

Abstract
Receptor tyrosine kinases (RTKs) are important drivers of cancers. In addition to genomic alterations, aberrant activation of WT RTKs plays an important role in driving cancer progression. However, the mechanisms underlying how RTKs drive prostate cancer remain incompletely characterized. Here we show that non-proteolytic ubiquitination of RTK regulates its kinase activity and contributes to RTK-mediated prostate cancer metastasis. TRAF4, an E3 ubiquitin ligase, is highly expressed in metastatic prostate cancer. We demonstrated here that it is a key player in regulating RTK-mediated prostate cancer metastasis. We further identified TrkA, a neurotrophin RTK, as a TRAF4-targeted ubiquitination substrate that promotes cancer cell invasion and found that inhibition of TrkA activity abolished TRAF4-dependent cell invasion. TRAF4 promoted K27- and K29-linked ubiquitination at the TrkA kinase domain and increased its kinase activity. Mutation of TRAF4-targeted ubiquitination sites abolished TrkA tyrosine autophosphorylation and its interaction with downstream proteins. TRAF4 knockdown also suppressed nerve growth factor (NGF) stimulated TrkA downstream p38 MAPK activation and invasion-associated gene expression. Furthermore, elevated TRAF4 levels significantly correlated with increased NGF-stimulated invasion–associated gene expression in prostate cancer patients, indicating that this signaling axis is significantly activated during oncogenesis. Our results revealed a posttranslational modification mechanism contributing to aberrant non-mutated RTK activation in cancer cells.
Keywords: Cell Biology, Genetics
Keywords: Prostate cancer, Protein kinases, Ubiquitin-proteosome system
Introduction
Ubiquitination is an important posttranslational modification regulating protein degradation, trafficking, and activity, as well as protein-protein interaction. Dysregulation of the ubiquitin pathways has been implicated in a number of diseases including cancer (1–5). Targeting the ubiquitination machinery has been considered to be an effective therapeutic strategy (3, 6, 7).
The RING domain E3 ubiquitin ligase TRAF4 is emerging as a key regulator in cancer development, metastasis, and chemoresistance (8–15). It was originally identified as a gene upregulated in metastatic breast cancer (16). TRAF4 belongs to the TRAF family, which consists of 7 members. They are adaptor/scaffold proteins that couple TNF receptors and interleukin receptors to downstream signaling pathways. Unlike other TRAFs, TRAF4 weakly interacts with very few TNF receptor family members (17). It also does not substantially contribute to the development and normal function of the immune system, except for facilitating immune cell migration (18). The biological function of TRAF4 has remained elusive. It is expressed at basal levels in most adult tissues (17) but is overexpressed and amplified in a variety of human cancers (11). We found that TRAF4 has a significantly higher expression level in metastatic prostate cancer compared with primary tumor and plays an important role in prostate cancer cell invasion.
After screening TRAF4 ubiquitination substrates in prostate tumors, we identified tyrosine receptor kinase A (TrkA, also named NTRK1) as a prominent substrate for TRAF4-mediated ubiquitination. TrkA is a receptor tyrosine kinase (RTK) that binds to nerve growth factor (NGF) at the cell membrane. It activates Ras/MAPK, PI3K, and PLCγ signaling pathways to promote cell survival, proliferation, and invasion (19). In addition to the nervous system, NGF is also abundant in prostate cancer, and its receptor has been linked to prostate cancer proliferation and metastasis (19–25). Targeting genetically altered constitutively active protein kinases has led to dramatic clinical responses in several cancers. Although TrkA-activating mutations through genomic rearrangement and deletion have been documented in a number of cancers (26–31), its mutations were not identified in prostate cancer (32). The pathways leading to aberrant activation of non-mutated TrkA have remained poorly understood. Herein we present evidence that TRAF4 promotes TrkA ubiquitination at its kinase domain through atypical K27 and K29 ubiquitin linkages. This posttranslational modification hyperactivates TrkA kinase activity and alters its phosphorylation status. Our study deciphered that the TRAF4-regulated signaling cascade is an important driver for prostate cancer metastasis.
Results
TRAF4 plays a role in prostate cancer metastasis.
To investigate the expression of TRAF4 in prostate tumor specimens, we used a prostate cancer tissue cDNA array consisting of 39 prostate cancer and 9 normal prostate tissue samples. TRAF4 mRNA was highly expressed in prostate tumors compared with normal tissues (Figure 1A). We also found that TRAF4 protein levels were significantly higher in 7 of 10 human prostate tumors compared with matched benign prostate tissues (Figure 1B). Since TRAF4 has been reported to be associated with cell migration and cancer metastasis (9, 15, 18, 33, 34), we also analyzed its expression in several publicly available prostate cancer datasets containing a substantial number of metastatic cancers (35–39). Consistent with our analysis of tumors (Figure 1, A and B), TRAF4 expression was significantly elevated in prostate tumors compared with adjoining prostate tissues (Figure 1, C–F). Interestingly, patients with metastatic disease all had enhanced levels of TRAF4 compared with those with localized tumors (Figure 1, C–F). These findings suggest the possibility of a critical role of TRAF4 in aggressive metastatic prostate cancers.
Figure 1. TRAF4 is overexpressed in prostate cancers.

(A) TRAF4 mRNA is upregulated in prostate tumors (n = 39) as compared with normal tissue (n = 9) in a prostate cancer tissue scan array (OriGene) as analyzed by RT-qPCR. Left: Relative TRAF4 mRNA expression in normal and tumor samples as normalized against β-actin. *P < 0.05 by Mann-Whitney U test. Right: Average fold induction of TRAF4 expression in tumor samples. *P < 0.05 by 2-tailed Student’s t test. (B) TRAF4 protein is upregulated in human prostate tumors (T) compared with matched benign tissue (N) samples (n = 10 each) as analyzed by Western blot. (C–F) TRAF4 is highly expressed in metastatic prostate cancers. The expression of TRAF4 was analyzed in 4 different prostate cancer datasets: Chandran and Yu, refs. 37, 38; Grasso, ref. 39; Taylor, ref. 35; Varambally, ref. 36 and presented as mean ± SEM. β-Actin was used as an internal control. *P < 0.05 by 1-way ANOVA with multiple comparisons test. PC, prostate cancer; CRPC, castration-resistant prostate cancer.
To determine whether TRAF4 plays a role in prostate cancer metastasis, we first analyzed the effect of TRAF4 knockdown in prostate cancer cell migration and invasion. TRAF4 knockdown was achieved using pooled siRNA or 2 different shRNAs. PC3 cells, a highly invasive prostate cancer line, were subjected to TRAF4 or control knockdown, followed by seeding at equal numbers in a Transwell invasion chamber either with (for invasion) or without Matrigel (for migration assays) in a serum-free medium for 12–16 hours. No significant difference in cell proliferation was observed under these conditions (data not shown). We found that the numbers of migratory and invasive PC3 cells were significantly decreased upon TRAF4 knockdown compared with non-targeting control (Figure 2, A and B, respectively). A similar observation was made in another metastatic prostate cancer line, DU145 (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI96060DS1).
Figure 2. TRAF4 plays a role in prostate cancer cell migration, invasion, and metastasis.

Knockdown of TRAF4 in PC3 cells inhibited cell migration (A) and invasion (B). Left panels: Cells migrated through migration chamber (A) or Matrigel (B) (n = 3). Images were obtained at ×100 magnification. Middle panels: Quantitation of cell numbers per field. Right panels: Levels of TRAF4 in control or TRAF4-knockdown cells as assessed by qRT-PCR. *P < 0.05 by Student’s t test versus siControl or siTRAF4 (A) and *P < 0.05 by 1-way ANOVA with Dunnett’s multiple comparisons test (B). Data are presented as mean ± SEM. (C) TRAF4 knockdown reduced prostate cancer cell colonization and metastasis in vivo. Top left: Representative bioluminescence images of SCID mice at different time periods after tail vein injection of PC3 shControl or shTRAF4 cells. Lower left: Quantitation of bioluminescence signals at the lung area in PC3 shControl- or shTRAF4-injected mice (n = 5). Right: Levels of TRAF4 in control or TRAF4-stable-knockdown cells as assessed by qRT-PCR. *P < 0.05 by 2-tailed Student’s t test. (D) TRAF4 knockdown reduced lung metastatic nodules. H&E-stained lung sections and graph showing the number of lung metastasis nodules after tail vein injection (n = 10). Data are presented as mean ± SEM. *P < 0.05 by 2-tailed Student’s t test. (E) H&E-stained bone sections (left) and immunohistochemistry using anti-luciferase antibody staining (right) confirming bone metastasis in the control group but not in the shTRAF4-knockdown group. Images were obtained at ×40 and ×100 (insets) magnification.
