/D.
→ What this article adds:
A novel mutation is reported as well as segregation analysis of this mutation within the family. In silico analysis of the novel mutation, it was shown that its pathogenicity leads to ARVC phenotype. The role of modifier genes is indicated using interactome analysis. Intrafamilial phenotypic variability, which is an important issue in genetic counseling in ARVC/D, is presented in an Iranian family.
Introduction
Arrhythmogenic ventricular cardiomyopathy (AVC or previously ARVC/D, arrhythmogenic right ventricular cardiomyopathy/dysplasia) is an inherited cardiac disease defined by abnormal structure and function of the right and possibly the left ventricle, resulting in the progressive fibrofatty replacement of myocardium (1). It has been estimated that ARVC/D frequency affects 1 in 1000 individuals, with a mean diagnosed age of 31 years (2). Clinical symptoms of the affected individuals include palpitations, syncope, sudden death, atypical chest pain, and dyspnea ventricular heart failure. Based on the 2010 task force criteria (3), ARVC/D is diagnosed by major and minor diagnostic criteria of echocardiographic, arrhythmic, and tissue abnormalities. Approximately 30% to 50% of the patients show a positive family history (4). Mutations in desmosome coding genes have been reported to cause ARVC in 40% to 50% of the cases (5). Desmosomes act as anchors between myocytes and intermediate filaments/cells and involve in Wnt/B-Catenin signaling. Plakophilin-2 protein, which participates in intercellular coupling and interacts with voltage-gated sodium channel complex, has an essential role in heart morphogenesis (6).
Heterozygous mutation in PKP2 gene, encoding Plakophilin-2, is the common cause of ARVC/D (7-9). More than 200 mutations in PKP2 gene have been reported so far, which are dominantly categorized as point mutations. Here, a chain termination variant in PKP2 gene is described for the first time that causes ARVC/D with low penetrance.
Methods
Clinical evaluations, ECG, 12-lead ECG, CMR, and SAECG were performed. Family history was also recorded. After obtaining the signed informed consent form, 5 mL peripheral blood was used for DNA extraction using standard methods. To date, mutations of 7 major genes including RYR2, TMEM43, DSP, PKP2, DSG2, DSC2, and JUP have been reported to cause ARVC/D. Genetic testing was performed. Briefly, coding regions of PKP2 gene (NM_001005242) (exons and intron boundaries) were amplified using PCR reactions (primers are available upon request), and direct sequencing was performed using BigDye protocol by sequencing analyzer ABI 3500 (PE Applied BioSystems, US).
Results
A 23-year-old male presented with an earlier history of syncope was referred to the hospital. Electrocardiogram (ECG) records showed a broad QRS tachycardia with left bundle branch block (LBBB) morphology and superior axis, with a rate of 260 beats/minute terminated by DC-shock (Fig. 1a). Medical history showed repeated episodes of sudden onset palpitation without syncope. A history of sudden cardiac arrest in the patient’s aunt was recorded.
Fig. 1.

(a) 12-Lead ECG of the patient showing VT with LBBB morphology and superior axis. (b) Resting 12-Lead ECG after admission to our hospital illustrating sinus rhythm and T-wave inversion in V1-V5.
The resting 12-lead ECG was consistent with T-wave inversion in leads V1-V5, with no bundle branch block patterns (Fig. 1b). Laboratory tests were within the normal ranges, from mild to moderate LV systolic dysfunction, with an estimated ejection fraction of 40% to 45%. The right ventricle (RV) was also severely enlarged, with moderate to severe dysfunction. Cardiac magnetic resonance imaging (CMR) of the patient revealed diffuse evidence of localized dyskinesia and microaneurismal formation in the anterior RV free wall, inferior RV wall, and the right ventricular outflow tract (RVOT) region in addition to localized LV myocardial thinning, and a few microaneurismal formation, especially in mid and apicolateral LV segments. Diffuse subepicardial to transmural fibrosis of RV inferior and anterior wall extended close to RVOT, and RV apex was determined in late gadolinium enhancement (LGE) images; moreover, LV inferoapical segment and a subepicardial fibrosis of mid lateral LV segment were found (Fig. 2).
Fig. 2.

CMR images depicting RV dilation and aneurysm formation (a) and diffuse fibrosis in RV and parts of LV (b and c) (videos are available upon request).
Signal-averaged ECG (SAECG) indicated the presence of late potentials. Taken together, the definite clinical diagnosis was made according to 2010 Task Force major and minor criteria for diagnosis of ARVC/D (3).
Sequencing of the PKP2 gene showed a new heterozygous stop codon mutation in Exon 3, c.504C>G (TAC>TAG), and p.Tyr168Ter (Fig. 3). This variant has not yet been reported in the literature. However, since the mutation leads to formation of a stop codon terminating the translation, the resulting truncated protein (p.Tyr168Ter) causes pathogenic effect.
Fig. 3.

