

Abstract
Interferonopathies are a subset of autoinflammatory disorders with a prominent type I IFN gene signature. Treatment of these patients has been challenging, given the lack of response to common autoinflammatory therapeutics including IL-1 and TNF blockade. JAK inhibitors (Jakinibs) are a family of small-molecule inhibitors that target the JAK/STAT signaling pathway and have shown clinical efficacy, with FDA and European Medicines Agency (EMA) approval for arthritic and myeloproliferative syndromes. Sanchez and colleagues repurposed baricitinib to establish a significant role for JAK inhibition as a novel therapy for patients with interferonopathies, demonstrating the power of translational rare disease research with lifesaving effects.
Interferonopathies, a new category of autoinflammatory diseases
The autoinflammatory disease category was developed to explain the pathogenesis of disorders, such as the hereditary fever syndromes, that did not fit squarely into the traditional classification of immune dysregulation disorders: immunodeficiency, autoimmunity, and allergy (1). Identification of the genetic and molecular basis for these rare inherited conditions defined these diseases as innate immune driven, exposed IL-1 as the central mediator, and led to the successful use of IL-1–targeted biologic therapies for these patients (2). This translational medicine success story (3), beginning with gene identification and elucidation of disease mechanisms followed by drug repurposing and investigator-initiated clinical trials, has expanded beyond these few disorders to encompass new groups of syndromes with different inflammatory mechanisms including the NF-κB or type I IFN pathways. The disorders, collectively known as the interferonopathies, as they share a common downstream mechanism involving type I IFNs, are the focus of a manuscript by Sanchez et al. in this month’s issue of the JCI (4).
The type I IFNs, including IFN-α and -β, are inflammatory cytokines implicated in antiviral responses and numerous hematologic and inflammatory diseases, from hepatitis to multiple sclerosis (5). Both cytokines bind to the heterodimeric IFN receptor (IFNAR) and signal through tyrosine protein kinase 2 (TYK2) and JAK1, with recruitment and formation of the STAT1-STAT2 dimer, leading to a downstream inflammatory cascade and disease-associated manifestations. The importance of JAK/STAT signaling in the pathogenesis of common inflammatory disorders is supported by the clinical development or approval of numerous specific small-molecule inhibitors in rheumatoid arthritis, psoriasis, and myelofibrosis (6). As with the application of IL-1 pathway inhibitors to hereditary fever disorders in the past decade (7), the availability of these targeted, oral small-molecule drugs has found new life as effective treatments for autoinflammation (Figure 1).
Figure 1. Disease mechanisms and new therapeutic strategy for the interferonopathies.
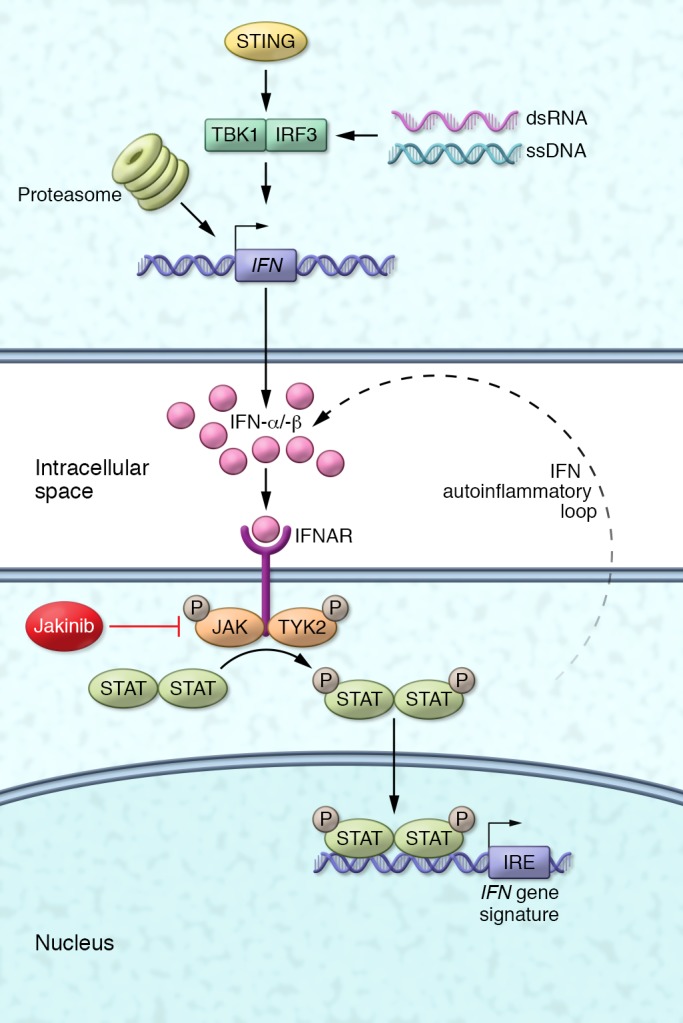
The interferonopathies including SAVI, CANDLE, and AGS are associated with overproduction of type I IFNs, IFN-α and IFN-β, through diverse mechanisms including pathways involving nucleic acid sensing and intracellular stress. These cytokines bind to the common IFN a/β receptor, leading to phosphorylation and activation of the JAK/STAT pathway, with phosphorylation and translocation of STAT proteins to the nucleus. The STATs subsequently stimulate transcription of several IFN-induced genes that are part of a clinically validated IFN gene signature. Like other autoinflammatory diseases, IFNs stimulate the further expression and release of IFNs, thereby amplifying inflammation. JAK inhibitors (Jakinibs) block the activation of JAK, preventing the expression of IFN-induced genes and the autoinflammatory loop. IRE, IFN-regulatory element; IRF3, IFN-regulatory factor 3; TBK1, TANK-binding kinase 1.
