

Abstract
Chronic inflammation is a risk factor for gastrointestinal cancer and other diseases. Most studies have focused on cytokines and chemokines as mediators connecting chronic inflammation to cancer, whereas the involvement of lipid mediators, including prostanoids, has not been extensively investigated. Prostanoids are among the earliest signaling molecules released in response to inflammation. Multiple lines of evidence suggest that prostanoids are involved in gastrointestinal cancer. In this Review, we discuss how prostanoids impact gastrointestinal cancer development. In particular, we highlight recent advances in our understanding of how prostaglandin E2 induces the immunosuppressive microenvironment in gastrointestinal cancers.
Introduction
Clinical and epidemiologic evidence indicates that chronic inflammation is a major risk factor for several gastrointestinal malignancies, including esophageal, gastric, colorectal, hepatic, and pancreatic cancer. For example, patients with persistent hepatitis B infection, Helicobacter pylori infection, or autoimmune disorders such as inflammatory bowel diseases (IBD) face an increased lifetime risk for liver cancer, gastric cancer (GC), or colorectal cancer (CRC), respectively. In addition, solid tumors themselves exhibit certain characteristics found in inflamed tissues, referred to as tumor-induced inflammation. The common pathological features of chronic inflammatory diseases and solid cancers include elevation of proinflammatory mediators such as cytokines, chemokines, and lipids, massive infiltration of deregulated immune cells, and recruitment of endothelial cells and fibroblasts (1–3). The observation that nonsteroidal antiinflammatory drugs (NSAIDs) reduce the incidence, metastasis, and mortality of various solid tumors (4–10), including gastrointestinal cancer, supports the concept that chronic inflammation promotes tumor initiation, growth, and progression. NSAIDs are the most commonly used drugs that help reduce inflammation and relieve fever and pain. It is well accepted that NSAIDs primarily target the cyclooxygenase enzymes COX-1 and COX-2 in reducing inflammation and relieving pain and/or fever.
COX-1 is constitutively expressed in most tissues and is thought to provide basal levels of prostanoids, a subgroup of eicosanoids including prostaglandins (PGs), thromboxanes, and prostacyclins that are important for tissue homeostasis and platelet function. In contrast, COX-2 is an immediate-early response gene that is usually absent in healthy tissues and organs, but is highly inducible at sites of inflammation and is overexpressed in certain cancers, such as those that arise in the gastrointestinal tract. For example, COX-2 expression is elevated in approximately 50% of colorectal adenomas and 85% of adenocarcinomas (11–13). Similarly, COX-2 overexpression is also observed in esophageal and gastric cancer (14, 15). Elevation of COX-2 expression is also associated with a shorter survival time among patients with CRC and esophageal cancer (16, 17). However, conflicting results have been reported in the association between COX-2 expression and survival in patients with GC (18). COX enzymes convert arachidonic acid into an endoperoxide intermediate that can be further metabolized to prostanoids, including PGs such as PGE2, PGD2, PGF2α, PGI2, and thromboxane A2 (TxA2) via specific PG synthases (Figure 1). Moreover, prostanoids exert their cellular functions by binding cell surface G protein–coupled receptors. These cell surface receptors are designated EP (EP1, EP2, EP3, and EP4) for the PGE2 receptors, DP1 and DP2 for the PGD2 receptor, FP for the PGF2α receptor, IP for the PGI2 receptor, and TP for the TxA2 receptor (Figure 1).
Figure 1. An overview of prostanoid synthesis pathways.
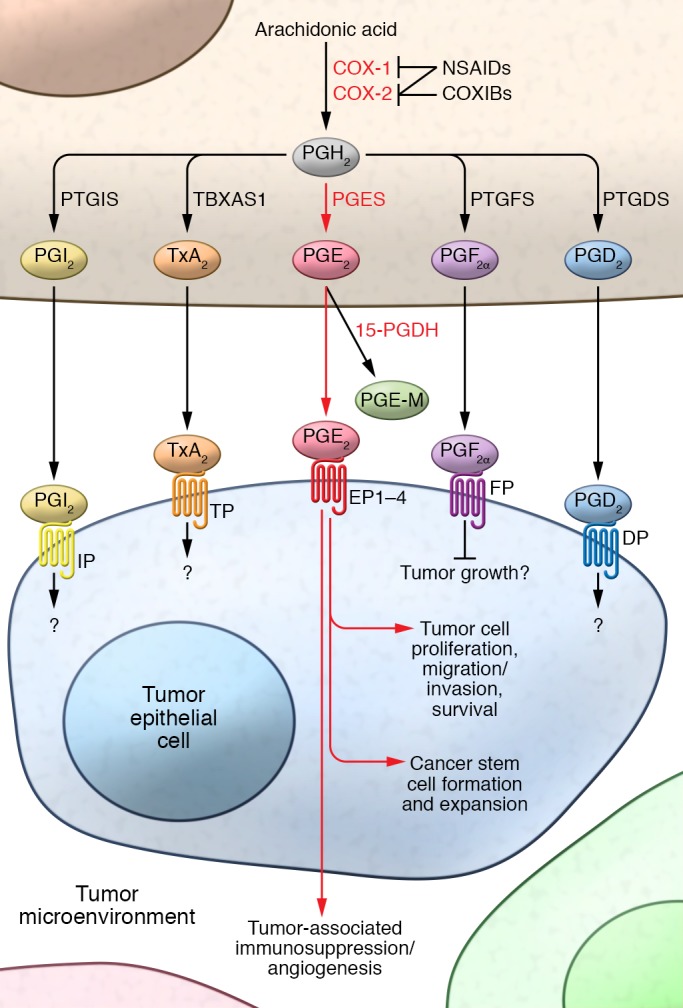
Free arachidonic acid can be metabolized to PGH2 by COX-1 and COX-2. NSAIDs inhibit activity of COX-1 and COX-2, whereas COXIBs inhibit activity of COX-2. PGH2 is sequentially metabolized to PGI2 by PTGIS, TxA2 by TBXAS1, PGE2 by PGES, PGF2α by PTGFS, and PGD2 by PTGDS. PGE2 binding at EP1–EP4 receptors is known to promote cancer development via multiple mechanisms (described in detail in the text and Figures 2 and 3). The roles of other prostanoids and their receptors in gastrointestinal cancer remain unclear (also detailed in the text).
The roles of prostanoids in acute inflammation have been recognized very early, and their levels are immediately elevated before leukocyte infiltration in acute inflammation. Among prostanoids, both PGE2 and PGI2 have been shown to induce acute inflammation in the majority of animal models (19). In contrast, PGD2 has been shown to suppress acute inflammation via binding to its DP receptors and via enzymatically independent generation of 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2) in animal models (20). 15d-PGJ2 mainly binds to PPARγ and directly inhibits the NF-κB signaling pathway (21, 22). Similarly, PGE2 and PGI2 also enhance chronic inflammation (23) and play a key role in arthritis and IBD (24, 25). The role of PGD2 in chronic inflammation is context-dependent. On the one hand, PGD2-derived 15d-PGJ2 inhibits adjuvant-induced arthritis in vivo (26). On the other hand, PGD2 facilitates allergic inflammation (25). The roles of PGF2α and TxA2 in inflammation remain unclear.
Although NSAIDs exhibit antitumor effects, the molecular mechanisms underlying their effects, especially aspirin, are not fully understood. Although other mechanisms have been proposed to explain the antitumor effects of these drugs, and “off-target” effects do exist, COX-1 and COX-2 remain primary targets. For example, celecoxib, belonging to a family of COX-2–selective inhibitors (COXIBs), was initially approved by the FDA for use as adjuvant therapy for patients with familial adenomatous polyposis (FAP), but is no longer recommended for that indication. However, long-term use of celecoxib and other COXIBs as well as nonselective NSAIDs (except for aspirin) at high doses is associated with unacceptable cardiovascular side effects in patients, especially in those with a history of atherosclerotic heart disease (27).
Unlike COXIBs and other nonselective NSAIDs, long-term daily use of aspirin is beneficial for prevention of both cancer and cardiovascular diseases. Daily use of aspirin has been shown to suppress polyp growth in FAP patients in two double-blind randomized controlled trials (28, 29) and to reduce risk of adenoma recurrence in patients with a history of colorectal adenomas in four randomized controlled trials (30–33). More intriguingly, recent observational and clinical studies revealed that daily use of aspirin was associated with a reduced risk of metastatic spread (5) and inhibited the spread of primary tumor cells to other organs after the diagnosis of localized disease, in particular CRC (7). Based on recent clinical and epidemiologic evidence, the United States Preventive Services Task Force endorsed aspirin as the first agent for CRC prevention in individuals between the ages of 50 and 69 with specific cardiovascular risk profiles (34). In addition, aspirin has been shown to enhance the efficacy of chemoradiation for patients with stage II–III rectal cancer (35), indicating that aspirin can be used as a neoadjuvant agent for certain cancer treatments. Aspirin has also been shown to reduce risk of esophageal cancer and GC (10, 36, 37). Moreover, epidemiologic studies have shown that regular use of aspirin specifically reduced risk of the subgroup of patients whose colon tumors expressed higher levels of COX-2 (38), and its use after the diagnosis of CRC at stages I, II, and III prolonged overall survival, especially among individuals whose tumors overexpressed COX-2 (39). These results suggest that antitumor effects of aspirin on CRC might depend on the presence of COX-2. In addition to COX-2 expression, PIK3CA mutation and HLA class I antigen expression levels also affect the efficacy of aspirin in improving the overall survival rate of CRC patients (40, 41). However, it is not clear how PIK3CA mutations and HLA class I antigen expression are involved in antitumor effects of aspirin.