We next examined the role of TRAF4 in prostate cancer cell metastatic potential in an experimental lung metastatic mouse model by injecting prostate tumor cells via tail vein. PC3 luciferase-expressing cells selected for stable knockdown of TRAF4 or non-targeting shRNA control were injected via tail vein into 4- to 5-week-old male NOD/SCID mice. Numbers of injected tumor cells in the circulation in the control and shTRAF4 groups were compared at 5 minutes after tail vein injection by bioluminescence imaging (BLI) of the mouse lung (0 week) (Figure 2C). Although the same number of cells was injected, the shTRAF4 cells showed higher luminescence intensities compared with shControl cells due to greater expression levels of the luciferase gene in shTRAF4 cells (Supplemental Figure 1B). Both groups showed a gradual decrease in luminescence signals 1–3 weeks after injection, indicating that most of the injected cells did not survive in the lung. Through 9 weeks, the luminescence signals at the lung areas of control shRNA–injected mice steadily increased (Figure 2C), indicating the growth of successfully colonized tumor cells. In contrast, the signals from the shTRAF4 cell–injected mice continued to fade. This difference is unlikely due to a difference in proliferation rates of tumor cells, since comparable Ki-67 staining intensities were found in the luminescence signal–positive tumor areas of the 2 mouse groups (Supplemental Figure 1C). These results suggest that the injected shTRAF4 cells had reduced ability to colonize into the lung compared with control cells. H&E staining of the lungs also showed a reduction in size and number (*P < 0.05) of metastatic nodules in TRAF4-knockdown cells (Figure 2D). We found that some of the shRNA control cell– but not shTRAF4 cell–injected mice developed luminescence signals 8–9 weeks after injection at areas in addition to the lung, an indication of further metastasis. Tumor bone metastasis was confirmed via immunohistochemistry using an anti-luciferase antibody in shRNA control mice (Figure 2E). However, we did not find any bone metastasis in shTRAF4-injected mice. These results suggest that TRAF4 plays an important in vivo role in prostate cancer cell metastasis.
The RING domain is critical for TRAF4-mediated cell invasion in prostate cancer cell lines.
Next we investigated the molecular mechanisms by which TRAF4 drives prostate cancer cell invasion and eventually systemic metastasis. TRAF4 is an E3 ubiquitin ligase containing a RING domain that plays an important role in TRAF4-mediated ubiquitination. To determine whether the E3 ubiquitin ligase activity of TRAF4 is important for its invasive function, we deleted the RING domain and compared the effect of this truncated protein on the invasion ability of a poorly invasive prostate cancer cell line, LNCaP, with that of the full-length TRAF4. LNCaP cell lines stably expressing vector control, TRAF4 WT, or TRAF4ΔRING mutant were used in an invasion assay. The expression levels of FLAG-TRAF4 WT and FLAG-TRAF4ΔRING mutant were comparable. As shown in Figure 3A, WT TRAF4 overexpression significantly increased cell invasion, confirming the role of TRAF4 in mediating cell invasion. In contrast, the RING domain deletion mutant of TRAF4 lost the ability to promote cell invasion (Figure 3A). These results suggest that the TRAF4 RING domain vital for its E3 ubiquitin ligase activity also is critical for driving prostate tumor cell invasion.
Figure 3. TRAF4-mediated TrkA ubiquitination is critical for cell invasion.

(A) Overexpression of TRAF4 WT but not the RING domain deletion mutant promoted LNCaP cell invasion. Left: LNCaP cells invaded through Matrigel were stained with crystal violet (n = 3). Images were obtained at ×100 magnification. Middle: Quantitation of invaded cells per field. *P < 0.05 by 1-way ANOVA. Right: Western blot analysis of the expression of TRAF4 WT or its mutant in LNCaP stable cells using a TRAF4-specific antibody. (B) TRAF4 overexpression enhanced TrkA ubiquitination. The human ubiquitin array kit was used to identify TRAF4 ubiquitination targets. Cell lysate from GFP- (control) or TRAF4-expressing adenovirus–infected PC3 cells were used on each array. Each dot represents the ubiquitination level of a target protein recognized by an anti-ubiquitin antibody. *P < 0.05 by 2-tailed Student’s t test. (C) Knockdown of TrkA using specific siRNAs decreased DU145 cell invasion (n = 3). Images were obtained at ×100 magnification. Right: Levels of TrkA in control or TrkA-knockdown cells as assessed by qRT-PCR. *P < 0.05 by 1-way ANOVA with Dunnett’s multiple comparisons test. (D) TrkA inhibition by GW441756 reduced prostate cancer cell colonization and metastasis in vivo. Left: Bioluminescence images of SCID mice at 0 or 9 weeks after tail vein injection of PC3 luciferase cells with or without GW441756 treatment. Right: Quantitation of the bioluminescence signals at the metastasis area in control or GW441756-injected mice 9 weeks after injection (n = 5 per group). Data are presented as mean ± SEM. *P < 0.05 by 2-tailed Student’s t test.
TRAF4 interacts with and ubiquitinates the neurotrophin receptor TrkA.
To identify a TRAF4-targeted ubiquitination substrate that mediates TRAF4’s ability to promote cell invasiveness, we performed an unbiased screen on a ubiquitin array, which measured the ubiquitination levels of 49 different proteins. Among them we found TrkA as one of the top-hit candidate proteins showing significantly enhanced ubiquitination upon TRAF4 overexpression in PC3 cells (Figure 3B). TrkA is a tyrosine kinase receptor that is activated upon binding to its ligand, NGF, a member of the neurotrophin family that regulates brain development and function. TrkA was previously reported to also regulate prostate cancer cell metastasis (19–25). To test the hypothesis that TrkA is an important downstream mediator of TRAF4-dependent prostate tumor cell invasion, we examined the invasion potential of DU145 or PC3 cells following TrkA depletion. Our data revealed that TrkA silencing significantly reduced cell invasion, similar to the effect of TRAF4 knockdown (Figure 3C and Supplemental Figure 2, A and B), indicating that TrkA likely is a potential TRAF4 ubiquitination substrate involved in cell invasion.
We further tested the role of TrkA in prostate cancer cell metastasis in vivo using a TrkA-specific inhibitor, GW441756 (40). PC3 luciferase-expressing cells were injected via tail vein into male NOD/SCID mice. The mice were then randomized into control and drug treatment groups. The drug treatment mice group received 10mg/kg GW441756 i.p. twice a week, while the control group received solvent only. We did not observe any significant change in mouse weight between the 2 groups (Supplemental Figure 2C). Nine weeks after injection, the TrkA inhibitor treatment group had significantly reduced tumor metastasis compared with the control group (Figure 3D). These results suggest that TrkA inhibition in PC3 cells has an inhibitory effect on prostate cancer metastasis.
To test whether TRAF4 can interact with TrkA to promote its ubiquitination in cells, we transiently transfected FLAG-tagged TrkA or empty vector along with HA-tagged TRAF4. Immunoprecipitation of FLAG-tagged TrkA revealed a direct association with HA-tagged TRAF4 compared with cells expressing only FLAG-vector as a control (Figure 4A). Next we carried out an ubiquitination assay in 293T cells by transiently transfecting HA-ubiquitin, FLAG-TrkA, and V5-TRAF4 or its RING domain deletion mutant. The levels of TrkA ubiquitination were detected through immunoprecipitation using a FLAG-specific antibody, followed by Western blot analysis using an HA-specific antibody. The results confirmed that the WT TRAF4 but not the RING domain deletion mutant promoted the ubiquitination of TrkA in cells (Figure 4B). To confirm that the TRAF4-TrkA interaction exists in prostate tumor cells, we immunoprecipitated endogenous TrkA from DU145 cells and identified a strong interaction with TRAF4 (Figure 4C). Since NGF stimulation triggers TrkA activation, we investigated whether TrkA ubiquitination is regulated by NGF induction and whether TRAF4 plays a role in the ubiquitination process of endogenous TrkA. As shown in Figure 4D, NGF treatment substantially increased TrkA ubiquitination, whereas TRAF4 knockdown dramatically reduced the levels of ubiquitinated TrkA. We also performed an in vitro ubiquitination assay using purified FLAG-TrkA from 293T cells, purified TRAF4, HA-ubiquitin, ubiquitin-activating enzyme UBE1, and ubiquitin-conjugating enzyme UbcH5a to demonstrate that TRAF4 can directly promote TrkA ubiquitination (Supplemental Figure 3A). These results suggest that TRAF4 plays an important role in NGF-induced TrkA ubiquitination in prostate cancer cells.
Figure 4. TRAF4 interacted with TrkA and promoted its ubiquitination.

(A) HA-TRAF4 interacted with FLAG-TrkA in transiently transfected 293T cells. Shown is a co-IP experiment using an anti-FLAG antibody for immunoprecipitation. (B) WT TRAF4 but not the RING domain deletion mutant promoted TrkA ubiquitination. 293T cells were cotransfected with constructs as indicated. FLAG-TrkA was immunoprecipitated with an anti-FLAG antibody, and the ubiquitinated TrkA was visualized by Western blot analysis using an anti-HA antibody. (C) Endogenous TrkA interacted with endogenous TRAF4 in DU145 cells. Shown is a co-IP experiment using a TrkA-specific antibody or IgG control for immunoprecipitation. (D) TRAF4 knockdown abolished NGF-induced TrkA ubiquitination. DU145 cells were transfected with control siRNA or siTRAF4 and HA-ubiquitin. Cells were then treated with 50 ng/ml NGF for 15 minutes before harvest. Ubiquitinated TrkA was detected using an anti-ubiquitin antibody in a Western blot analysis from cell lysates immunoprecipitated with an anti-TrkA antibody. Ub, ubiquitin. (E) TRAF4 overexpression promoted TrkA ubiquitination at the cell membrane. 293T cells were cotransfected with TrkA and HA-Ub in the absence or presence of TRAF4 cotransfection. Cytosolic and membrane fraction were isolated and subjected to immunoprecipitation using an anti-FLAG antibody, and the ubiquitinated TrkA was visualized by Western blot analysis using an anti-HA antibody. (F) TRAF4-mediated TrkA polyubiquitination through K27- or K29-linked ubiquitin chain. K6–K63 represent the ubiquitin mutant with all lysine mutations except the indicated number of lysine.