JAK inhibition as a therapy for interferonopathies
The group, led by Goldbach-Mansky, applied their tried-and-true model of methodical patient phenotyping to enroll 18 severely affected pediatric and adult patients with three defined autoinflammatory disorders including CANDLE (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature) (8–11), SAVI (stimulator of IFN genes–associated [STING-associated] vasculopathy with onset in infancy) (12), and Aicardi Goutières syndrome (AGS) (13), as well as patients with undefined interferonopathies that were identified by a recently validated panel of 25 genes related to type I IFNs (14). Each of the subjects had been treated unsuccessfully with corticosteroids and other immunosuppressive drugs or targeted biologics. Patients were treated with baricitinib under a dose optimization protocol. Efficacy measures during the five-year study included symptom diary and pain scores, quality of life, reduction in corticosteroid dose, growth parameters, lung function, bone density, inflammatory markers, metabolic markers, hematologic indices, autoantibodies, serum cytokines, and the IFN signature. It should be noted that a similar comprehensive protocol was the basis for FDA licensing and orphan disease approval of anakinra for neonatal-onset multisystem inflammatory disease (NOMID), without the traditional large-scale, placebo-controlled study typical of pivotal clinical trials (15). In the current manuscript (4), the authors further establish this patient-focused approach as the definitive method for therapeutic intervention in rare diseases.
Most of the patients in this study showed significant improvement from baseline in many of the efficacy measures, despite the fact that it was almost impossible to assess the placebo response. Overall, the CANDLE patients showed more improvement in their inflammatory markers and IFN signature than did the SAVI and AGS patients, demonstrating a disease-specific responsiveness that may elucidate previously unappreciated subtleties in IFN signaling. Interestingly, two of the patients with higher initial IFN signatures and undefined interferonopathies improved significantly, suggesting that genotype is not necessary to tailor treatment decisions. However, manipulation of a key inflammatory pathway is not without risks. Adverse events were characterized primarily by increased infections, ranging from mild upper respiratory infections to bacteremia, osteomyelitis, and BK viremia requiring antibiotic therapy or discontinuation of the drug.
Gene signatures define disease pathogenesis and therapy
IFNs are classically known for their role in antiviral and antitumor responses and act directly or indirectly on phagocytic cells, DCs, as well as B cells, T cells, and NK cells. Only recently, however, has their role in autoinflammation generated increased interest in the manipulation of these pathways beyond chronic viral infections such as hepatitis or HIV. Despite the seemingly different inflammatory outcomes of infection, autoimmunity, and now autoinflammation, the combined study of this full spectrum of immunologic disease has powered our knowledge of the IFN response genes and downstream mediators. Goldbach-Mansky and colleagues have harnessed this information into a clinically useful IFN signature based on gene expression that helps to differentiate this group of diseases from other autoinflammatory syndromes (14). Consistent with the origins of autoinflammation, this translational approach has not only defined this new class, but it also clearly demonstrates that additional disease-causing genes remain to be identified.