Since NSAIDs are known to cause gastrointestinal and/or cardiovascular side effects, one of the ways to avoid these side effects would be to target only the COX-2–derived prostanoids that mediate the tumor-promoting effects of COX-2. In this Review, we highlight our current understanding of the role of specific prostanoids in gastrointestinal cancer. Understanding how these bioactive lipids regulate tumor formation, growth, progression, and metastasis may provide a rationale for developing novel and more effective strategies in cancer prevention and treatment that avoid side effects associated with NSAID use.
Prostanoids and gastrointestinal cancer
The biological functions of COX-1/2 enzymes depend on which COX-derived prostanoids are produced in cancers. Among prostanoids, PGE2 is the most abundant in human gastrointestinal cancers, including CRC and GC (42, 43). More importantly, only PGE2 and PGI2 levels are elevated in CRC specimens as compared with matched normal tissues (44). The steady-state accumulation of PGE2 in tumor tissues depends on the relative rates of COX-2/PGE2 synthase–dependent biosynthesis and 15-hydroxyprostaglandin dehydrogenase–dependent (15-PGDH–dependent) degradation (Figure 1). 15-PGDH first converts PGE2 into an inactive 15-keto PGE2 that is then further metabolized to a stable end metabolite (PGE-M) in a series of steps. 15-PGDH is highly expressed in normal tissues but is ubiquitously lost in many human cancers, including CRC, GC, and esophageal cancer (45–48). Since measurement of the urinary PGE2 metabolite PGE-M is an effective way to quantify systemic PGE2 production in vivo, much work has been done to evaluate whether urinary PGE-M levels could serve as a promising biomarker for predicting cancer risk and prognosis. Emerging epidemiologic evidence and a phase II biomarker study showed that urinary PGE-M levels were associated with an increased risk of developing CRC and GC (49–53). These results suggest that urinary PGE-M could be used as a biomarker for predicting gastrointestinal cancer risk and prognosis. More importantly, epidemiologic evidence revealed a strong inverse association between aspirin use and levels of urinary PGE-M in healthy humans (54) and breast cancer patients (55, 56). Moreover, a recent study showed that low-dose aspirin (100 mg/d for 7 days) reduced PGE2 levels in human colorectal mucosa by 46% (57). In addition, one epidemiologic study showed that regular use of aspirin more effectively reduced CRC risk in patients with higher 15-PGDH expression compared with low 15-PGDH expression in colonic mucosa (58). These findings further support the hypothesis that PGE2 mediates some of the tumor-promoting effects of COX-2 as well as the notion that the COX-2/PGE2 pathway is a legitimate target for cancer prevention and treatment.
Direct evidence that PGE2 promotes tumor growth came from animal studies. In mouse models of FAP and/or sporadic CRC, PGE2 treatment dramatically increased both small and large intestinal adenoma burden in ApcMin/+ mice and significantly enhanced azoxymethane-induced (AOM-induced) colon tumor incidence and multiplicity (59, 60). Furthermore, elevating endogenous PGE2 by genetically deleting 15-Pgdh promotes colon tumor growth in ApcMin/+ and AOM mouse models (61). PGE2 also reverses the antitumor effects of an NSAID in ApcMin/+ mice (62), suggesting that PGE2 is one of the important NSAID targets for cancer prevention and treatment. In accordance with the above results, inhibition of endogenous PGE2 by genetic deletion of microsomal PGE2 synthase 1 (mPges-1) suppresses intestinal tumor formation and growth in Apc-mutant and AOM models (63, 64). In mouse models of GC, simultaneous overexpression of COX-2 and mPGES-1 in gastric epithelial cells was sufficient to induce gastric tumor formation (65). Moreover, deletion of EP1 or EP4, but not EP3, attenuates AOM-induced aberrant crypt foci (66, 67). In ApcΔ716 mice, loss of EP2, but not EP1 and EP3, reduces intestinal tumor burden (68). Interestingly, one report showed that loss of EP3 promoted colon tumor development in AOM-treated mice (69). In a mouse model of colitis-associated CRC, loss of EP2 reduced the number of colon tumors, whereas deletion of EP1 or EP3 increased colon tumor numbers (70). Collectively, these results demonstrate that PGE2 promotes intestinal tumorigenesis via EP2 and EP4, but not EP3. The role of EP1 in CRC remains unclear.
In evaluating the role of other PGs in CRC, contradictory results have been reported in mouse models of CRC. For PGD2, loss of hematopoietic PGD2 synthase (PTGDS) accelerated intestinal tumor growth in ApcMin/+ mice (71), and deletion of Ptgds in mast cells enhanced colitis-associated tumorigenesis in an AOM/dextran sulfate sodium (DSS) model (72). In addition, deletion of DP resulted in an increase of intestinal tumor numbers in ApcMin/+ and AOM/DSS-treated mice (70, 73). In contrast, ApcMin/+ mice expressing transgenic human hematopoietic PTGDS exhibited fewer intestinal adenomas than controls (71). These results suggest that PGD2 serves as a tumor suppressor in CRC. However, one study showed that disruption of DP did not affect colon tumor formation in AOM-treated mice (74). For PGI2, one report showed that loss of PGI2 synthase (PTGIS) facilitated colon carcinogenesis in AOM-treated mice (75). However, the results that loss of IP did not affect colon tumor formation in AOM/DSS–treated mice (70, 74) do not support the antitumor effect of PGI2 in the colon via the IP receptor. For PGF2α and TxA2, one study showed that disruption of FP or TP did not affect colon tumor burden in AOM-treated mice (74), whereas loss of TP increased the number of colon tumors in AOM/DSS–treated mice (70). More intriguingly, the expression of DP, FP, and IP receptors is reduced in human CRC specimens as compared with adjacent normal colon tissues (76). Clearly, the question of whether PGD2, PGI2, PGF2α, TxA2, and their receptors are involved in gastrointestinal cancer needs to be further investigated.
To understand the mechanisms underlying effects of PGE2 on cancer development, researchers have been investigating precisely how PGE2 promotes tumor formation, growth, progression, and metastasis. Numerous reports suggest that PGE2 promotes cancer development via multiple mechanisms, including regulation of tumor epithelial cell biology, promotion of tumor-associated angiogenesis, and suppression of tumor immunity (Figure 1).
PGE2 and tumor epithelial cells
The mechanisms by which PGE2 promotes tumor epithelial cell proliferation, survival, and migration/invasion as well as tumor-associated angiogenesis have been reviewed in detail elsewhere (refs. 22, 77; and Figure 1). Here we highlight emerging evidence indicating that PGE2 may be a targetable link between chronic inflammation and tumor initiation (Figure 2). PGE2 has been shown to promote intestinal tumor initiation and growth by silencing certain tumor suppressor and DNA repair genes via DNA methylation (78). In addition, cancer stem cells (CSCs) are thought to be responsible for tumor initiation, growth, metastatic spread, relapse, and recurrence. The observation that the expression of stem cell factors (i.e., CD44, LGR5, SOC-2, and OCT4) is associated with a worse prognosis in CRC (79) supports this hypothesis. It is also believed that chemotherapy/radiation resistance is due to the presence of CSCs that are not being properly targeted (80). Moreover, the observation that chemotherapeutic and/or radiation therapy enhances COX-2 expression and PGE2 production in cancers prompted investigators to postulate that PGE2 regulates CSC biology. NSAIDs have been shown to eliminate oncogenic intestinal stem cells via inducing apoptosis in ApcMin/+ mice (81) and to inhibit sphere formation in human colorectal carcinoma cells in vitro (82). Strikingly, PGE2 promotes colonic CSC formation and expansion as well as liver metastasis by activating NF-κB via EP4-dependent PI3K/MAPK pathways in vivo (83, 84). Similarly, simultaneous overexpression of COX-2 and mPGES-1 in the gastric epithelial cells is sufficient to induce CD44+ slow-cycling tumor cell expansion in vivo (85), indicating that PGE2 induces gastric CSC expansion. In addition, PGE2 released following chemotherapy-induced apoptotic tumor cells promotes neighboring CSC repopulation in a xenograft model of bladder cancer (86). Collectively, these results suggest that reduction of PGE2 levels and/or inhibition of PGE2 signaling pathways may not only suppress tumor cell proliferation, survival, and migration/invasion, but may also eliminate CSCs. Targeting CSCs may thus present a novel therapeutic approach for cancer patients.
Figure 2. PGE2 regulation of tumor initiation.