TrkA undergoes internalization following activation at the cell membrane. To determine where the TRAF4-mediated ubiquitination event happens, we isolated the cytosolic and membrane protein fractions from cells transfected with FLAG-TrkA with or without TRAF4 overexpression and then examined TrkA ubiquitination. Although TrkA was located at both cytoplasm and cell membrane, TRAF4-mediated TrkA ubiquitination mainly occurred at the cell membrane (Figure 4E).
Ubiquitination chain linkage involves one or more of 7 ubiquitin lysine residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63). The type of ubiquitin linkage determines the fate of proteins in the cell. The most common ubiquitin linkage is K48, and it is usually associated with protein degradation. Interestingly, we did not observe any substantial change in the TrkA protein level upon TRAF4 overexpression in the absence of proteasome inhibitors, suggesting the unlikelihood that TRAF4-mediated TrkA ubiquitination promotes its protein degradation (Supplemental Figure 4A, right panel). It has been reported previously that TRAF4 is capable of mediating K63-linked ubiquitination (8, 9), suggesting that TRAF4 may target TrkA ubiquitination through nonclassical ubiquitin linkage. To determine which lysine linkage is involved in TRAF4-mediated TrkA ubiquitination, we utilized a series of ubiquitin mutants that only contain one of the 7 lysine residues, while all other lysine residues are mutated into arginine residues. As shown in Figure 4F, the ubiquitin mutants containing only K27 or K29 residue were able to promote TrkA ubiquitination similar to the WT ubiquitin, suggesting that TRAF4-mediated TrkA ubiquitination occurs through the K27 and K29 ubiquitin linkages.
TrkA plays a role in TRAF4-promoted cell invasion.
To understand how TRAF4 promoted cell invasion and how TrkA plays a role in this function, we carried out a reverse phase protein array (RPPA) study using cell lysates from PC3 cells expressing 2 different shTRAF4 or control shRNA plasmids. A total of 213 antibodies recognizing different proteins or protein phosphorylation forms were analyzed in the RPPA study. Among the proteins that were downregulated or had reduced phosphorylation levels in TRAF4-knockdown cells compared with control cells, we found a number of epithelial-mesenchymal transition– (EMT-) and invasion-associated proteins (Table 1). We further validated the RPPA results using quantitative real-time PCR (qRT-PCR) to examine the effect of siTRAF4 on the expression of these target genes (Figure 5A). Interestingly, many of them, including COX2, Slug, IL-6, and integrin β1, were also upregulated upon NGF stimulation (Figure 5B). To determine whether TrkA plays a role in TRAF4-regulated expression of these target genes, we knocked down TrkA using siRNAs, followed by measurement of target gene expression. TrkA-knockdown cells showed a pattern of reduced gene expression of TRAF4 target genes, as was observed with siTRAF4 treatment (Figure 5C). We also found that knockdown of TRAF4 abolished NGF-stimulated gene upregulation (Figure 5D), confirming that TRAF4 is important for NGF-induced gene expression.
Table 1. TRAF4 regulates genes involved in cell migration/invasion.
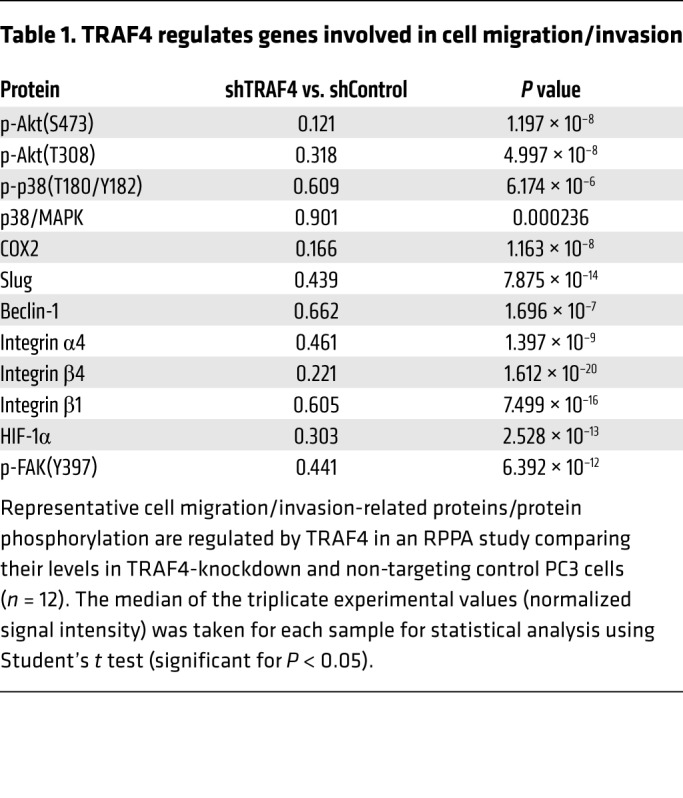
Figure 5. TrkA plays an important role in TRAF4-promoted cell invasion.

(A) Validation of the RPPA data for some of the invasion-related genes after TRAF4 knockdown using qRT-PCR. Right: Levels of TRAF4 in control or TRAF4-knockdown cells as assessed by qRT-PCR. *P < 0.05 by 1-way ANOVA with Dunnett’s multiple comparisons test. n = 3. (B) Expression of TRAF4-regulated genes at different time points following NGF stimulation as assessed by qRT-PCR. n = 3. (C) TrkA knockdown also downregulates TRAF4-regulated invasion-related genes. *P < 0.05 by 1-way ANOVA with Dunnett’s multiple comparisons test. n = 3. (D) Knockdown of TRAF4 inhibited NGF-stimulated gene expression. n = 3. (E) TrkA-selective inhibitor treatment abolished TRAF4-stimulated cell invasion (n = 3). Images were obtained at ×100 magnification. PC3 cells were infected with GFP or TRAF4 adenovirus and then treated with or without 0.5 µM GW441756 for 2 days before seeding in an invasion chamber. *P < 0.05 by 1-way ANOVA with Tukey’s multiple comparisons test. Data are presented as mean ± SEM.
Since TrkA is a tyrosine kinase, we then determined whether inhibition of TrkA kinase activity affects TRAF4-mediated cell invasion. We first examined whether the TrkA-selective inhibitor GW441756 affects cell growth in our experimental condition. An MTT assay was performed on PC3 cells treated with a series of concentrations of GW441756 for 2 days. No significant effect of the inhibitor treatment on cell growth was found (Supplemental Figure 3B). We next chose a 0.5-μM concentration of GW441756 to examine its effect on TRAF4-promoted cell invasion. As shown in Figure 5E, overexpression of TRAF4 significantly increased PC3 cell invasion. Treatment with GW441756 for 2 days abolished the TRAF4-mediated effects. These results substantiate the hypothesis that the interplay of TRAF4 and TrkA regulates cell invasion–associated gene expression and cell invasion.
Lysine residues at the TrkA kinase domain are responsible for TRAF4-mediated TrkA ubiquitination.
Since TRAF4 regulates TrkA ubiquitination, we next investigated the ubiquitination target sites in TrkA to understand the importance of this posttranslational modification on its function. TrkA is a 140-kDa transmembrane receptor containing extracellular domains involved in NGF binding, a transmembrane domain (TM), a juxtamembrane domain (JM), a tyrosine kinase domain (TK), and a short C-terminal domain (CT). A schematic representation of these domains is shown in Figure 6A. To determine which domain is targeted by TRAF4 for ubiquitination, we generated different deletion mutants of TrkA and then tested their relative ubiquitination levels in TRAF4-overexpressing cells (Figure 6A). TrkA ubiquitination was abolished upon deletion of the TK domain (ΔTK and ΔJM vs. WT and ΔCT). These results suggested that the TK domain is likely the region targeted by TrkA ubiquitination.
Figure 6. TRAF4 ubiquitinated 3 lysine residues present in the kinase domain of TrkA.

(A) Deletion of the tyrosine kinase domain (TK) of TrkA abolished its ubiquitination. Upper panel: Schematic representation of TrkA and its deletion mutants. Lower panels: Ubiquitination levels of different TrkA deletion mutants. FLAG-TrkA and the mutants were cotransfected with TRAF4 and HA-Ub into 293T cells. The ubiquitinated TrkA was immunoprecipitated using a FLAG antibody and then detected using an anti-HA antibody in the Western blot. (B) Mutation of K523, K544, and K547 residues at the TK domain abolished TrkA ubiquitination. (C) TRAF4 hyperactivated TrkA WT but not the K523_544_547R mutant in an in vitro kinase assay. Left: Purified FLAG-TrkA in vitro kinase activity using a poly(Glu4, Tyr1) synthetic peptide as a substrate. The activity was measured through an ADP-Glo Kinase assay. Right: Protein levels of purified TrkA, its mutant, and PKCδ used in the kinase assay with or without TRAF4 overexpression as demonstrated by Western blotting using an anti-FLAG antibody. Data are presented as mean ± SEM. n = 3. *P < 0.05 by 1-way ANOVA.