Translational impact and future challenges
Beyond the interferonopathies, use of this gene expression methodology to evaluate diseases from a biologic pathway perspective provides opportunities to expand our knowledge of other immunologic syndromes and to apply targeted therapy. Immunodysregulatory disorders are increasingly identified, in which there are contradictory clinical or laboratory features of autoinflammation or autoimmunity in association with immunodeficiency, resulting in seemingly paradoxical therapeutic approaches. Furthermore, the use of immunologic gene signatures in patients’ blood may help to guide unconventional sequential or combination therapy in difficult-to-treat, complex inflammatory diseases such as hemophagocytic lymphohistiocytosis (HLH) (16) and macrophage-activation syndrome (MAS) (17). This exciting novel approach to therapy opens up a new frontier for personalized medicine.
Given the relatively recent description of the interferonopathies, future challenges include defining long-term treatment–related complications, unrecognized manifestations of disease, and potential changes in response to therapy over time. As additional patients are identified, genotypic-phenotypic variability and better-characterized therapeutic responses will likely come to light. However, with this report, Sanchez et al. fill a critical current niche of reducing morbidity and mortality in patients with interferonopathy.
Acknowledgments
Our studies are supported by the National Institute of Child Health and Human Development (NICHD), NIH (5K08HD075830, to LB); the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH (R01 DK113592, to HH); and the National Heart, Lung, and Blood Institute (NHLBI), NIH (HL140898, to HH).
Version 1. 06/11/2018
Electronic publication
Version 2. 07/02/2018
Print issue publication
Footnotes
Conflict of interest: HMH is a consultant and speaker for Novartis and has received research funds from Jecure.
Reference information: J Clin Invest. 2018;128(7):2760–2762. https://doi.org/10.1172/JCI121526.
See the related article at JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies.
Contributor Information
Hal M. Hoffman, Email: hahoffman@ucsd.edu.
Lori Broderick, Email: lbroderick@ucsd.edu.
References
- 1.Stoffels M, Kastner DL. Old dogs, new tricks: monogenic autoinflammatory disease unleashed. Annu Rev Genomics Hum Genet. 2016;17:245–272. doi: 10.1146/annurev-genom-090413-025334. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman HM, Broderick L. The role of the inflammasome in patients with autoinflammatory diseases. J Allergy Clin Immunol. 2016;138(1):3–14. doi: 10.1016/j.jaci.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Holzinger D, Kessel C, Omenetti A, Gattorno M. From bench to bedside and back again: translational research in autoinflammation. Nat Rev Rheumatol. 2015;11(10):573–585. doi: 10.1038/nrrheum.2015.79. [DOI] [PubMed] [Google Scholar]
- 4.Montealegre Sanchez GA, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. 2018;128(7):3041–3052. doi: 10.10172/JCI98814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282(28):20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 6.O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311–328. doi: 10.1146/annurev-med-051113-024537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu Rev Med. 2014;65:223–244. doi: 10.1146/annurev-med-061512-150641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agarwal AK, et al. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87(6):866–872. doi: 10.1016/j.ajhg.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arima K, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci U S A. 2011;108(36):14914–14919. doi: 10.1073/pnas.1106015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitamura A, et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest. 2011;121(10):4150–4160. doi: 10.1172/JCI58414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012;64(3):895–907. doi: 10.1002/art.33368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crow YJ, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38(8):910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 14.Kim H, et al. Development of a validated interferon score using nanostring technology. J Interferon Cytokine Res. 2018;38(4):171–185. doi: 10.1089/jir.2017.0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldbach-Mansky R, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1β inhibition. N Engl J Med. 2006;355(6):581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brisse E, Matthys P, Wouters CH. Understanding the spectrum of haemophagocytic lymphohistiocytosis: update on diagnostic challenges and therapeutic options. Br J Haematol. 2016;174(2):175–187. doi: 10.1111/bjh.14144. [DOI] [PubMed] [Google Scholar]
- 17.Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12(5):259–268. doi: 10.1038/nrrheum.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]