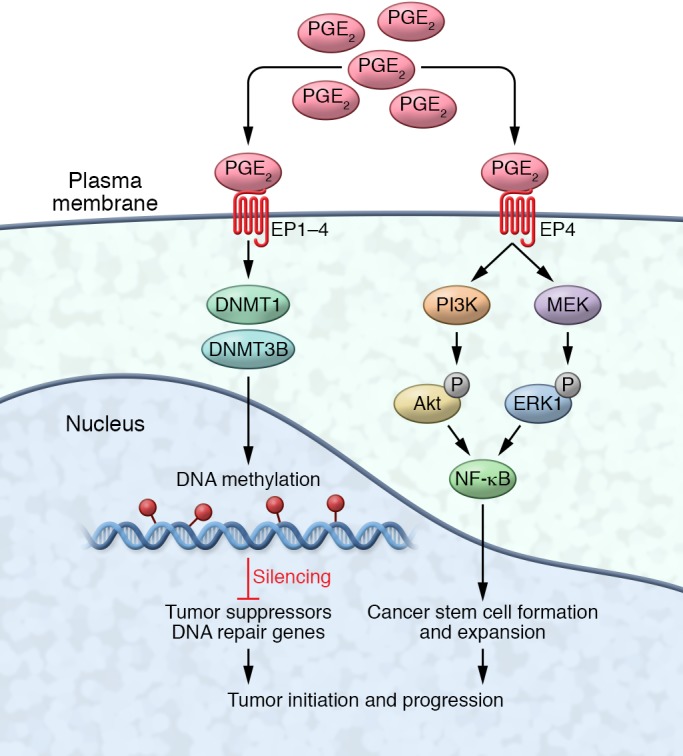
PGE2 promotes tumor initiation and growth via DNA methylation and cancer stem cells. PGE2 binds to its receptors (EP1–EP4) to suppress certain tumor suppressor and DNA repair genes by DNA methylation via induction of expression of the DNA methyltransferases DNMT1 and DNMT3B. PGE2 also promotes cancer stem cell formation and expansion by activating NF-κB via EP4-dependent PI3K/MAPK pathways.
PGE2 and tumor-associated immunosuppression
The role of PGE2 in regulating immunity and host defense against viral, fungal, and bacterial pathogens has been reviewed in detail elsewhere (87). Here we focus on the role of PGE2 in tumor-associated immunosuppression. The tumor microenvironment not only supports tumor growth, progression, and spread by angiogenesis, but also allows tumor cells to evade host immunosurveillance. This tumor-associated immunosuppression is characterized by enhancement of immunosuppressive cells, a defect of antigen-presenting cell function, a shift from Th1 to Th2 and Th17 immune responses, and impairment of cytotoxic activity of CD8+ T and natural killer (NK) cells. Reversing immunosuppression remains one of the major challenges in cancer immunotherapy. It is becoming increasingly evident that PGE2 has a broader impact on tumor-associated immunosuppression than previously thought (Figure 3). However, the mechanisms by which PGE2 induces tumor-associated immunosuppression remain largely unclear. Understanding the mechanisms underlying PGE2 induction of tumor-associated immunosuppression may provide a rationale for developing more effective therapeutic strategies to subvert tumor-induced immunosuppression for patients with gastrointestinal cancer.
Figure 3. A model of PGE2-regulated tumor-associated immunosuppression.

PGE2 regulates immunosuppressive cells and their functions by (a) inducing MDSC differentiation and production of PD-L1 and arginase I; (b) shifting macrophages from M1 to M2, inducing PD-L1 expression, and reducing macrophage phagocytosis; and (c) inducing differentiation and migration of Tregs. PGE2 regulates DCs and their functions through inhibition of differentiation and maturation; induction of T cell tolerance and IL-23 expression; and induction of dedifferentiation of DCs to MDSCs. PGE2 regulates Th cells and their functions by inducing differentiation and recruitment of Th17, and shifting Th cells from Th1 to Th2. PGE2 regulates CD8+ T cells and their functions by induction of proliferation and tumor antigen–specific tolerance and reduction of CD8+ T cell cytotoxicity. PGE2 regulates NKs and their functions via suppression of cell activation and proliferation and induction of cell apoptosis.
PGE2 and immunosuppressive cells
Myeloid-derived suppressor cells.
In healthy individuals, immature myeloid cells differentiate into mature myeloid cells including macrophages, DCs, and granulocytes. However, this normal physiological process is interrupted in cancer patients. Myeloid-derived suppressor cells (MDSCs) are a heterogeneous group of immature myeloid cells that have been demonstrated to contribute to cancer immunosuppression by suppression of effector T cell activation, proliferation, trafficking, and viability; by inhibition of NKs; and by activation/expansion of regulatory T cells (Tregs) (88). The levels of MDSCs in the blood and/or tumor tissue correlated with clinical cancer stage, metastatic tumor burden, or poor survival in patients with colon, esophageal, gastric, or pancreatic cancer (89–93). Animal studies have demonstrated that MDSCs mediate one of the protumor effects of chronic inflammation. For example, depletion of MDSCs attenuated colitis-associated tumorigenesis in a mouse model of IBD-associated carcinogenesis (94). Along the same lines, transfer of MDSCs promoted chronic colonic inflammation and colitis-associated tumor development via suppression of colonic CD8+ T cell cytotoxicity against tumor cells in a mouse model of colitis-associated carcinogenesis (95). Moreover, liver-infiltrating MDSCs formed a premetastatic niche that ultimately promoted liver metastases without involvement of T and NK cells in a mouse model of metastatic spread of CRC (96). MDSCs isolated from premetastatic livers of immunodeficient NSG mice bearing cecal tumors inhibited colorectal carcinoma cell apoptosis induced by serum deprivation in cell culture without cell-cell interaction of malignant cells and MDSCs, suggesting that MDSCs must secrete factors that promote tumor cell survival (96). In addition, MDSCs have also been shown to directly enhance CSC formation and protect proliferating tumor cells from senescence without involvement of T and NK cells in vivo (97, 98).
Multiple studies have shown that inhibition of COX-2 suppressed tumorigenesis by inhibiting tumor-associated MDSC infiltration in mouse models of CRC and glioma (99, 100) as well as in mice with implanted mesothelioma and mammary carcinoma (101, 102). Moreover, PGE2 promoted tumor progression via inducing the development of MDSCs from bone marrow myeloid progenitor cells, whereas inhibition of PGE2 signaling by deletion of EP2 or its antagonists blocked this differentiation in mice with implanted 4T1 murine mammary carcinoma tumors (102). An EP4 antagonist, E7046, has been shown to reduce tumor-infiltrating MDSCs and to enhance the antitumor effect of anti–CTLA-4 antibodies in syngeneic mouse models of cancer (103), indicating that EP4 mediates the effect of PGE2 on MDSCs. An in vitro study showed that PGE2 blocked differentiation of monocytes into DCs and promoted MDSC development (104). Moreover, PGE2 enhanced immunosuppressive function by inducing MDSC-specific hypermethylation via DNMT3A (105) and by inducing PD-L1 expression (106) and arginase I expression (107) in vitro. However, the role of PGE2 in enhancement of gastrointestinal tumor-infiltrating MDSCs is still largely unknown, and the mechanisms by which PGE2 regulates MDSC differentiation, expansion, and immunosuppressive functions are also not fully understood.
Regulatory T cells.
Tregs are essential for suppressing immune responses and maintaining self-tolerance by regulating the activity of other immune cells. The frequency of Tregs (CD4+CD25+Foxp3+) is elevated in the peripheral blood and in primary tumors of CRC and GC patients (108, 109). Tumor-infiltrating Tregs are also associated with GC progression and a poor survival rate (110, 111). There is a positive correlation between PGE2 levels and the numbers of Foxp3+ Tregs in the peripheral blood, tumor tissues, and draining lymph nodes of CRC patients (112). In addition, Foxp3 expression in tumor-infiltrating Tregs correlates with COX-2 expression and PGE2 levels in GC (109).
In mouse models of cancer, inhibition of COX-2 by celecoxib resulted in reduction of tumor burden and proportion of Tregs in intestinal lamina propria lymphocytes in ApcMin/+ mice (113). Deletion of mPges-1 attenuated AOM-induced tumor formation with reduction of Tregs in the colon-draining mesenteric lymph nodes (64). In addition, treatment with an EP4 antagonist resulted in a decreased number of Tregs in the peripheral lymph nodes after UV irradiation (114). An EP1 antagonist inhibited tumor growth with reduction of tumor-infiltrating Tregs in a colon tumor implantation model (115). Consistent with these findings, PGE2 promoted tumor growth with induction of Treg expansion and activity in a mouse model of lung cancer (116). In vitro studies further demonstrate that PGE2 induces Treg differentiation and migration. For example, PGE2 can directly enhance the differentiation of naive CD4+ T cells into Tregs (117). PGE2 secreted from breast cancer cells directly induces Treg migration via EP2 and EP4 (118). In addition, PGE2 indirectly attracts Tregs via induction of CCL22 in mature DCs (119). Collectively, these studies indicate that PGE2 enhances tumor formation and growth via tumor-infiltrating Tregs (Figure 3).
Macrophages.
Macrophages are highly plastic and can be activated in two polarization states: classically M1 (Th1 response) or alternatively M2 (Th2 response), depending on the context of their microenvironment. Tumor-associated macrophages (TAMs) resemble an M2-like phenotype and are a major subpopulation of tumor-infiltrating immune cells (120). Multiple lines of evidence indicate that TAMs promote cancer progression and metastasis by supporting tumor-associated angiogenesis, enhancing tumor cell migration/invasion and intravasation, and suppressing immunosurveillance (121). For example, TAMs contribute to immunosuppression by suppressing CD8+ T cell cytotoxic activity via stimulation of expression of immune checkpoint receptor ligands such as PD-L1 and B7-H4 and/or via recruitment of Tregs (122, 123). TAMs are recognized as a poor prognostic sign in CRC (124). Moreover, a meta-analysis of 55 studies indicated that high density of TAMs correlated with overall poor survival of GC (125).