Next we determined the ubiquitination sites in the TK domain. Ubiquitin ligases often target multiple neighboring lysine residues for ubiquitination (41–43). There are 10 lysine residues present in the TK domain. Three of them (K523, K544, and K547) are located close to each other to form a lysine cluster in the crystal structure (Supplemental Figure 3C and ref. 44). To test whether this lysine cluster is the TRAF4-targeted ubiquitination site, we used site-directed mutagenesis to generate lysine-to-argine mutants of TrkA. Mutation of 2 of these 3 lysine residues to arginine (K523_44R or K544_47R) markedly reduced TRAF4-mediated ubiquitination, while mutation of all 3 lysine residues completely abolished the ubiquitination (Figure 6B). These results suggested that TRAF4 targets TrkA ubiquitination at a lysine cluster containing K523, K544, and K547 residues.
Interestingly, the 3 lysine residues we identified are located in proximity to the kinase activation loop (Supplemental Figure 3C). It is likely that posttranslational modification at the lysine cluster affects the kinase activity. We next determined whether TRAF4 directly regulates TrkA kinase activity through an in vitro kinase assay. FLAG-tagged WT TrkA or the K523_44_47R mutant was purified from TRAF4-overexpressing cells or vector control cells using a FLAG antibody. A FLAG-tagged serine kinase, PKCδ, was purified following the same procedure to serve as a negative control. Comparable levels of TrkA, its ubiquitin mutant, and PKCδ were then used in an in vitro luminescence kinase assay utilizing a poly(Glu4, Tyr1) synthetic peptide as a substrate (Figure 6C). The luminescence value from the PKCδ kinase reaction was used as a background reading, and subsequent kinase assay readings were adjusted according to it. We found that TRAF4 overexpression significantly enhanced the in vitro kinase activity of WT TrkA, whereas TRAF4 had no effect on the kinase activity of the TrkA ubiquitination–deficient mutant (Figure 6C). The K547 residue was previously reported to be an important site for ATP binding, and the K547A mutant of TrkA was considered as a kinase-dead mutant (45). It is not clear whether or not K547R mutation could behave similarly to K547A mutation to inactivate the kinase activity. To avoid the potential complication caused by K547R mutation, we also tested the in vitro kinase activity of the K523_44R double mutant, which had substantially reduced ubiquitination compared with the WT TrkA (Figure 6B). Similar to the triple mutation, the double mutation abolished TRAF4-mediated enhancement of TrkA in vitro kinase activity (Supplemental Figure 3D). These results suggest that TRAF4-mediated TrkA ubiquitination at the lysine cluster (K523_44_47) adjacent to the kinase domain hyperactivates TrkA kinase activity.
TRAF4-mediated ubiquitination affects TrkA tyrosine phosphorylation.
Next we assessed how TRAF4-mediated ubiquitination affects the NGF-induced signal transduction cascade. The ubiquitination mainly occurs at the cell membrane (Figure 4E). It was reported previously that TrkA ubiquitination at a different site regulates the receptor internalization (46). We did not observe a major change in TrkA subcellular localization when TRAF4 was overexpressed (Supplemental Figure 4A), suggesting that TRAF4-mediated ubiquitination does not have a marked effect on receptor trafficking. When TrkA is activated, several of its tyrosine residues are phosphorylated. The tyrosine phosphorylation could either release autoinhibition that is important for kinase activity (Y674/675) or serve as a docking site for binding to adaptor proteins to activate downstream signaling cascades (Y490 or Y785). Y674/675 phosphorylation correlates positively with the TrkA kinase activity, and it precedes the phosphorylation of other Tyr residues (47). To test whether TRAF4-mediated ubiquitination affects TrkA tyrosine phosphorylation, we analyzed the levels of TrkA tyrosine phosphorylation in the absence or presence of TRAF4 overexpression. As shown in Figure 7A, TRAF4 overexpression increased the levels of TrkA tyrosine phosphorylation at Y674/675, Y490, and Y785 sites in the presence of NGF, consistent with its ability to enhance TrkA kinase activity (Figure 6C). More importantly, mutation of the 3 TRAF4 targeted ubiquitination sites (K523_544_547R) abolished the tyrosine phosphorylation of TrkA. We observed a higher basal level of TrkA WT phosphorylation in the absence of TRAF4 overexpression compared with phosphorylation of the mutant (Figure 7A, lane 1 vs. lane 3). This is probably due to the presence of endogenous TRAF4. A similar result was observed for the K523_544R mutant (Supplemental Figure 4B). Interestingly, K547R single mutation appears to have had minor effects on TrkA phosphorylation levels, suggesting that K547R mutation by itself did not abolish its kinase activity (Supplemental Figure 4C) in contrast to the K547A mutation, as previously reported (48). These results suggest that TRAF4-mediated TrkA ubiquitination regulates TrkA tyrosine phosphorylation.
Figure 7. TRAF4-mediated TrkA ubiquitination regulated NGF-stimulated TrkA signaling cascade.

(A) TRAF4 overexpression increased the phosphorylation level of WT TrkA but not the K523_544_547R mutant in the presence of NGF. Specific tyrosine phosphorylation antibodies were used in the Western blot analysis in cells treated with 50 ng/ml NGF for 15 minutes. (B) Mutation of the K523, K544, and K547 residues reduced the association of Shc protein with TrkA. FLAG-TrkA WT or its mutant was transfected into DU145 cells, and the interaction between Shc protein and TrkA was determined in a co-IP experiment using a FLAG antibody for immunoprecipitation. (C) TRAF4 knockdown substantially reduced NGF-induced p38 MAPK phosphorylation (pT180/Y182). PC3 cells were treated with and without NGF (50 ng/ml) for 10 minutes after 18 hours of serum starvation. (D) The level of TRAF4 expression correlated with Slug gene expression in a prostate cancer patient cDNA array. (E) The level of TRAF4 expression correlated with the IL-6 gene expression in a prostate cancer patient cDNA array. Pearson’s correlations for D and E for fold change (log2) in gene expression as determined by qRT-PCR (r = 0.852, P < 0.0001 and r = 0.890, P = 2.53 × 10–14, respectively). (F) Working model of the role of TRAF4 in TrkA signaling and prostate cancer cell invasion. In low-TRAF4-expressing cells, NGF induce limited TrkA ubiquitination, resulting in low levels of TrkA kinase signaling. In TRAF4-overexpressing cells, high levels of TRAF4 significantly increase TrkA K27- and K29-linked ubiquitination at K523, K544, and K547 in the kinase domain upon NGF stimulation. This ubiquitination enhances TrkA kinase activity, its tyrosine phosphorylation levels, and the recruitment of downstream adaptor proteins, resulting in a hyperactivated TrkA signaling cascade. Consequently, NGF-responsive invasion-associated targeted gene transcription is upregulated to promote cell migration and invasion.
Upon NGF stimulation, the binding of phosphatases, such as SHP-1, to TrkA is transiently induced, which counterbalances TrkA activation (49). This could affect the phosphorylation levels of TrkA. To determine whether TRAF4-mediated ubiquitination affects phosphatase recruitment, we performed a co-IP experiment using a TrkA-specific antibody. As shown in Supplemental Figure 4D, no substantial difference in the association of SHP-1 with TrkA was found whether in the absence and presence of TRAF4 overexpression. Next, we asked whether mutation of the ubiquitination sites affects the ability of TrkA to recruit adaptor proteins. Y490 phosphorylation serves as a docking site for recruiting the Shc adaptor protein and is necessary for subsequent activation of downstream Ras/MAPK and PI3K pathways (49–51). A co-IP experiment was performed to examine the interaction between Shc and TrkA or its mutant in the presence of NGF. There are 3 Shc isoforms, p66, p52, and p46. P52 and p46 but not p66 play a role in mediating growth factor signaling. As shown in Figure 7B, p52 and p46 but not p66 were indeed associated with FLAG-TrkA WT. The K523_544_547R mutant, however, had a substantially reduced ability to interact with p52/p46 Shc. TrkA dimerizes upon NGF stimulation. The reason we did not observe complete loss of the interaction between Shc and the TrkA mutant is likely due to heterodimerization between the mutant TrkA and endogenous WT receptor.
In the RPPA analysis (Table 1), we found that p38 MAPK phosphorylation levels were downregulated when TRAF4 was knocked down. p38 MAPK plays an important role in EMT, invasion, extravasation, and organ colonization during cancer metastasis (11, 44). Since TrkA ubiquitination mutant has reduced interaction with the Shc adaptor protein, we examined whether TRAF4 regulates Shc downstream p38 kinase activation upon NGF induction. As shown in Figure 7C, the level of p38 phosphorylation (T180/Y182) increased when cells were stimulated with NGF. TRAF4 knockdown substantially reduced NGF-induced p38 phosphorylation. This result confirms that TRAF4 plays a role in regulating NGF-induced TrkA signaling pathways.
The levels of TrkA phosphorylation and its downstream p38 phosphorylation were also substantially reduced in metastatic mouse tumors derived from tail vein–injected shTRAF4-PC3 cells compared with controls (Supplemental Figure 4E), suggesting that TRAF4 expression levels regulate TrkA phosphorylation and its downstream signaling in vivo.
Taken together, our results suggested that TRAF4-mediated TrkA ubiquitination is important for TrkA function.
Correlation of TRAF4 expression and TrkA-regulated gene transcription in human prostate cancer samples.
To determine whether the regulation of TRAF4 on TrkA signaling also exists in human prostate cancer patients, we analyzed the expression of TRAF4 and 2 of the TrkA-regulated genes involved in cancer metastasis, Slug and IL-6, in a prostate cancer cDNA array. As shown in Figure 7, D and E, a significant correlation between expression levels of TRAF4 and Slug and of TRAF4 and IL-6 was observed (r = 0.852, P < 0.0001 and r = 0.890, P = 2.53 × 10–14, respectively). These results suggest that TRAF4 also can regulate TrkA signaling in human prostate cancers.