(a) Pedigree of the family. Individual III2 and II2 had heterozygous c.504C>G. (b) Electropherograms of the patient showed a heterozygous stop codon mutation in Exon 3, c.504C>G, TAC>TAG, and p.Tyr168Ter.
Cascade screening of first-degree family members of this individual for the new variant showed that his mother is heterozygote, but the mutation was not found in his brother and his father. His mother underwent a comprehensive cardiological work-up and ARVC was not diagnosed in this individual.
Protein–protein interaction analysis was performed using STRING10 to find whether the proteins interact with Plakophilin 2.
Discussion
A 23-year-old male (III2) was presented with diagnosed ARVC due to a novel chain termination mutation in the PKP2 gene. ARVC was diagnosed according to the 2010 Task Force criteria. Genetic testing was performed for his family, which showed mutation in his mother (II2), following an autosomal-dominant inheritance.
Genetic testing can be used for possible ARVC patients (1 major or 2 minor criteria of the Heart Rhythm Society/European Heart Rhythm Association’s Expert Consensus Statement). Mutation identification would be useful for screening other family members, so that effective life style and predictive management could be used.
PKP2 mutations are the most common cause of ARVC in some populations; mutation frequency has been reported to range from 10% to 78% in ARVC patients (9,10). To date, nearly 200 mutations have been described to be pathogenic in PKP2 gene (http://www.hgmd.cf.ac.uk/). c.504C>G (p.Tyr168Ter) in PKP2 gene, which is a nonsense mutation creating a stop codon at 168 position of the protein. Of 68 reported substitution mutations, 24 are nonsense (http://www.hgmd.cf.ac.uk/). In general, nonsense mutations are responsible for nearly11% of all mutations leading to inherited disease and approximately 20% of disease-associated point mutations within coding regions (11).
As mentioned, p.Tyr168Ter in PKP2 was observed in both the son and his mother, but the mother’s condition was apparently normal. The patient underwent successful ICD implantation for secondary prophylaxis of sudden cardiac death and was discharged from the hospital in a good condition after 2 days. To find out why his mother had no diseased phenotype, we considered several hypotheses; however, she might have shown very small degrees of the phenotype, so she was underdiagnosed. On the other hand, this mutation may have a reduced penetrance. There are different factors affecting the penetrance of a mutation, such as lifestyle, genetic variations, sport activity, and gender (12-16). However, interaction analysis of PKP2 protein by STRING database Version 10.0 (17) was performed to check the probable modifier genes. STRING is a database of known and predicted proteins and showed that PKP2 gene is related to desmoplakin [DSP], Desmoglein 2 [DSG2], Desmocollin 2 [DSC2], Desmoglein 1 [DSG1], gap junction protein, Alpha 1 (GJA1), polymerase (RNA) III [POLR3A], MAP/microtubule affinity-regulating kinase 3 [MARK3A], sodium channel, voltage-gated, type V, alpha subunit (SCN5A), and Desmocollin 1 (DSC1) (Fig. 4). DSP is a major high molecular weight protein of desmosomes that plays a role in assembling the desmosomal cadherin- plakoglobin complexes to discrete transmembrane domains and anchor desmosomal intermediate filaments. DSG2, DSC2, DSG1, and DSC1 are components of intercellular desmosome junctions. DSG1 and DSC1 may also contribute to epidermal cell positioning by mediating differential adhesiveness between cells that express different isoforms. Therefore, variants of these genes leading to different isoforms of a protein may influence and affect the penetrance and expression of this mutation. Hence, other related factors, eg, various isoforms of the protein involved in desmosome junctions, could influence or compensate for Plakophilin 2 deficiency caused by this nonsense variant; in other words, although Plakophilin 2 had a defective function in the mother, other related proteins could compensate this deficiency (of Plakophilin 2, having Tyr168Ter mutation), while no such phenomenon was found in her son. However, this hypothesis should be investigated in this family. Finding this gene is an interesting idea because it may have a significant impact on unravelling the involved factors in NMD process in this individual and can open a new window to the treatment of the disease. Further studies are needed to find the prevalence of PKP2 mutations and modifier genes.
Fig. 4.
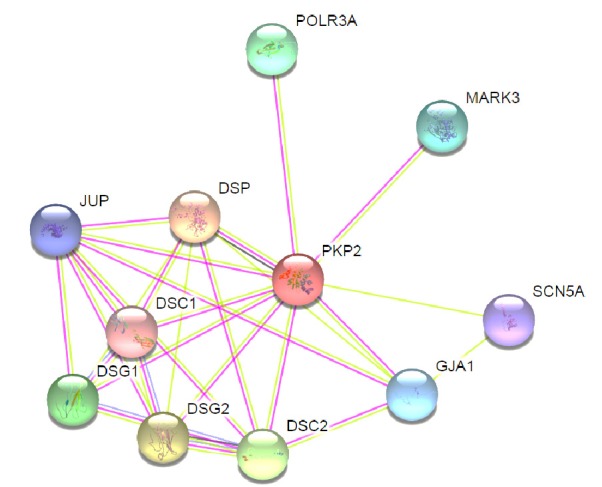
Protein–protein interaction network of Plakophilin 2 with STRING10. Plakophilin 2 is one of several proteins that make up desmosomes that provide strength to the myocardium and are involved in signaling between neighboring cells. Predicted functional partners are as follow: DSP: desmoplakin; DSG2: desmoglein 2; DSC2: desmocollin 2; DSG1: desmoglein 1; DSC1: desmocollin 1; POLR3A: polymerase (RNA) III, (DNA directed) polypeptide A; GJA1: gap junction protein, alpha 1; MARK3A: MAP/microtubule affinity-regulating kinase 3; SCN5A: sodium channel, voltage-gated, type V, alpha subunit.