Treatment with celecoxib resulted in reduction of polyp burden and conversion of TAMs from M2 to M1 in ApcMin/+ mice (126). In a colon tumor implantation model, overexpression of 15-PGDH in tumor tissue is sufficient to redirect the differentiation of intratumoral CD11b cells from immunosuppressive M2-oriented TAMs to M1 macrophages (127). It has been reported that macrophages express EP2 and EP4, but not EP1 or EP3 (128), and that EP3 and EP4 have higher affinity for PGE2 than EP1 and EP2 (129). Therefore, deletion of EP4 in myeloid cells resulted in a reduction of tumor burden in ApcMin/+ mice (130). An EP4 antagonist, E7046, has also been shown to shift TAMs from M2 to M1 macrophages and to enhance the antitumor effect of anti–CTLA-4 antibodies in syngeneic murine models of cancer (103). In vitro studies showed that PGE2 promoted M2 macrophage polarization via a CREB/CRTC pathway in bone marrow–derived macrophages (131) and eliminates CD8+ T cells by inducing PD-L1 expression in TAMs (106). PGE2 also inhibited macrophage phagocytosis in vitro (132). In addition to macrophage function, H. pylori and PGE2 cooperated to upregulate CCL2, which recruited macrophages to gastric tumors (133). Collectively, these results demonstrate that PGE2 promotes tumor growth via induction of M2 macrophages (Figure 3).
PGE2 and antigen-presenting cells
Dendritic cells.
Professional antigen-presenting cells include dendritic cells (DCs), macrophages, and B cells. Among antigen-presenting cells, DCs are central to the host immune response to tumor antigens. Since little is known about the role of B cells in gastrointestinal cancer and other solid tumors, we will focus on DCs here. Circulating DC levels and activity are reduced in CRC patients as compared with healthy controls, and this correlates with the stage of disease (134, 135). Moreover, highly mature tumor-infiltrating DCs correlate negatively with tumor stage in patients with CRC (136) and are associated with better survival in GC patients (137). DCs include both conventional DCs (cDCs) and plasmacytoid DCs (pDCs). The studies evaluating levels of circulating cDCs and pDCs in CRC patients have resulted in conflicting data. One report showed that the levels of pDCs, but not cDCs, in blood were reduced in CRC patients (138). In contrast, another study showed that levels of both circulating cDCs and pDCs were reduced in CRC patients (139). Further studies with large numbers of patients are necessary to clarify this discrepancy.
In tumor implantation models of colon cancers, PGE2 promoted tumor growth by suppressing differentiation of DCs from bone marrow progenitors (140). Indeed, PGE2 suppresses DC differentiation and maturation in vitro and in vivo (141, 142). Moreover, PGE2 inhibits the antigen presentation ability of bone marrow–derived DCs by reduction of MHC II expression and upregulation of IL-10 via EP2 and EP4 (143). PGE2 has also been shown to switch the function of DCs from induction of immunity to T cell tolerance via upregulation of CD25 and indoleamine 2,3-dioxygenase (IDO), a rate-limiting enzyme in the kynurenine pathway (144). Furthermore, PGE2 shifted the IL-12/IL-23 balance in DCs signaling via EP2 and EP4 receptors in favor of IL-23, which in turn increases the number of Th17 cells in vitro (145). More interestingly, PGE2 has recently been shown to redirect the differentiation of human DCs into monocytic MDSCs (146). Further work is necessary to determine whether PGE2 promotes tumorigenesis by inhibition of DC differentiation, maturation, and function in spontaneous mouse models of gastrointestinal cancer (Figure 3).
PGE2 and T cells
CD4+ T helper cells.
T helper (Th) cells include Th1, Th2, and Th17 cells. Th1 and Th2 cells are characterized by secretion of Th1 cytokines (IFN-γ, TNF-α, and IL-2) and Th2 cytokines (IL-4, IL-10, and IL-6), respectively, whereas Th17 cells are characterized by secretion of IL-17. High Th1/Th2 ratios in tumor tissues are associated with better overall survival in CRC patients (147). In addition, tumor-infiltrating Th1 cells are associated with a positive prognosis, whereas intratumoral Th17 cells are associated with a poor prognosis in CRC (148, 149). Similarly, high Th1/Th2 ratios in peripheral blood are associated with a positive postoperative prognosis, whereas high circulating Th17 cells correlate with tumor progression and poor survival in GC patients (150, 151).
Although an in vivo study indicated that Th17 cells promoted intestinal tumor burden (152), little is known about the impact of PGE2 on the imbalance of Th1/Th2 response and Th17 cells in the tumor microenvironment. In vitro studies showed that PGE2 shifted CD4+ T cells to Th2 cells by downregulation of Th1 cytokines and upregulation of Th2 cytokines (153, 154). However, another study revealed that low concentrations of PGE2 induced Th1 differentiation and high concentrations inhibited Th1 differentiation (155). In addition, PGE2 exacerbated inflammation and disease severity by increasing infiltration of Th17 cells into the colonic tissue in a murine model of IBD (145). Moreover, an EP4 antagonist was found to reduce accumulation of both Th1 and Th17 cells in regional lymph nodes and suppressed disease progression in an animal model of chronic inflammation (155). Indeed, in vitro studies revealed that PGE2 facilitated IL-23–induced differentiation of Th17 from naive T cells (156). PGE2 also directly promotes differentiation of memory CD4+ T cells to Th17 cells by induction of IL-17 expression and reduction of IFN-γ expression (157). Clearly, further research is needed to determine whether PGE2 promotes gastrointestinal tumorigenesis via Th cells.
CD8+ cytotoxic T cells.
The density of tumor-infiltrating CD8+ T cells is associated with better survival of CRC and GC patients (148, 158). Although the role of PGE2 in regulation of tumor-associated CD8+ T cells in vivo remains unclear, one in vivo study showed that PGE2 suppressed cytotoxic T lymphocyte (CTL) survival and function during chronic lymphocytic choriomeningitis virus infection (159). Moreover, a number of in vitro studies have demonstrated that PGE2 inhibits CD8+ T cell proliferation and impairs the CD8+ CTL function. PGE2 can directly inhibit CD8+ T cell proliferation by promoting replicative senescence (160). PGE2 also suppresses the cytotoxic activity of CD8+ T cells by upregulation of CD94 and the NKG2A C-type lectin receptor complex (161) or by attenuating T cell receptor–induced IFN-γ release (162). Moreover, PGE2 produced by metastatic renal carcinoma cells shifted CD8+ CTLs toward tumor antigen–specific tolerance during interaction of CTLs and tumor cells (163). Clearly, these in vitro results need to be confirmed in animal models of gastrointestinal cancer.
PGE2 and innate leukocytes
Innate leukocytes include NK cells, mast cells (MCs), and phagocytic cells. The role of MCs in gastrointestinal cancer remains elusive, because contradictory results have been reported in human CRC specimens and mouse models of CRC. For example, tumor-infiltrating MCs have been shown to correlate with either positive or negative prognoses in CRC (164–166). Similarly, elimination of MCs resulted in reduction of tumors in ApcΔ468 mice and mice treated with carcinogenic 1,2-dimethylhydrazine (167, 168), indicating that MCs promote polyp formation. In contrast, absence of MCs led to induction of tumors in ApcMin/+ mice (169), suggesting that MCs inhibit tumor formation. Therefore, more work needs to be completed in this area before any definitive conclusions can be made.
NK cells.
NK cells are able to recognize and kill transformed or virus-infected cells but spare normal cells in the absence of antigen-specific priming. Interestingly, one in vitro result showed that NK cells selectively recognized and killed colonic cancer stem cells (CSCs) (170). Suppressed NK cell activity has been found in human CRC and is an important prognostic factor for the development of distant metastases (171, 172). Similarly, tumor-infiltrating NK cell levels are associated with an improved survival in GC (173). More intriguingly, intratumoral NK cell levels are negatively correlated with levels of COX-2 expression in GC (173).
Although there are no available in vivo data showing that PGE2 suppresses NK cell cytotoxicity in gastrointestinal cancer, in vivo studies demonstrated that treatment of rats with PGE2 inhibited NK cell activity and enhanced lung metastases (174) and reversed enhancement of NK cell activity by an NSAID (175). Modulation of EP4 receptor signaling mediated the effects of PGE2 on promotion of breast cancer metastasis and suppression of NK cell function in a syngeneic murine model of metastatic breast cancer (176). Substantial in vitro evidence has further demonstrated that PGE2 suppresses NK cell function via multiple mechanisms. PGE2 suppressed NK cytotoxicity by inhibiting NK receptors via a cAMP/PKA pathway (177). Moreover, PGE2 not only directly inhibited NK cell production of IFN-γ, which is essential for NK cell functions, but also attenuated IL-12–induced or IL-18–induced IFN-γ expression in NK cells via EP2 receptor (178, 179). In addition to NK function, PGE2 secreted from GC cells also inhibited NK cell proliferation and induced apoptosis (173). Taken together, these findings suggest that PGE2 not only suppresses NK functions, but also inhibits NK cell proliferation and survival (Figure 3). More work is needed to evaluate whether PGE2 promotes gastrointestinal tumorigenesis via suppression of NK cells.