Altogether, our results demonstrate that TRAF4 promoted TrkA ubiquitination through K27 and K29 linkages at the tyrosine kinase domain. This posttranslational modification enhanced TrkA kinase activity, its tyrosine phosphorylation levels, and subsequent downstream signaling activation to promote cancer metastasis.
Discussion
Malfunction of the ubiquitination system can contribute substantially to cancer development and metastasis. In addition to the well-studied proteasome-dependent protein degradation, non-proteolytic ubiquitination is emerging as a pivotal player in cancers. Herein we demonstrated that the RING domain E3 ubiquitin ligase TRAF4 is highly expressed in metastatic prostate cancers and plays an important role in regulating prostate cancer invasion and metastasis. Its E3 ubiquitin ligase activity is essential for promoting cell invasion. We further revealed that TrkA, a member of the receptor tyrosine kinase superfamily, is a ubiquitination substrate of TRAF4 that mediates the effect of TRAF4 on prostate cancer cell invasion.
Receptor tyrosine kinases are important signaling molecules that regulate cell proliferation, survival, differentiation, apoptosis, and migration. Aberrant activation of tyrosine kinases has been linked to a variety of cancers. In addition to genomic alterations, the activation of non-mutated kinases can contribute to cancer development and metastasis, especially in cancers with a low mutation rates such as prostate cancer (52–55). Global tyrosine phosphorylation levels are significantly increased in advanced prostate cancers even in the absence of kinase somatic mutations (56, 57). It was reported that several WT tyrosine kinases, including Trk family members TrkB (NTRK2) and TrkC (NTRK3), can drive prostate cancer bone and visceral metastasis in vivo (58). Thus, mechanisms other than genomic alterations are important for aberrantly activating tyrosine kinases in prostate cancers. Our study here underscores a role for non-proteolytic ubiquitination in aberrant activation of WT TrkA. Emerging evidence has documented the function of neurotrophins and their receptors in prostate cancer development and metastasis (21, 22, 24, 58, 59). Our present results provide strong evidence supporting the functional role of the TRAF4/TrkA axis in prostate cancer cell invasion.
Ubiquitination of several receptor tyrosine kinases has been reported previously, but the ubiquitination (mainly K48- or K63-linked) was associated with receptor protein turnover or receptor trafficking (46, 60–65). Similar to other RTKs, TrkA was found to be ubiquitinated by the RING-type E3 ubiquitin ligase c-Cbl (64) and the HECT-type ligase Nedd4-2, which promote its degradation through the proteasome (62). TRAF6 promotes K63-linked TrkA ubiquitination at the juxtamembrane domain and subsequent receptor internalization (46). Our results demonstrate that TRAF4-mediated ubiquitination occurs at the tyrosine kinase domain and it hyperactivates TrkA kinase activity, suggesting that TRAF4-targeted ubiquitination regulates TrkA function through a novel mechanism apart from other E3 ubiquitin ligases.
Interestingly, TRAF4 promoted TrkA ubiquitination through atypical K27- and K29-linked ubiquitin chains (Figure 4F). Unlike the well-studied K48 and K63 polyubiquitin chain, the functional roles of K27 and K29 ubiquitin linkages are less clear. It appears that they are nonproteolytic, and they recently have been implicated in protein-protein association/dissociation, negative regulation of protein degradation, and protein aggregation (66–70). It is not clear how the ubiquitination precisely affects TrkA kinase activity. The cellular localization of TrkA and its interaction with phosphatase SHP-1 were not substantially changed upon TRAF4 overexpression (Supplemental Figure 4, A and D). We found that TrkA is ubiquitinated at the K523, K544, and K547 residues (Figure 6B). These lysine residues are located adjacent to each other in the N-lobe of the kinase domain, which is close to the center of the kinase active site: the DFG motif at the activation loop and the ATP-binding site (Supplemental Figure 3C and ref. 44). Conformational change of the activation loop, especially the position of the DFG motif, upon ligand binding is essential for tyrosine kinase activation (71). Protein-protein interaction is one of the mechanisms to stabilize the active conformation of the activation loop of RTK or serine/threonine kinases during activation. Binding of cyclins to CDKs induces conformational change of the CDK kinase domain and stabilizes the active activation loop conformation (71). Similarly, one of the EGFR molecules in the ligand-bound EGFR dimer serves as an activator kinase to stabilize the active conformation of its partner through dimerization (71). It is possible that the K27- and K29-linked ubiquitin chain conjugation at the K523, K544, and K547 sites stabilizes the active conformation of the TrkA activation loop, functioning in a similar manner to cyclin/CDK and EGFR. The second possibility is that the positive charges introduced by the ubiquitin modification at these positions affect the position of the Asp residue at the DFG motif and stabilize the active conformation of the activation loop, or stabilize ATP binding, to sustain the kinase activation. Consistent with these speculations, we found that tyrosine phosphorylation (Y674/675) at the kinase domain, which is associated with kinase activation, was abolished when these lysine residues were mutated (Figure 7A).
It is known that NGF binds and activates TrkA, which autophosphorylates tyrosine residues in TrkA (Figure 7F). This activates several important signaling pathways, such as PI3K/Akt, PLC-γ, and MAPK, which regulate migration, invasion, and metastasis. We found that overexpression of TRAF4 increased TrkA tyrosine phosphorylation in the presence of NGF. The TrkA ubiquitination mutant not only was defective in tyrosine phosphorylation, but also had reduced ability to interact with downstream adaptor proteins. Consequently, we found that NGF-induced p38 MAPK phosphorylation and the expression of downstream invasion-associated genes were suppressed in TRAF4-knockdown cells. All of these results support the notion that TRAF4-mediated TrkA ubiquitination is required for TrkA phosphorylation and subsequent pathway activation (Figure 7F).
We also investigated whether other RTKs may be regulated by TRAF4 in a manner similar to TrkA, which might indicate that regulation of kinase activity by ubiquitination could be more generally applicable. We explored this concept using an array containing multiple receptor tyrosine kinases and found that TRAF4 overexpression increased ubiquitination of several receptor tyrosine kinases in addition to TrkA (Supplemental Figure 4F). These new substrate kinases need to be further explored as additional therapeutic targets in prostate cancer.
Taken together, our study not only expands knowledge of the role of TRAF4 in prostate cancer metastasis, but also provides a potential novel drug target for treating aggressive prostate cancers. Unlike genomic alteration or protein overexpression, WT tyrosine kinase activation other than kinase overexpression is not easily diagnosed in patients. The study we present here reveals a potential biomarker that could help in the prediction of TrkA activation in cancer patients.
Methods
Animal and human studies.
All animal experiments were performed in accordance with the Institutional Animal Care and Use Committees (IACUCs) at Baylor College of Medicine. For in vivo studies, 5- to 6-week-old male SCID mice (The Jackson Laboratory) were used for experimental lung metastasis assays. Human tissue samples were obtained from the Human Tissue Acquisition and Pathology Core of the Dan L. Duncan Comprehensive Cancer Center and were collected from fresh radical prostatectomy specimens after informed consent was obtained under an Institutional Review Board–approved protocol. Cancer samples contained a minimum of 70% cancer, and benign tissues were free of cancer on pathologic examination.
Cell culture.
The human prostate cancer cell lines LNCaP, PC3, and DU145 and HEK293T cells were obtained from ATCC. PC3, DU145, and LNCaP cells were maintained in RPMI 1640 medium containing 10% FBS, 2 mM l-glutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin at 37°C and 5% CO2. The human embryonic kidney epithelial cell line HEK293T was maintained in DMEM supplemented with 10% FBS. For NGF treatment experiments, cells were maintained in serum-free culture medium for the stated periods of time supplemented with 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin and then treated with NGF (50 ng/ml) for the specified periods of time.
Reagents and antibodies.
Human NGF-β (catalog 300-174P) was obtained from Gemini Bio-Products. Primary antibodies were as follows: anti-pTrkA(Y785) (catalog 4168), anti-pTrkA(Y674/675) (catalog 4621), anti-p38 (catalog 9212), anti–p-p38 (catalog 9211), anti-Shc (catalog 2432), anti–SHP-1 (catalog 3759), anti–Na,K-ATPase (catalog 3010) from Cell Signaling Technology; anti-TRAF4 (catalog sc-10776), anti–HA-probe (catalog sc-805), anti-Ub (catalog sc-8017), anti-GAPDH (catalog sc-32233) from Santa Cruz Biotechnology Inc.; anti-TrkA (catalog 06-574), anti-Shc (catalog 06-203) from EMD Millipore; anti-pTrkA(Y490) (catalog ab85130), anti–Ki-67 (catalog 66155) from Abcam. HRP-conjugated secondary anti-mouse (catalog 1706516) or anti-rabbit (catalog 1706515) antibodies were obtained from Bio-Rad. Monoclonal ANTI-FLAG M2-peroxidase (HRP) antibody (catalog 8592A), EZview Red ANTI-FLAG M2 Affinity Gel (catalog F2426) were obtained from Sigma-Aldrich. TRAF4 adenovirus (catalog VH819961) was obtained from Vigene Biosciences. GFP adenovirus was produced in the Gene Vector Core at Baylor College of Medicine. GW441756 (catalog 141051) was purchased from Abcam.