Alternatively, phenotypic variability may be due to effects of modifying factors including environmental and genetic agents interacting with deficient Plakophilin 2. Modifier factors may usually influence the expression, age at onset, and progression of the disease. It is likely that the existence of an unknown modifier effect, such as delE120 mutation in GJB2 gene, may modulate this scenario (14). Nonsense mutations result in a premature termination codon and most often lead to complete loss of protein and reduce mRNA levels because of the nonsense-mediated mRNA decay (NMD). NMD, as an mRNA quality-control mechanism, starts selective degradation of mutant transcripts and improves cellular surveillance (18). In the studied family, NMD may decrease the severity of disease in his mother.
Conclusion
This study, as a first report of PKP2 mutation in an Iranian family, highlights the role of modifier genes involved in ARVC. Finding the cause of phenotypic variability due to this variant within the family may shed light on the road of treatment approach for ARVC. Also, PKP2 mutation may play a major role in the development of disease in our population.
Acknowledgments
We thank the family who participated in this study and we also thank all staff of Cardiogenetic Research Laboratory, Rajaie Cardiovascular Center for their assistance.
Conflict of Interests
The authors declare that they have no competing interests.
Cite this article as: Mahdieh N, Saedi S, Soveizi M, Rabbani B, Najafi N, Maleki M. A novel PKP2 mutation and intrafamilial phenotypic variability in ARVC/D. Med J Islam Repub Iran. 2018 (6 Feb);32:5. https://doi.org/10.14196/mjiri.32.5
References
- 1.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G. et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215–8. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C. et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226–33. doi: 10.1016/s0735-1097(00)00997-9. [DOI] [PubMed] [Google Scholar]
- 3.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA. et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31:806–14. doi: 10.1093/eurheartj/ehq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinamonti B, Brun F, Mestroni L, Sinagra G. Arrhythmogenic right ventricular cardiomyopathy: From genetics to diagnostic and therapeutic challenges. World J Cardiol. 2014;6:1234–44. doi: 10.4330/wjc.v6.i12.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quarta G, Muir A, Pantazis A, Syrris P, Gehmlich K, Garcia-Pavia P. et al. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation. 2011;123:2701–9. doi: 10.1161/CIRCULATIONAHA.110.976936. [DOI] [PubMed] [Google Scholar]
- 6.Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW. et al. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. 2004;167:149–60. doi: 10.1083/jcb.200402096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalal D, Molin LH, Piccini J, Tichnell C, James C, Bomma C. et al. Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation. 2006;113:1641–9. doi: 10.1161/CIRCULATIONAHA.105.568642. [DOI] [PubMed] [Google Scholar]
- 8.Syrris P, Ward D, Asimaki A, Sen-Chowdhry S, Ebrahim HY, Evans A. et al. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:356–64. doi: 10.1161/CIRCULATIONAHA.105.561654. [DOI] [PubMed] [Google Scholar]
- 9.van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA. et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–8. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- 10.Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ. et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol. 2011;57:2317–27. doi: 10.1016/j.jacc.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29:1037–47. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- 12.Cutting GR. Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci. 2010;1214:57–69. doi: 10.1111/j.1749-6632.2010.05879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dipple KM, McCabe ER. Modifier genes convert "simple" Mendelian disorders to complex traits. Mol Genet Metab. 2000;71:43–50. doi: 10.1006/mgme.2000.3052. [DOI] [PubMed] [Google Scholar]
- 14.Mahdieh N, Bagherian H, Shirkavand A, Sharafi M, Zeinali S. High level of intrafamilial phenotypic variability of non-syndromic hearing loss in a Lur family due to delE120 mutation in GJB2 gene. Int J Pediatr Otorhinolaryngol. 2010;74:1089–91. doi: 10.1016/j.ijporl.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 15.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H. et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–7. doi: 10.1016/j.jacc.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E. et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6:533–42. doi: 10.1161/CIRCGENETICS.113.000288. [DOI] [PubMed] [Google Scholar]
- 17.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurosaki T, Maquat LE. Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci. 2016;129:461–7. doi: 10.1242/jcs.181008. [DOI] [PMC free article] [PubMed] [Google Scholar]