Summary
Our focus on prostanoids indicates that PGE2 has been shown to promote gastrointestinal tumor formation, progression, and metastasis by multiple mechanisms. In addition to the direct effect of PGE2 on tumor cell proliferation, survival, and migration/invasion, PGE2 has been shown to promote CRC initiation, growth, and metastasis by silencing certain tumor suppressor and DNA repair genes via DNA methylation and by induction of CSC formation and expansion. Strikingly, PGE2 is also one of the tumor-associated immunosuppressive mediators that help to stimulate immunosuppression by enhancement of immunosuppressive cells, a defect in antigen-presenting cell function, a shift from Th1 to Th2 and Th17 immune responses, and/or impairment of functions of CD8+ cytotoxic T cells and NK cells, resulting in escape of tumor cells from effective immunosurveillance. Therefore, more selective pharmacologic inhibitors of PGE2 signaling not only target tumor cells, including CSC, but also subvert tumor-induced immunosuppression. It is clear that effective therapies should include elimination of tumor cells, especially CSCs, inhibition of tumor-associated angiogenesis, and subversion of tumor-induced immunosuppression. Clinical studies are warranted to evaluate the efficacy and toxicity of these inhibitors, such as EP2 and EP4 antagonists, in gastrointestinal cancer.
Acknowledgments
This work is supported, in part, by NIH R01 DK47297, NCI R01 CA184820, and P01 CA77839. We thank the National Colorectal Cancer Research Alliance for its generous support (to RND).
Version 1. 05/07/2018
Electronic publication
Version 2. 07/02/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(7):2732–2742.https://doi.org/10.1172/JCI97953.
References
- 1.Neuman MG. Immune dysfunction in inflammatory bowel disease. Transl Res. 2007;149(4):173–186. doi: 10.1016/j.trsl.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 2.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117(3):514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sands BE. Inflammatory bowel disease: past, present, and future. J Gastroenterol. 2007;42(1):16–25. doi: 10.1007/s00535-006-1995-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology. 2009;17(2):55–67. doi: 10.1007/s10787-009-8049-8. [DOI] [PubMed] [Google Scholar]
- 5.Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012;13(5):518–527. doi: 10.1016/S1470-2045(12)70112-2. [DOI] [PubMed] [Google Scholar]
- 6.Rothwell PM, et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet. 2012;379(9826):1602–1612. doi: 10.1016/S0140-6736(11)61720-0. [DOI] [PubMed] [Google Scholar]
- 7.Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379(9826):1591–1601. doi: 10.1016/S0140-6736(12)60209-8. [DOI] [PubMed] [Google Scholar]
- 8.Wang D, Dubois RN. The role of anti-inflammatory drugs in colorectal cancer. Annu Rev Med. 2013;64:131–144. doi: 10.1146/annurev-med-112211-154330. [DOI] [PubMed] [Google Scholar]
- 9.Drew DA, Cao Y, Chan AT. Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat Rev Cancer. 2016;16(3):173–186. doi: 10.1038/nrc.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong P, et al. The effects of anti-inflammatory drug treatment in gastric cancer prevention: an update of a meta-analysis. J Cancer. 2016;7(15):2247–2257. doi: 10.7150/jca.16524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107(4):1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 12.Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1(1):11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- 13.Marnett LJ, DuBois RN. COX-2: a target for colon cancer prevention. Annu Rev Pharmacol Toxicol. 2002;42:55–80. doi: 10.1146/annurev.pharmtox.42.082301.164620. [DOI] [PubMed] [Google Scholar]
- 14.Saukkonen K, et al. Cyclooxygenase-2 and gastric carcinogenesis. APMIS. 2003;111(10):915–925. doi: 10.1034/j.1600-0463.2003.1111001.x. [DOI] [PubMed] [Google Scholar]
- 15.Yu HP, et al. Cyclooxygenase-2 expression in squamous dysplasia and squamous cell carcinoma of the esophagus. Cancer Lett. 2003;198(2):193–201. doi: 10.1016/S0304-3835(03)00340-9. [DOI] [PubMed] [Google Scholar]
- 16.Ogino S, et al. Cyclooxygenase-2 expression is an independent predictor of poor prognosis in colon cancer. Clin Cancer Res. 2008;14(24):8221–8227. doi: 10.1158/1078-0432.CCR-08-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Z, Yang Y, Zhao Y, Huang Y. The prognostic value of cyclooxygenase-2 expression in patients with esophageal cancer: evidence from a meta-analysis. Onco Targets Ther. 2017;10:2893–2901. doi: 10.2147/OTT.S134599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng J, Fan XM. Role of cyclooxygenase-2 in gastric cancer development and progression. World J Gastroenterol. 2013;19(42):7361–7368. doi: 10.3748/wjg.v19.i42.7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108(1):15–23. doi: 10.1172/JCI13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scher JU, Pillinger MH. The anti-inflammatory effects of prostaglandins. J Investig Med. 2009;57(6):703–708. doi: 10.2310/JIM.0b013e31819aaa76. [DOI] [PubMed] [Google Scholar]
- 21.Straus DS, et al. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc Natl Acad Sci U S A. 2000;97(9):4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang D, DuBois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10(3):181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends Pharmacol Sci. 2012;33(6):304–311. doi: 10.1016/j.tips.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Wang D, Mann JR, DuBois RN. The role of prostaglandins and other eicosanoids in the gastrointestinal tract. Gastroenterology. 2005;128(5):1445–1461. doi: 10.1053/j.gastro.2004.09.080. [DOI] [PubMed] [Google Scholar]
- 25.Narumiya S. Prostanoids and inflammation: a new concept arising from receptor knockout mice. J Mol Med. 2009;87(10):1015–1022. doi: 10.1007/s00109-009-0500-1. [DOI] [PubMed] [Google Scholar]
- 26.Kawahito Y, et al. 15-Deoxy-Δ(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106(2):189–197. doi: 10.1172/JCI9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuzick J, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10(5):501–507. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- 28.Burn J, et al. A randomized placebo-controlled prevention trial of aspirin and/or resistant starch in young people with familial adenomatous polyposis. Cancer Prev Res (Phila) 2011;4(5):655–665. doi: 10.1158/1940-6207.CAPR-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishikawa H, et al. Preventive effects of low-dose aspirin on colorectal adenoma growth in patients with familial adenomatous polyposis: double-blind, randomized clinical trial. Cancer Med. 2013;2(1):50–56. doi: 10.1002/cam4.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Logan RF, Grainge MJ, Shepherd VC, Armitage NC, Muir KR, ukCAP Trial Group Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology. 2008;134(1):29–38. doi: 10.1053/j.gastro.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 31.Baron JA, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348(10):891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 32.Benamouzig R, et al. Daily soluble aspirin and prevention of colorectal adenoma recurrence: one-year results of the APACC trial. Gastroenterology. 2003;125(2):328–336. doi: 10.1016/S0016-5085(03)00887-4. [DOI] [PubMed] [Google Scholar]
- 33.Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348(10):883–890. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 34.Bibbins-Domingo K, U.S. Preventive Services Task Force Aspirin Use for the Primary Prevention of Cardiovascular Disease and Colorectal Cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2016;164(12):836–845. doi: 10.7326/M16-0577. [DOI] [PubMed] [Google Scholar]
- 35.Restivo A, et al. Aspirin as a neoadjuvant agent during preoperative chemoradiation for rectal cancer. Br J Cancer. 2015;113(8):1133–1139. doi: 10.1038/bjc.2015.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang S, et al. Cyclooxygenase inhibitors use is associated with reduced risk of esophageal adenocarcinoma in patients with Barrett’s esophagus: a meta-analysis. Br J Cancer. 2014;110(9):2378–2388. doi: 10.1038/bjc.2014.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sivarasan N, Smith G. Role of aspirin in chemoprevention of esophageal adenocarcinoma: a meta-analysis. J Dig Dis. 2013;14(5):222–230. doi: 10.1111/1751-2980.12047. [DOI] [PubMed] [Google Scholar]
- 38.Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med. 2007;356(21):2131–2142. doi: 10.1056/NEJMoa067208. [DOI] [PubMed] [Google Scholar]
- 39.Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2009;302(6):649–658. doi: 10.1001/jama.2009.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao X, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367(17):1596–1606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reimers MS, et al. Expression of HLA class I antigen, aspirin use, and survival after a diagnosis of colon cancer. JAMA Intern Med. 2014;174(5):732–739. doi: 10.1001/jamainternmed.2014.511. [DOI] [PubMed] [Google Scholar]
- 42.Rigas B, Goldman IS, Levine L. Altered eicosanoid levels in human colon cancer. J Lab Clin Med. 1993;122(5):518–523. [PubMed] [Google Scholar]
- 43.Uefuji K, Ichikura T, Mochizuki H. Cyclooxygenase-2 expression is related to prostaglandin biosynthesis and angiogenesis in human gastric cancer. Clin Cancer Res. 2000;6(1):135–138. [PubMed] [Google Scholar]
- 44.Mal M, Koh PK, Cheah PY, Chan EC. Ultra-pressure liquid chromatography/tandem mass spectrometry targeted profiling of arachidonic acid and eicosanoids in human colorectal cancer. Rapid Commun Mass Spectrom. 2011;25(6):755–764. doi: 10.1002/rcm.4926. [DOI] [PubMed] [Google Scholar]