Reverse transcription and quantitative real-time PCR.
Total RNA was extracted from the indicated cells by using an RNeasy Mini Kit (QIAGEN). RNA concentration and purity were measured by a NanoDrop 2000 UV-Vis Spectrophotometer (Thermo Fisher Scientific). 2 μg total RNA was used to generate cDNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche). Real-time PCR was performed using SYBR green PCR master mix (Life Technologies, Thermo Fisher Scientific). Primers used are listed in Supplemental Tables 1 and 2. For all RT-qPCR analysis, β-actin was used to normalize RNA input, and expression levels were calculated according to the comparative Ct method (ΔΔCT).
Construction of expression vectors and TrkA mutants.
The TRAF4 TrkA cDNA was cloned into FLAG-tagged pSG5 expression vector. All TrkA deletion as well as lysine mutants were also cloned into FLAG-tagged pSG5 expression vector. In addition, TRAF4 was cloned into HA-tagged pCM5 expression vector. SHP-1 (catalog 8572), WT ubiquitin, and its mutant constructs were obtained from Addgene. TRAF4 shRNA was cloned into pLenti6/TR vector (Thermo Fisher Scientific). Primers used for cloning are listed in Supplemental Tables 1 and 2.
Transfection and lentivirus infection.
Cells were transfected with plasmid DNA using Lipofectamine 3000 and siRNA using Lipofectamine RNAiMAX transfection reagent (both from Thermo Fisher Scientific) following the manufacturer’s protocol. Virus packaging was performed in HEK293T cells after cotransfection of plasmid with the packaging plasmid psPAX2 and envelope plasmid pMD2.G using Lipofectamine 3000. Viruses were harvested 48 hours after transfection, and viral titers were determined. Target cells were infected with recombinant lentivirus-transducing units in the presence of 8 μg/ml Polybrene (Sigma-Aldrich).
Immunoblotting.
Cells were harvested and protein was extracted from cells as previously described (72). The protein concentration was determined using a protein assay kit (Bio-Rad), and samples were separated in SDS polyacrylamide gels, with various concentrations depending on the molecular weight of the protein under investigation. After probing with a primary antibody, the membrane was incubated with a secondary antibody conjugated with HRP. Finally, signal intensity was determined using the enhanced chemiluminescence reagents. Endogenous GAPDH was used as the internal control.
MTS assay.
The CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) reagent (catalog G358A) was obtained from Promega, and the assay was performed according to the manufacturer’s instructions. Briefly, cells were seeded in a 96-well plate and treated with or without the TrkA inhibitor GW441756. The plate was incubated at 37°C in a humidified, 5% CO2 atmosphere. 20 μl CellTiter 96 AQueous One Solution Reagent was added to each well containing 100 μl media and again incubated for 3 hours. Absorbance was measured at 490 nm using a microplate reader.
Cell migration and invasion assay.
Cell invasion activity was determined in vitro using a BD BioCoat tumor invasion system (catalog 354483; BD Biosciences), which contains an 8-μm polyethylene terephthalate membrane with a thin layer of reconstituted Matrigel basement membrane matrix per the manufacturer’s protocol. In brief, cells were harvested, resuspended in serum-free medium, and then transferred to the hydrated Matrigel chambers (~25,000 cells per well). The chambers were then incubated for 16 hours in culture medium with 10% FBS in the bottom chambers before examination. The cells on the upper surface were scraped and washed away, whereas the invaded cells on the lower surface were fixed and stained with 0.05% crystal violet for 2 hours. Finally, invaded cells were counted under a microscope, and the relative number was determined. Cell migration assay was performed by following the same procedures as in the cell invasion assay, except that a modified 2-chamber Transwell system was used in the migration assay (catalog 354578; BD Biosciences).
Ubiquitination screen assay.
Targets of TRAF4-mediated ubiquitination were determined using an R&D Systems Proteome Profiler Human Ubiquitin Array Kit (catalog ARY027), which consists of 49 different protein samples, and a Proteome Profiler Human Phospho-RTK Array Kit (catalog ARY001B) consisting of 49 human receptor tyrosine kinases. Relative expression levels of ubiquitination of human proteins in samples were determined as per the manufacturer’s protocol. Briefly, the human ubiquitin array nitrocellulose membranes spotted with 49 different antibodies to human ubiquitin target proteins were incubated with prepared cell lysates for 1 hour on a rocking platform shaker. After thorough washing to remove unbound proteins, the membrane was incubated with biotinylated pan–anti-ubiquitin detection antibody cocktail at 4˚C overnight. The next day the membrane was washed thoroughly, followed by addition of streptavidin-HRP. The signal produced at each capture spot corresponding to the relative amount of ubiquitinated protein bound was exposed to autoradiography film and analyzed.
In vitro ubiquitination assay.
FLAG-TrkA was transiently transfected into 293T cells. The protein was then purified from 293T cell lysates using anti-FLAG M2 beads and eluted from the beads using 3X FLAG peptide (Sigma-Aldrich). The purified protein was incubated with 100 ng UBE1, 150 ng UbcH5a, and 5 μg HA-ubiquitin (Boston Biochem) in the absence or presence of 500 ng TRAF4 (Novus Biologicals) with ubiqutination buffer (50 mM Tris-Cl, pH 7.4, 2 mM ATP, 5 mM MgCl2, 2 mM DTT) at 30°C for 90 minutes. The incubation mixture was then subjected to immunoprecipitation using an anti-FLAG antibody, followed by Western blot analysis using an anti-HA antibody.
Tail vein injection and IVIS imaging.
The animal studies were conducted in accordance with NIH animal use guidelines, and the experimental protocol was approved by the Baylor College of Medicine Animal Care Research Committee. To evaluate the role of TRAF4 in tumor metastasis, luciferase-positive PC3 cells (control and TRAF4-knockdown by shRNA) were injected into SCID mice through the tail vein (1 × 106 cells per mouse). To study the effect of TrkA inhibition in tumor metastasis, we used a TrkA-specific inhibitor, GW441756. Luciferase-positive PC3 cells were injected into SCID mice through the tail vein (1 × 106 cells per mouse). Thereafter, mice were treated with i.p. injection twice a week with solvent control or GW441756 (10 mg/kg). Tumor metastasis in mice was assessed via in vivo bioluminescence measurement using the IVIS Imaging System (PerkinElmer). For the luciferase detection imaging, 200 μl of 15 mg/ml d-luciferin (Caliper Life Sciences) in PBS was injected i.p. before imaging. The photometry of the tumor was calculated by Living Image 3.1.0 software (Caliper Life Sciences), and the results were used to generate the tumor metastasis progression. Nine weeks after injection, mice were sacrificed, lungs were collected and fixed in Bouin’s solution, and images were captured.
RPPA analysis.
RPPA assays were carried out as described previously with minor modifications (73). Protein lysates were prepared from cultured cells with modified Tissue Protein Extraction Reagent (TPER; Pierce) and a cocktail of protease and phosphatase inhibitors (Roche Life Science) (73). The lysates were diluted into 0.5 mg/ml total protein in SDS sample buffer and denatured on the same day. The Aushon 2470 Arrayer (Aushon BioSystems) with a 40-pin (185 μm) configuration was used to spot samples and control lysates onto nitrocellulose-coated slides (Grace Bio-Labs) using an array format of 960 lysates/slide (2,880 spots/slide). The slides were processed as described (73) and probed with a set of 213 antibodies against total and phosphoproteins using an automated slide stainer (Autolink 48, Dako). Each slide was incubated with one specific primary antibody, and a negative control slide was incubated with antibody diluent instead of primary antibody. Primary antibody binding was detected using a biotinylated secondary antibody, followed by streptavidin-conjugated IRDye 680 fluorophore (LI-COR Biosciences). Total protein content of each spotted lysate was assessed by fluorescence staining with Sypro Ruby Protein Blot Stain according to the manufacturer’s instructions (Molecular Probes).
Fluorescence-labeled slides were scanned on a GenePix AL4200 scanner, each slide, along with its accompanying negative control slide, was scanned at an appropriate photomultiplier (PMT) to obtain optimal signal for this specific set of samples. The images were analyzed with GenePix Pro 7.0 (Molecular Devices). Total fluorescence signal intensities of each spot were obtained after subtraction of the local background signal for each slide and were then normalized for variation in total protein, background, and nonspecific labeling using a group-based normalization method as described previously (Chang et al., 2015). For each spot on the array, the background-subtracted foreground signal intensity was subtracted by the corresponding signal intensity of the negative control slide (omission of primary antibody) and then normalized to the corresponding signal intensity of total protein for that spot. Each image, along with its normalized data, was carefully evaluated for quality through manual inspection and control samples. Antibody slides that failed the quality inspection were either repeated at the end of the staining runs or removed before data reporting. A total of 213 antibodies remained in the list. A complete list of validated antibodies can be found in Supplemental Table 3.
The median of the triplicate experimental values (normalized signal intensity) was taken for each sample for subsequent statistical analysis. We determined significantly changed proteins between experimental groups by employing Student’s t test (significant for P < 0.05).
Kinase assay.