- 45.Backlund MG, et al. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem. 2005;280(5):3217–3223. doi: 10.1074/jbc.M411221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-β-induced suppressor of human gastrointestinal cancers. Proc Natl Acad Sci U S A. 2004;101(50):17468–17473. doi: 10.1073/pnas.0406142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seo SH, et al. Correlation of 15-prostagladin dehydrogenase expression with clinicopathological factors and survival rate in gastric adenocarcinoma. Int J Surg. 2015;13:96–101. doi: 10.1016/j.ijsu.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 48.Yang GT, Wang J, Xu TZ, Sun XF, Luan ZY. Expression of PGDH correlates with cell growth in both esophageal squamous cell carcinoma and adenocarcinoma. Asian Pac J Cancer Prev. 2015;16(3):997–1000. doi: 10.7314/APJCP.2015.16.3.997. [DOI] [PubMed] [Google Scholar]
- 49.Shrubsole MJ, et al. Urinary prostaglandin E2 metabolite and risk for colorectal adenoma. Cancer Prev Res (Phila) 2012;5(2):336–342. doi: 10.1158/1940-6207.CAPR-11-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson JC, et al. Urine PGE-M: a metabolite of prostaglandin E2 as a potential biomarker of advanced colorectal neoplasia. Clin Gastroenterol Hepatol. 2006;4(11):1358–1365. doi: 10.1016/j.cgh.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 51.Cai Q, et al. Prospective study of urinary prostaglandin E2 metabolite and colorectal cancer risk. J Clin Oncol. 2006;24(31):5010–5016. doi: 10.1200/JCO.2006.06.4931. [DOI] [PubMed] [Google Scholar]
- 52.Dong LM, et al. Urinary prostaglandin E2 metabolite and gastric cancer risk in the Shanghai women’s health study. Cancer Epidemiol Biomarkers Prev. 2009;18(11):3075–3078. doi: 10.1158/1055-9965.EPI-09-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang T, et al. A prospective study of urinary prostaglandin E2 metabolite, helicobacter pylori antibodies, and gastric cancer risk. Clin Infect Dis. 2017;64(10):1380–1386. doi: 10.1093/cid/cix106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murphey LJ, et al. Quantification of the major urinary metabolite of PGE2 by a liquid chromatographic/mass spectrometric assay: determination of cyclooxygenase-specific PGE2 synthesis in healthy humans and those with lung cancer. Anal Biochem. 2004;334(2):266–275. doi: 10.1016/j.ab.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 55.Morris PG, et al. Increased levels of urinary PGE-M, a biomarker of inflammation, occur in association with obesity, aging, and lung metastases in patients with breast cancer. Cancer Prev Res (Phila) 2013;6(5):428–436. doi: 10.1158/1940-6207.CAPR-12-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim S, Taylor JA, Milne GL, Sandler DP. Association between urinary prostaglandin E2 metabolite and breast cancer risk: a prospective, case-cohort study of postmenopausal women. Cancer Prev Res (Phila) 2013;6(6):511–518. doi: 10.1158/1940-6207.CAPR-13-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patrignani P, et al. Low-dose aspirin acetylates cyclooxygenase-1 in human colorectal mucosa: implications for the chemoprevention of colorectal cancer. Clin Pharmacol Ther. doi: 10.1002/cpt. [published online ahead of print January 31, 2017]. https://doi.org/10.1002/cpt.639. [DOI] [PubMed] [Google Scholar]
- 58.Fink SP, et al. Aspirin and the risk of colorectal cancer in relation to the expression of 15-hydroxyprostaglandin dehydrogenase (HPGD) Sci Transl Med. 2014;6(233):233re2. doi: 10.1126/scitranslmed.3008481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawamori T, Uchiya N, Sugimura T, Wakabayashi K. Enhancement of colon carcinogenesis by prostaglandin E2 administration. Carcinogenesis. 2003;24(5):985–990. doi: 10.1093/carcin/bgg033. [DOI] [PubMed] [Google Scholar]
- 60.Wang D, et al. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell. 2004;6(3):285–295. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 61.Myung SJ, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A. 2006;103(32):12098–12102. doi: 10.1073/pnas.0603235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, Whelan J. Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in Apc(Min/+) mice. Cancer Res. 2002;62(2):403–408. [PubMed] [Google Scholar]
- 63.Nakanishi M, et al. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68(9):3251–3259. doi: 10.1158/0008-5472.CAN-07-6100. [DOI] [PubMed] [Google Scholar]
- 64.Nakanishi M, et al. Selective PGE(2) suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev Res (Phila) 2011;4(8):1198–1208. doi: 10.1158/1940-6207.CAPR-11-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. EMBO J. 2004;23(7):1669–1678. doi: 10.1038/sj.emboj.7600170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Watanabe K, et al. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res. 1999;59(20):5093–5096. [PubMed] [Google Scholar]
- 67.Mutoh M, et al. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 2002;62(1):28–32. [PubMed] [Google Scholar]
- 68.Sonoshita M, et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in ApcΔ716 knockout mice. Nat Med. 2001;7(9):1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 69.Shoji Y, et al. Downregulation of prostaglandin E receptor subtype EP3 during colon cancer development. Gut. 2004;53(8):1151–1158. doi: 10.1136/gut.2003.028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma X, Aoki T, Tsuruyama T, Narumiya S. Definition of prostaglandin E2-EP2 signals in the colon tumor microenvironment that amplify inflammation and tumor growth. Cancer Res. 2015;75(14):2822–2832. doi: 10.1158/0008-5472.CAN-15-0125. [DOI] [PubMed] [Google Scholar]
- 71.Park JM, et al. Hematopoietic prostaglandin D synthase suppresses intestinal adenomas in ApcMin/+ mice. Cancer Res. 2007;67(3):881–889. doi: 10.1158/0008-5472.CAN-05-3767. [DOI] [PubMed] [Google Scholar]
- 72.Iwanaga K, et al. Mast cell-derived prostaglandin D2 inhibits colitis and colitis-associated colon cancer in mice. Cancer Res. 2014;74(11):3011–3019. doi: 10.1158/0008-5472.CAN-13-2792. [DOI] [PubMed] [Google Scholar]
- 73.Tippin BL, et al. Intestinal tumor suppression in ApcMin/+ mice by prostaglandin D2 receptor PTGDR. Cancer Med. 2014;3(4):1041–1051. doi: 10.1002/cam4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mutoh M, et al. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 2002;62(1):28–32. [PubMed] [Google Scholar]
- 75.Sasaki Y, et al. Genetic-deletion of cyclooxygenase-2 downstream prostacyclin synthase suppresses inflammatory reactions but facilitates carcinogenesis, unlike deletion of microsomal prostaglandin E synthase-1. Sci Rep. 2015;5:17376. doi: 10.1038/srep17376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gustafsson A, et al. Prostanoid receptor expression in colorectal cancer related to tumor stage, differentiation and progression. Acta Oncol. 2007;46(8):1107–1112. doi: 10.1080/02841860701403061. [DOI] [PubMed] [Google Scholar]
- 77.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55(1):115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xia D, Wang D, Kim SH, Katoh H, DuBois RN. Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat Med. 2012;18(2):224–226. doi: 10.1038/nm.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ong CW, et al. The prognostic value of the stem-like group in colorectal cancer using a panel of immunohistochemistry markers. Oncotarget. 2015;6(14):12763–12773. doi: 10.18632/oncotarget.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 81.Qiu W, et al. Chemoprevention by nonsteroidal anti-inflammatory drugs eliminates oncogenic intestinal stem cells via SMAC-dependent apoptosis. Proc Natl Acad Sci U S A. 2010;107(46):20027–20032. doi: 10.1073/pnas.1010430107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moon CM, et al. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int J Cancer. 2014;134(3):519–529. doi: 10.1002/ijc.28381. [DOI] [PubMed] [Google Scholar]
- 83.Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E promotes colorectal cancer stem cell expansion metastasis in mice. Gastroenterology. 2015;149(7):1884–1895.e4. doi: 10.1053/j.gastro.2015.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bellamkonda K, Chandrashekar NK, Osman J, Selvanesan BC, Savari S, Sjölander A. The eicosanoids leukotriene D4 and prostaglandin E2 promote the tumorigenicity of colon cancer-initiating cells in a xenograft mouse model. BMC Cancer. 2016;16:425. doi: 10.1186/s12885-016-2466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ishimoto T, et al. CD44+ slow-cycling tumor cell expansion is triggered by cooperative actions of Wnt and prostaglandin E2 in gastric tumorigenesis. Cancer Sci. 2010;101(3):673–678. doi: 10.1111/j.1349-7006.2009.01430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kurtova AV, et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517(7533):209–213. doi: 10.1038/nature14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martinez-Colon GJ, Moore BB. Prostaglandin E2 as a regulator of immunity to pathogens. Pharmacol Ther. doi: 10.1016/j.pharmthera.2017.12. [published online ahead of print December 22, 2017]. https://doi.org/10.1016/j.pharmthera.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Duffy A, et al. Comparative analysis of monocytic and granulocytic myeloid-derived suppressor cell subsets in patients with gastrointestinal malignancies. Cancer Immunol Immunother. 2013;62(2):299–307. doi: 10.1007/s00262-012-1332-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60(10):1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sun HL, et al. Increased frequency and clinical significance of myeloid-derived suppressor cells in human colorectal carcinoma. World J Gastroenterol. 2012;18(25):3303–3309. doi: 10.3748/wjg.v18.i25.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang L, Chang EW, Wong SC, Ong SM, Chong DQ, Ling KL. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol. 2013;190(2):794–804. doi: 10.4049/jimmunol.1202088. [DOI] [PubMed] [Google Scholar]
- 93.Zhang B, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS One. 2013;8(2):e57114. doi: 10.1371/journal.pone.0057114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Poh TW, et al. Downregulation of hematopoietic MUC1 during experimental colitis increases tumor-promoting myeloid-derived suppressor cells. Clin Cancer Res. 2013;19(18):5039–5052. doi: 10.1158/1078-0432.CCR-13-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell. 2013;24(5):631–644. doi: 10.1016/j.ccr.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res. 2017;77(13):3655–3665. doi: 10.1158/0008-5472.CAN-16-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cui TX, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. 2013;39(3):611–621. doi: 10.1016/j.immuni.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Di Mitri D, et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature. 2014;515(7525):134–137. doi: 10.1038/nature13638. [DOI] [PubMed] [Google Scholar]
- 99.Talmadge JE, Hood KC, Zobel LC, Shafer LR, Coles M, Toth B. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int Immunopharmacol. 2007;7(2):140–151. doi: 10.1016/j.intimp.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 100.Fujita M, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011;71(7):2664–2674. doi: 10.1158/0008-5472.CAN-10-3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Veltman JD, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. 2010;10:464. doi: 10.1186/1471-2407-10-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67(9):4507–4513. doi: 10.1158/0008-5472.CAN-06-4174. [DOI] [PubMed] [Google Scholar]
- 103.Albu DI, et al. EP4 antagonism by E7046 diminishes myeloid immunosuppression and synergizes with Treg-reducing IL-2-Diphtheria toxin fusion protein in restoring anti-tumor immunity. Oncoimmunology. 2017;6(8):e1338239. doi: 10.1080/2162402X.2017.1338239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Obermajer N, Kalinski P. Generation of myeloid-derived suppressor cells using prostaglandin E2. Transplant Res. 2012;1(1):15. doi: 10.1186/2047-1440-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rodríguez-Ubreva J, et al. Prostaglandin E2 leads to the acquisition of DNMT3A-dependent tolerogenic functions in human myeloid-derived suppressor cells. Cell Rep. 2017;21(1):154–167. doi: 10.1016/j.celrep.2017.09.018. [DOI] [PubMed] [Google Scholar]
- 106.Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci U S A. 2017;114(5):1117–1122. doi: 10.1073/pnas.1612920114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rodriguez PC, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202(7):931–939. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bacić D, Uravić M, Bacić R, Sutić I, Petrosić N. Augmentation of regulatory T cells (CD4+CD25+Foxp3+) correlates with tumor stage in patients with colorectal cancer. Coll Antropol. 2011;35(suppl 2):65–68. [PubMed] [Google Scholar]
- 109.Yuan XL, et al. Elevated expression of Foxp3 in tumor-infiltrating Treg cells suppresses T-cell proliferation and contributes to gastric cancer progression in a COX-2-dependent manner. Clin Immunol. 2010;134(3):277–288. doi: 10.1016/j.clim.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 110.Kashimura S, et al. CD83(+) dendritic cells and Foxp3(+) regulatory T cells in primary lesions and regional lymph nodes are inversely correlated with prognosis of gastric cancer. Gastric Cancer. 2012;15(2):144–153. doi: 10.1007/s10120-011-0090-9. [DOI] [PubMed] [Google Scholar]
- 111.Shen Z, et al. Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J Cancer Res Clin Oncol. 2010;136(10):1585–1595. doi: 10.1007/s00432-010-0816-9. [DOI] [PubMed] [Google Scholar]
- 112.Yaqub S, et al. Regulatory T cells in colorectal cancer patients suppress anti-tumor immune activity in a COX-2 dependent manner. Cancer Immunol Immunother. 2008;57(6):813–821. doi: 10.1007/s00262-007-0417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Faluyi OO, Fitch P, Howie SEM. An increased CD25-positive intestinal regulatory T lymphocyte population is dependent upon Cox-2 activity in the Apcmin/+ model. Clin Exp Immunol. 2018;191(1):32–41. doi: 10.1111/cei.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Soontrapa K, et al. Prostaglandin E2-prostaglandin E receptor subtype 4 (EP4) signaling mediates UV irradiation-induced systemic immunosuppression. Proc Natl Acad Sci U S A. 2011;108(16):6668–6673. doi: 10.1073/pnas.1018625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.O’Callaghan G, Ryan A, Neary P, O’Mahony C, Shanahan F, Houston A. Targeting the EP1 receptor reduces Fas ligand expression and increases the antitumor immune response in an in vivo model of colon cancer. Int J Cancer. 2013;133(4):825–834. doi: 10.1002/ijc.28076. [DOI] [PubMed] [Google Scholar]
- 116.Sharma S, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res. 2005;65(12):5211–5220. doi: 10.1158/0008-5472.CAN-05-0141. [DOI] [PubMed] [Google Scholar]
- 117.Baratelli F, et al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol. 2005;175(3):1483–1490. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 118.Karavitis J, Hix LM, Shi YH, Schultz RF, Khazaie K, Zhang M. Regulation of COX2 expression in mouse mammary tumor cells controls bone metastasis and PGE2-induction of regulatory T cell migration. PLoS One. 2012;7(9):e46342. doi: 10.1371/journal.pone.0046342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Muthuswamy R, Urban J, Lee JJ, Reinhart TA, Bartlett D, Kalinski P. Ability of mature dendritic cells to interact with regulatory T cells is imprinted during maturation. Cancer Res. 2008;68(14):5972–5978. doi: 10.1158/0008-5472.CAN-07-6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 121.Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15(2):73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kuang DM, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206(6):1327–1337. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kryczek I, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203(4):871–881. doi: 10.1084/jem.20050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Isidro RA, Appleyard CB. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am J Physiol Gastrointest Liver Physiol. 2016;311(1):G59–G73. doi: 10.1152/ajpgi.00123.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang QW, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One. 2012;7(12):e50946. doi: 10.1371/journal.pone.0050946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nakanishi Y, et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis. 2011;32(9):1333–1339. doi: 10.1093/carcin/bgr128. [DOI] [PubMed] [Google Scholar]
- 127.Eruslanov E, et al. Altered expression of 15-hydroxyprostaglandin dehydrogenase in tumor-infiltrated CD11b myeloid cells: a mechanism for immune evasion in cancer. J Immunol. 2009;182(12):7548–7557. doi: 10.4049/jimmunol.0802358. [DOI] [PubMed] [Google Scholar]
- 128.Treffkorn L, Scheibe R, Maruyama T, Dieter P. PGE2 exerts its effect on the LPS-induced release of TNF-α, ET-1, IL-1α, IL-6, and IL-10 via the EP2 and EP4 receptor in rat liver macrophages. Prostaglandins Other Lipid Mediat. 2004;74(1-4):113–123. doi: 10.1016/j.prostaglandins.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 129.Abramovitz M, et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483(2):285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- 130.Chang J, et al. Prostaglandin E receptor 4 (EP4) promotes colonic tumorigenesis. Oncotarget. 2015;6(32):33500–33511. doi: 10.18632/oncotarget.5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A. 2015;112(51):15642–15647. doi: 10.1073/pnas.1519644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. 2004;173(1):559–565. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- 133.Oshima H, et al. Prostaglandin E(2) signaling and bacterial infection recruit tumor-promoting macrophages to mouse gastric tumors. Gastroenterology. 2011;140(2):596–607.e7. doi: 10.1053/j.gastro.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 134.Della Porta M, et al. Dendritic cells and vascular endothelial growth factor in colorectal cancer: correlations with clinicobiological findings. Oncology. 2005;68(2–3):276–284. doi: 10.1159/000086784. [DOI] [PubMed] [Google Scholar]
- 135.Huang A, Gilmour JW, Imami N, Amjadi P, Henderson DC, Allen-Mersh TG. Increased serum transforming growth factor-β1 in human colorectal cancer correlates with reduced circulating dendritic cells and increased colonic Langerhans cell infiltration. Clin Exp Immunol. 2003;134(2):270–278. doi: 10.1046/j.1365-2249.2003.02295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gulubova MV, Ananiev JR, Vlaykova TI, Yovchev Y, Tsoneva V, Manolova IM. Role of dendritic cells in progression and clinical outcome of colon cancer. Int J Colorectal Dis. 2012;27(2):159–169. doi: 10.1007/s00384-011-1334-1. [DOI] [PubMed] [Google Scholar]
- 137.Ananiev J, Gulubova MV, Manolova IM. Prognostic significance of CD83 positive tumor-infiltrating dendritic cells and expression of TGF-beta 1 in human gastric cancer. Hepatogastroenterology. 2011;58(110-111):1834–1840. doi: 10.5754/hge10320. [DOI] [PubMed] [Google Scholar]