The TrkA kinase assay was performed using the TrkA kinase enzyme system (catalog V2931) and ADP-Glo Kinase Assay kit (catalog V6930; Promega) as per the manufacturer’s protocol. To compare the kinase activity of TrkA with and without TRAF4 overexpression and TrkA ubiquitin mutant with WT TrkA, 293T cells were transfected with different plasmids using Lipofectamine 3000. Cells were grown in complete media, followed by serum starvation for 4 hours. Thereafter, cells were briefly induced with NGF before harvesting. Cell lysis was performed using M-PER mammalian protein extraction reagent (Thermo Fisher Scientific). Overexpressed FLAG-tagged proteins were immunoprecipitated using EZview Red ANTI-FLAG M2 Affinity Gel. After thoroughly washing the beads with wash buffer (1× PBS, 1 mM DTT, 1× protease inhibitor), protein was eluted by incubating the beads at 4°C for 30 minutes with 100 μl of 3X FLAG peptide at a 100-ng/ml working concentration. An equal amount of protein was used to compare the kinase activity using the TrkA kinase enzyme system and ADP-Glo Kinase Assay kit. The TrkA reaction utilizes ATP and generates ADP. Then the ADP-Glo reagent terminates the kinase reaction and depletes the remaining ATP. Finally, the kinase detection reagent converts ADP to ATP, and the newly synthesized ATP emits light using the luciferase/luciferin reaction. The light generated correlates to the amount of ADP generated in the kinase or ATPase assay, which is indicative of kinase activity.
Statistics.
Unless otherwise indicated, all results represent mean ± SEM, and statistical comparisons between different groups were performed using the 2-tailed Student’s t test or 1-way ANOVA with multiple comparisons corrections. For all statistical analyses, differences of P < 0.05 were considered statistically significant, and experiments were repeated at least 3 times. GraphPad Prism software version 4.0/7.0 (GraphPad Software) was used for data analysis.
Study approval.
All animal experiments were approved by the Institutional Animal Care and Use Committees (IACUCs) at Baylor College of Medicine. Human tissue samples were obtained from the Human Tissue Acquisition and Pathology Core of the Dan L. Duncan Comprehensive Cancer Center and were collected from fresh radical prostatectomy specimens after obtaining written informed consent under an Institutional Review Board–approved protocol.
Author contributions
RS, BWO, and PY conceived and designed the experiments. RS and DK performed cell studies. RS, JS, SD, and HS performed animal studies. SH and DPE performed RPPA analysis. MMI provided human prostate tumor samples. RS, BWO, and PY interpreted the data and wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Supplementary Material
Acknowledgments
The authors acknowledge joint participation by the Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine (BCM) and the TRAF4 and Castration-Resistant Prostate Cancer Program. This work was also supported by grants from the Department of Defense (W81XWH-15-1-0536 to PY and W81XWH-16-1-0297 to SD), NCI Cancer Center Support grant P30CA125123 (BCM Gene Vector Core and Antibody-Based Proteomics Core), and the Dan L. Duncan Comprehensive Cancer Center (P30 CA125123) supporting Human Tissue Acquisition and Pathology. In addition, this work was supported in part by a Cancer Prevention & Research Institute of Texas Proteomics and Metabolomics Core Facility Support Award (RP170005 to DPE and SH). We thank Kimal Rajapakshe and Cristian Coarfa for RPPA data processing and normalization. We thank Fuli Jia and Danli Wu of the Antibody-Based Proteomics Core/Shared Resource for their excellent technical assistant in performing RPPA experiments.
Version 1. 05/01/2018
In-Press Preview
Version 2. 06/18/2018
Electronic publication
Version 3. 07/02/2018
Print issue publication
Footnotes
DK’s present address is: University of Texas Southwestern Medical School, Dallas, Texas, USA.
SD’s present address is: Department of Cell Stress Biology, Roswell Park Comprehensive Cancer Center, Buffalo, New York, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(7):3129–3143.https://doi.org/10.1172/JCI96060.
Contributor Information
Ramesh Singh, Email: Ramesh.Singh@bcm.edu.
Dileep Karri, Email: Dileep.Karri@UTSouthwestern.edu.
Hong Shen, Email: Hong.Shen@bcm.edu.
Jiangyong Shao, Email: jiangyongshao@yahoo.com.
Subhamoy Dasgupta, Email: sdasgupt@bcm.edu.
Shixia Huang, Email: shixiah@bcm.edu.
Dean P. Edwards, Email: deane@bcm.edu.
Michael M. Ittmann, Email: mittmann@bcm.edu.
Bert W. O’Malley, Email: berto@bcm.edu.
Ping Yi, Email: pyi@bcm.edu.
References
- 1.Shi D, Grossman SR. Ubiquitin becomes ubiquitous in cancer: emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets. Cancer Biol Ther. 2010;10(8):737–747. doi: 10.4161/cbt.10.8.13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell. 2008;14(1):10–21. doi: 10.1016/j.ccr.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Landré V, Rotblat B, Melino S, Bernassola F, Melino G. Screening for E3-ubiquitin ligase inhibitors: challenges and opportunities. Oncotarget. 2014;5(18):7988–8013. doi: 10.18632/oncotarget.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu X, et al. Ubiquitin-conjugating enzyme Ubc13 controls breast cancer metastasis through a TAK1-p38 MAP kinase cascade. Proc Natl Acad Sci U S A. 2014;111(38):13870–13875. doi: 10.1073/pnas.1414358111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang G, et al. K63-linked ubiquitination in kinase activation and cancer. Front Oncol. 2012;2:5. doi: 10.3389/fonc.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Y. Targeting E3 ubiquitin ligases for cancer therapy. Cancer Biol Ther. 2003;2(6):623–629. [PubMed] [Google Scholar]
- 7.Navon A, Ciechanover A. The 26 S proteasome: from basic mechanisms to drug targeting. J Biol Chem. 2009;284(49):33713–33718. doi: 10.1074/jbc.R109.018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W, et al. TRAF4 is a critical molecule for Akt activation in lung cancer. Cancer Res. 2013;73(23):6938–6950. doi: 10.1158/0008-5472.CAN-13-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L, et al. TRAF4 promotes TGF-β receptor signaling and drives breast cancer metastasis. Mol Cell. 2013;51(5):559–572. doi: 10.1016/j.molcel.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 10.Yi P, Xia W, Wu RC, Lonard DM, Hung MC, O’Malley BW. SRC-3 coactivator regulates cell resistance to cytotoxic stress via TRAF4-mediated p53 destabilization. Genes Dev. 2013;27(3):274–287. doi: 10.1101/gad.203760.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camilleri-Broët S, et al. TRAF4 overexpression is a common characteristic of human carcinomas. Oncogene. 2007;26(1):142–147. doi: 10.1038/sj.onc.1209762. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Wei D, Wang W, Shen B, Xu S, Cao Y. TRAF4 enhances oral squamous cell carcinoma cell growth, invasion and migration by Wnt-β-catenin signaling pathway. Int J Clin Exp Pathol. 2015;8(9):11837–11846. [PMC free article] [PubMed] [Google Scholar]
- 13.Yang K, Wang F, Han JJ. TRAF4 promotes the growth and invasion of colon cancer through the Wnt/β-catenin pathway. Int J Clin Exp Pathol. 2015;8(2):1419–1426. [PMC free article] [PubMed] [Google Scholar]
- 14.Ren HY, et al. Cytoplasmic TRAF4 contributes to the activation of p70s6k signaling pathway in breast cancer. Oncotarget. 2015;6(6):4080–4096. doi: 10.18632/oncotarget.2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao W, Wang X, Cai Q, Gao S, Wang J, Zhang P. TRAF4 enhances osteosarcoma cell proliferation and invasion by Akt signaling pathway. Oncol Res. 2014;22(1):21–28. doi: 10.3727/096504014X14077751730351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Régnier CH, et al. Presence of a new conserved domain in CART1, a novel member of the tumor necrosis factor receptor-associated protein family, which is expressed in breast carcinoma. J Biol Chem. 1995;270(43):25715–25721. doi: 10.1074/jbc.270.43.25715. [DOI] [PubMed] [Google Scholar]
- 17.Kedinger V, Rio MC. TRAF4, the unique family member. Adv Exp Med Biol. 2007;597:60–71. doi: 10.1007/978-0-387-70630-6_5. [DOI] [PubMed] [Google Scholar]
- 18.Cherfils-Vicini J, et al. Characterization of immune functions in TRAF4-deficient mice. Immunology. 2008;124(4):562–574. doi: 10.1111/j.1365-2567.2008.02810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molloy NH, Read DE, Gorman AM. Nerve growth factor in cancer cell death and survival. Cancers (Basel) 2011;3(1):510–530. doi: 10.3390/cancers3010510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arrighi N, et al. Nerve growth factor signaling in prostate health and disease. Growth Factors. 2010;28(3):191–201. doi: 10.3109/08977190903578678. [DOI] [PubMed] [Google Scholar]
- 21.Geldof AA, De Kleijn MA, Rao BR, Newling DW. Nerve growth factor stimulates in vitro invasive capacity of DU145 human prostatic cancer cells. J Cancer Res Clin Oncol. 1997;123(2):107–112. doi: 10.1007/BF01269888. [DOI] [PubMed] [Google Scholar]
- 22.Walch ET, Marchetti D. Role of neurotrophins and neurotrophins receptors in the in vitro invasion and heparanase production of human prostate cancer cells. Clin Exp Metastasis. 1999;17(4):307–314. doi: 10.1023/A:1006652605568. [DOI] [PubMed] [Google Scholar]
- 23.Festuccia C, et al. Tyrosine kinase inhibitor CEP-701 blocks the NTRK1/NGF receptor and limits the invasive capability of prostate cancer cells in vitro. Int J Oncol. 2007;30(1):193–200. [PubMed] [Google Scholar]
- 24.Warrington RJ, Lewis KE. Natural antibodies against nerve growth factor inhibit in vitro prostate cancer cell metastasis. Cancer Immunol Immunother. 2011;60(2):187–195. doi: 10.1007/s00262-010-0934-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weeraratna AT, et al. Pan-trk inhibition decreases metastasis and enhances host survival in experimental models as a result of its selective induction of apoptosis of prostate cancer cells. Clin Cancer Res. 2001;7(8):2237–2245. [PubMed] [Google Scholar]
- 26.Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature. 1986;319(6056):743–748. doi: 10.1038/319743a0. [DOI] [PubMed] [Google Scholar]
- 27.Kozma SC, Redmond SM, Fu XC, Saurer SM, Groner B, Hynes NE. Activation of the receptor kinase domain of the trk oncogene by recombination with two different cellular sequences. EMBO J. 1988;7(1):147–154. doi: 10.1002/j.1460-2075.1988.tb02794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bongarzone I, et al. High frequency of activation of tyrosine kinase oncogenes in human papillary thyroid carcinoma. Oncogene. 1989;4(12):1457–1462. [PubMed] [Google Scholar]
- 29.Butti MG, Bongarzone I, Ferraresi G, Mondellini P, Borrello MG, Pierotti MA. A sequence analysis of the genomic regions involved in the rearrangements between TPM3 and NTRK1 genes producing TRK oncogenes in papillary thyroid carcinomas. Genomics. 1995;28(1):15–24. doi: 10.1006/geno.1995.1100. [DOI] [PubMed] [Google Scholar]
- 30.Pierotti MA, Greco A. Oncogenic rearrangements of the NTRK1/NGF receptor. Cancer Lett. 2006;232(1):90–98. doi: 10.1016/j.canlet.2005.07.043. [DOI] [PubMed] [Google Scholar]
- 31.Reuther GW, Lambert QT, Caligiuri MA, Der CJ. Identification and characterization of an activating TrkA deletion mutation in acute myeloid leukemia. Mol Cell Biol. 2000;20(23):8655–8666. doi: 10.1128/MCB.20.23.8655-8666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.George DJ, Suzuki H, Bova GS, Isaacs JT. Mutational analysis of the TrkA gene in prostate cancer. Prostate. 1998;36(3):172–180. doi: 10.1002/(SICI)1097-0045(19980801)36:3<172::AID-PROS5>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 33.Wu RF, Xu YC, Ma Z, Nwariaku FE, Sarosi GA, Jr, Terada LS. Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol. 2005;171(5):893–904. doi: 10.1083/jcb.200507004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rousseau A, Wilhelm LP, Tomasetto C, Alpy F. The phosphoinositide-binding protein TRAF4 modulates tight junction stability and migration of cancer cells. Tissue Barriers. 2014;2(4):e975597. doi: 10.4161/21688370.2014.975597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varambally S, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8(5):393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Chandran UR, et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu YP, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22(14):2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 39.Grasso CS, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wood ER, Kuyper L, Petrov KG, Hunter RN, Harris PA, Lackey K. Discovery and in vitro evaluation of potent TrkA kinase inhibitors: oxindole and aza-oxindoles. Bioorg Med Chem Lett. 2004;14(4):953–957. doi: 10.1016/j.bmcl.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Baldi L, Brown K, Franzoso G, Siebenlist U. Critical role for lysines 21 and 22 in signal-induced, ubiquitin-mediated proteolysis of I kappa B-alpha. J Biol Chem. 1996;271(1):376–379. doi: 10.1074/jbc.271.1.376. [DOI] [PubMed] [Google Scholar]
- 42.Mattiroli F, Sixma TK. Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat Struct Mol Biol. 2014;21(4):308–316. doi: 10.1038/nsmb.2792. [DOI] [PubMed] [Google Scholar]
- 43.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79(1):13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 44.Bertrand T, et al. The crystal structures of TrkA and TrkB suggest key regions for achieving selective inhibition. J Mol Biol. 2012;423(3):439–453. doi: 10.1016/j.jmb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 46.Geetha T, Jiang J, Wooten MW. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol Cell. 2005;20(2):301–312. doi: 10.1016/j.molcel.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 47.Segal RA, et al. Differential utilization of Trk autophosphorylation sites. J Biol Chem. 1996;271(33):20175–20181. doi: 10.1074/jbc.271.33.20175. [DOI] [PubMed] [Google Scholar]
- 48.Dixon SJ, MacDonald JI, Robinson KN, Kubu CJ, Meakin SO. Trk receptor binding and neurotrophin/fibroblast growth factor (FGF)-dependent activation of the FGF receptor substrate (FRS)-3. Biochim Biophys Acta. 2006;1763(4):366–380. doi: 10.1016/j.bbamcr.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 49.Uren RT, Turnley AM. Regulation of neurotrophin receptor (Trk) signaling: suppressor of cytokine signaling 2 (SOCS2) is a new player. Front Mol Neurosci. 2014;7:39. doi: 10.3389/fnmol.2014.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Obermeier A, Lammers R, Wiesmuller KH, Jung G, Schlessinger J, Ullrich A. Identification of Trk binding sites for SHC and phosphatidylinositol 3′-kinase and formation of a multimeric signaling complex. J Biol Chem. 1993;268(31):22963–22966. [PubMed] [Google Scholar]
- 51.Ong SH, et al. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol Cell Biol. 2000;20(3):979–989. doi: 10.1128/MCB.20.3.979-989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gallick GE, Corn PG, Zurita AJ, Lin SH. Small-molecule protein tyrosine kinase inhibitors for the treatment of metastatic prostate cancer. Future Med Chem. 2012;4(1):107–119. doi: 10.4155/fmc.11.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Varkaris A, Katsiampoura AD, Araujo JC, Gallick GE, Corn PG. Src signaling pathways in prostate cancer. Cancer Metastasis Rev. 2014;33(2-3):595–606. doi: 10.1007/s10555-013-9481-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu J, Yu E. Insulin-like growth factor receptor-1 (IGF-IR) as a target for prostate cancer therapy. Cancer Metastasis Rev. 2014;33(2-3):607–617. doi: 10.1007/s10555-013-9482-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Acevedo VD, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12(6):559–571. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 56.Drake JM, et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc Natl Acad Sci U S A. 2012;109(5):1643–1648. doi: 10.1073/pnas.1120985109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drake JM, et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci U S A. 2013;110(49):E4762–E4769. doi: 10.1073/pnas.1319948110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faltermeier CM, et al. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proc Natl Acad Sci U S A. 2016;113(2):E172–E181. doi: 10.1073/pnas.1521674112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weeraratna AT, Arnold JT, George DJ, DeMarzo A, Isaacs JT. Rational basis for Trk inhibition therapy for prostate cancer. Prostate. 2000;45(2):140–148. doi: 10.1002/1097-0045(20001001)45:2<140::AID-PROS8>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 60.Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell. 2006;21(6):737–748. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 61.Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003;5(5):461–466. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- 62.Arévalo JC, et al. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron. 2006;50(4):549–559. doi: 10.1016/j.neuron.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 63.Sehat B, Andersson S, Girnita L, Larsson O. Identification of c-Cbl as a new ligase for insulin-like growth factor-I receptor with distinct roles from Mdm2 in receptor ubiquitination and endocytosis. Cancer Res. 2008;68(14):5669–5677. doi: 10.1158/0008-5472.CAN-07-6364. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi Y, et al. Ligand-induced downregulation of TrkA is partly regulated through ubiquitination by Cbl. FEBS Lett. 2011;585(12):1741–1747. doi: 10.1016/j.febslet.2011.04.056. [DOI] [PubMed] [Google Scholar]
- 65.Haglund K, Dikic I. The role of ubiquitylation in receptor endocytosis and endosomal sorting. J Cell Sci. 2012;125(pt 2):265–275. doi: 10.1242/jcs.091280. [DOI] [PubMed] [Google Scholar]
- 66.Fei C, et al. Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/β-catenin signaling. Mol Cell Biol. 2013;33(20):4095–4105. doi: 10.1128/MCB.00418-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou HL, Geng C, Luo G, Lou H. The p97-UBXD8 complex destabilizes mRNA by promoting release of ubiquitinated HuR from mRNP. Genes Dev. 2013;27(9):1046–1058. doi: 10.1101/gad.215681.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Birsa N, et al. Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J Biol Chem. 2014;289(21):14569–14582. doi: 10.1074/jbc.M114.563031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu J, et al. Rhbdd3 controls autoimmunity by suppressing the production of IL-6 by dendritic cells via K27-linked ubiquitination of the regulator NEMO. Nat Immunol. 2014;15(7):612–622. doi: 10.1038/ni.2898. [DOI] [PubMed] [Google Scholar]
- 70.Nucifora FC, et al. Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1. Nat Commun. 2016;7:11792. doi: 10.1038/ncomms11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011;42(1):9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh R, et al. Regulation of alternative splicing of Bcl-x by BC200 contributes to breast cancer pathogenesis. Cell Death Dis. 2016;7(6):e2262. doi: 10.1038/cddis.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang CH, et al. Mammary stem cells and tumor-initiating cells are more resistant to apoptosis and exhibit increased DNA repair activity in response to DNA damage. Stem Cell Reports. 2015;5(3):378–391. doi: 10.1016/j.stemcr.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.