- 138.Orsini G, et al. Quantification of blood dendritic cells in colorectal cancer patients during the course of disease. Pathol Oncol Res. 2014;20(2):267–276. doi: 10.1007/s12253-013-9691-4. [DOI] [PubMed] [Google Scholar]
- 139.Bellik L, et al. Role of conventional treatments on circulating and monocyte-derived dendritic cells in colorectal cancer. Clin Immunol. 2006;121(1):74–80. doi: 10.1016/j.clim.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 140.Yang L, et al. Cancer-associated immunodeficiency and dendritic cell abnormalities mediated by the prostaglandin EP2 receptor. J Clin Invest. 2003;111(5):727–735. doi: 10.1172/JCI16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stock A, Booth S, Cerundolo V. Prostaglandin E2 suppresses the differentiation of retinoic acid-producing dendritic cells in mice and humans. J Exp Med. 2011;208(4):761–773. doi: 10.1084/jem.20101967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ahmadi M, Emery DC, Morgan DJ. Prevention of both direct and cross-priming of antitumor CD8+ T-cell responses following overproduction of prostaglandin E2 by tumor cells in vivo. Cancer Res. 2008;68(18):7520–7529. doi: 10.1158/0008-5472.CAN-08-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Harizi H, Grosset C, Gualde N. Prostaglandin E2 modulates dendritic cell function via EP2 and EP4 receptor subtypes. J Leukoc Biol. 2003;73(6):756–763. doi: 10.1189/jlb.1002483. [DOI] [PubMed] [Google Scholar]
- 144.von Bergwelt-Baildon MS, et al. CD25 and indoleamine 2,3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: additional mechanisms of T-cell inhibition. Blood. 2006;108(1):228–237. doi: 10.1182/blood-2005-08-3507. [DOI] [PubMed] [Google Scholar]
- 145.Sheibanie AF, et al. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23-->IL-17 axis. J Immunol. 2007;178(12):8138–8147. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 146.Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011;118(20):5498–5505. doi: 10.1182/blood-2011-07-365825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nizri E, et al. Analysis of histological and immunological parameters of metastatic lymph nodes from colon cancer patients reveals that T-helper 1 type immune response is associated with improved overall survival. Medicine (Baltimore) 2016;95(45):e5340. doi: 10.1097/MD.0000000000005340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 149.Tosolini M, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011;71(4):1263–1271. doi: 10.1158/0008-5472.CAN-10-2907. [DOI] [PubMed] [Google Scholar]
- 150.Ubukata H, Motohashi G, Tabuchi T, Nagata H, Konishi S, Tabuchi T. Evaluations of interferon-γ/interleukin-4 ratio and neutrophil/lymphocyte ratio as prognostic indicators in gastric cancer patients. J Surg Oncol. 2010;102(7):742–747. doi: 10.1002/jso.21725. [DOI] [PubMed] [Google Scholar]
- 151.Liu T, et al. Increased circulating Th22 and Th17 cells are associated with tumor progression and patient survival in human gastric cancer. J Clin Immunol. 2012;32(6):1332–1339. doi: 10.1007/s10875-012-9718-8. [DOI] [PubMed] [Google Scholar]
- 152.Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(12):5540–5544. doi: 10.1073/pnas.0912675107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Snijdewint FG, Kaliński P, Wierenga EA, Bos JD, Kapsenberg ML. Prostaglandin E2 differentially modulates cytokine secretion profiles of human T helper lymphocytes. J Immunol. 1993;150(12):5321–5329. [PubMed] [Google Scholar]
- 154.Hilkens CM, Vermeulen H, van Neerven RJ, Snijdewint FG, Wierenga EA, Kapsenberg ML. Differential modulation of T helper type 1 (Th1) and T helper type 2 (Th2) cytokine secretion by prostaglandin E2 critically depends on interleukin-2. Eur J Immunol. 1995;25(1):59–63. doi: 10.1002/eji.1830250112. [DOI] [PubMed] [Google Scholar]
- 155.Yao C, et al. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15(6):633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 156.Boniface K, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. 2009;206(3):535–548. doi: 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Napolitani G, Acosta-Rodriguez EV, Lanzavecchia A, Sallusto F. Prostaglandin E2 enhances Th17 responses via modulation of IL-17 and IFN-gamma production by memory CD4+ T cells. Eur J Immunol. 2009;39(5):1301–1312. doi: 10.1002/eji.200838969. [DOI] [PubMed] [Google Scholar]
- 158.Lee HE, et al. Prognostic implications of type and density of tumour-infiltrating lymphocytes in gastric cancer. Br J Cancer. 2008;99(10):1704–1711. doi: 10.1038/sj.bjc.6604738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen JH, et al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med. 2015;21(4):327–334. doi: 10.1038/nm.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chou JP, Ramirez CM, Ryba DM, Koduri MP, Effros RB. Prostaglandin E2 promotes features of replicative senescence in chronically activated human CD8+ T cells. PLoS One. 2014;9(6):e99432. doi: 10.1371/journal.pone.0099432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zeddou M, et al. Prostaglandin E2 induces the expression of functional inhibitory CD94/NKG2A receptors in human CD8+ T lymphocytes by a cAMP-dependent protein kinase A type I pathway. Biochem Pharmacol. 2005;70(5):714–724. doi: 10.1016/j.bcp.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 162.Ganapathy V, Gurlo T, Jarstadmarken HO, von Grafenstein H. Regulation of TCR-induced IFN-γ release from islet-reactive non-obese diabetic CD8(+) T cells by prostaglandin E(2) receptor signaling. Int Immunol. 2000;12(6):851–860. doi: 10.1093/intimm/12.6.851. [DOI] [PubMed] [Google Scholar]
- 163.Basingab FS, Ahmadi M, Morgan DJ. IFNγ-dependent interactions between ICAM-1 and LFA-1 counteract prostaglandin E2-mediated inhibition of antitumor CTL responses. Cancer Immunol Res. 2016;4(5):400–411. doi: 10.1158/2326-6066.CIR-15-0146. [DOI] [PubMed] [Google Scholar]
- 164.Kashiwase Y, Inamura H, Morioka J, Igarashi Y, Kawai-Kowase K, Kurosawa M. Quantitative analysis of mast cells in benign and malignant colonic lesions: immunohistochemical study on formalin-fixed, paraffin-embedded tissues. Allergol Immunopathol (Madr) 2008;36(5):271–276. doi: 10.1016/S0301-0546(08)75222-4. [DOI] [PubMed] [Google Scholar]
- 165.Gulubova M, Vlaykova T. Prognostic significance of mast cell number and microvascular density for the survival of patients with primary colorectal cancer. J Gastroenterol Hepatol. 2009;24(7):1265–1275. doi: 10.1111/j.1440-1746.2007.05009.x. [DOI] [PubMed] [Google Scholar]
- 166.Mehdawi L, Osman J, Topi G, Sjölander A. High tumor mast cell density is associated with longer survival of colon cancer patients. Acta Oncol. 2016;55(12):1434–1442. doi: 10.1080/0284186X.2016.1198493. [DOI] [PubMed] [Google Scholar]
- 167.Gounaris E, et al. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci U S A. 2007;104(50):19977–19982. doi: 10.1073/pnas.0704620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wedemeyer J, Galli SJ. Decreased susceptibility of mast cell-deficient Kit(W)/Kit(W-v) mice to the development of 1, 2-dimethylhydrazine-induced intestinal tumors. Lab Invest. 2005;85(3):388–396. doi: 10.1038/labinvest.3700232. [DOI] [PubMed] [Google Scholar]
- 169.Sinnamon MJ, Carter KJ, Sims LP, Lafleur B, Fingleton B, Matrisian LM. A protective role of mast cells in intestinal tumorigenesis. Carcinogenesis. 2008;29(4):880–886. doi: 10.1093/carcin/bgn040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tallerico R, et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol. 2013;190(5):2381–2390. doi: 10.4049/jimmunol.1201542. [DOI] [PubMed] [Google Scholar]
- 171.Espí A, Arenas J, García-Granero E, Martí E, Lledó S. Relationship of curative surgery on natural killer cell activity in colorectal cancer. Dis Colon Rectum. 1996;39(4):429–434. doi: 10.1007/BF02054059. [DOI] [PubMed] [Google Scholar]
- 172.Kondo E, et al. Preoperative natural killer cell activity as a prognostic factor for distant metastasis following surgery for colon cancer. Dig Surg. 2003;20(5):445–451. doi: 10.1159/000072714. [DOI] [PubMed] [Google Scholar]
- 173.Li T, et al. Gastric cancer cells inhibit natural killer cell proliferation and induce apoptosis via prostaglandin E2. Oncoimmunology. 2016;5(2):e1069936. doi: 10.1080/2162402X.2015.1069936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yakar I, et al. Prostaglandin e(2) suppresses NK activity in vivo and promotes postoperative tumor metastasis in rats. Ann Surg Oncol. 2003;10(4):469–479. doi: 10.1245/ASO.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 175.Voth R, Chmielarczyk W, Storch E, Kirchner H. Induction of natural killer cell activity in mice by injection of indomethacin. Nat Immun Cell Growth Regul. 1986;5(6):317–324. [PubMed] [Google Scholar]
- 176.Ma X, et al. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE-mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology. 2013;2(1):e22647. doi: 10.4161/onci.22647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Martinet L, Jean C, Dietrich G, Fournié JJ, Poupot R. PGE2 inhibits natural killer and γδ T cell cytotoxicity triggered by NKR and TCR through a cAMP-mediated PKA type I-dependent signaling. Biochem Pharmacol. 2010;80(6):838–845. doi: 10.1016/j.bcp.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 178.Mailliard RB, et al. IL-18-induced CD83+CCR7+ NK helper cells. J Exp Med. 2005;202(7):941–953. doi: 10.1084/jem.20050128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Walker W, Rotondo D. Prostaglandin E2 is a potent regulator of interleukin-12- and interleukin-18-induced natural killer cell interferon-gamma synthesis. Immunology. 2004;111(3):298–305. doi: 10.1111/j.1365-2567.2004.01810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]