

Abstract
Chemoreflex sensitization produced by chronic intermittent hypoxia (CIH) in rats is attenuated by angiotensin II type 1 receptor (AT1R) blockade. AT1R blockade and xanthine oxidase inhibition both ameliorate CIH-induced endothelial dysfunction. We hypothesized treatment with losartan and allopurinol would reduce chemoreflex sensitivity and improve hypoxic vasodilation in patients with obstructive sleep apnoea (OSA). Eighty-six hypertensive patients with apnoea-hypopnoea index ≥25 events/hr and no other cardiovascular, pulmonary, renal, or metabolic disease were randomly assigned to receive allopurinol, losartan, or placebo for 6 weeks. Treatment with other medications and/or continuous positive airway pressure (CPAP) remained unchanged. Tests of chemoreflex sensitivity and hypoxic vasodilation were performed during wakefulness before and after treatment. Ventilation (pneumotachography), muscle sympathetic nerve activity (microneurography), heart rate (electrocardiography), arterial oxygen saturation (pulse oximetry), blood pressure (sphygmomanometry), forearm blood flow (venous occlusion plethysmography), and cerebral flow velocity (transcranial Doppler ultrasound) were measured during eupneic breathing and graded reductions in inspired O2 tension. Losartan and allopurinol lowered arterial pressure measured during eupneic breathing and exposure to acute hypoxia. Neither drug altered the slopes of ventilatory, sympathetic, or cardiovascular responses to acute hypoxia. We conclude that losartan and allopurinol are viable pharmacotherapeutic adjuncts for achieving blood pressure control in hypertensive OSA patients, even those who are adequately treated with CPAP.
Keywords: chemoreflex, renin-angiotensin system, sympathetic nervous system
Introduction
Chemoreflex sensitivity is enhanced following exposure to chronic intermittent hypoxia (CIH) in experimental animals (Marcus et al., 2010; Prabhakar et al., 2015; Del Rio et al., 2016; Morgan et al., 2016a), healthy humans (Pialoux et al., 2009; Tamisier et al., 2011), and humans with obstructive sleep apnoea (OSA) (Narkiewicz et al., 1999; Mansukhani et al., 2015). Increased generation of reactive oxygen species such as superoxide is thought to contribute importantly to this sensitization (Pialoux et al., 2009; Del Rio et al., 2010; Lim et al., 2014; Semenza & Prabhakar, 2015; Morgan et al., 2016b). Potential sources of superoxide ion in the carotid body and other components of the extended chemoreflex arc include angiotensin II and xanthine oxidase. In the present study, we sought to determine whether angiotensin II type 1 receptor (AT1R) blockade with losartan and/or xanthine oxidase inhibition with allopurinol reduces chemoreflex sensitivity in humans with OSA. Impairments in local vascular regulation have also been described with CIH and OSA (Carlson et al., 1996; Phillips et al., 2004; El Solh et al., 2006; Reichmuth et al., 2009; Dopp et al., 2011). Therefore, another goal was to test the hypothesis that allopurinol and losartan, drugs that prevent CIH-induced endothelial dysfunction in rats (Dopp et al., 2011; Marcus et al., 2012), alter vasodilatory responses to hypoxia in humans with OSA. Because chemoreflex hypersensitivity and vascular dysfunction are putative contributors to OSA-associated hypertension (Fletcher et al., 1992a; Del Rio et al., 2014; Prabhakar, 2016; Khayat et al., 2018), we evaluated the effects of allopurinol and losartan on blood pressure.
Methods
Ethical approval
Studies conformed to the standards set by the latest version of the Declaration of Helsinki. All subjects provided written informed consent prior to participation. The protocol was approved by the University of Wisconsin Health Sciences Institutional Review Board (HS IRB 2012-0036). Other referral sites deferred oversight to the University of Wisconsin Health Sciences IRB. This research was registered at clinicaltrials.gov (NCT01637623) prior to recruitment of subjects.
Research design
We conducted a randomized comparison (parallel design; 1:1:1 allocation ratio) of the effects of allopurinol, losartan, and placebo on chemoreflex sensitivity, hypoxic vasodilation, and blood pressure. The primary outcome measure was muscle sympathetic nerve activity (MSNA) response to acute hypoxic exposure. Secondary outcome measures included hyperoxic inhibition of MSNA, and ventilatory and vasodilatory responses to hypoxia.
Subjects
Males and females, aged 21-65, were recruited at four medical centers in Wisconsin: UW Health Wisconsin Sleep in Madison, Gundersen Lutheran in LaCrosse, Aurora BayCare in Green Bay, and Marshfield Clinic in Marshfield. Subjects were required to have an apnoea-hypopnoea index ≥25 events/hr (determined at the referral site) and either a clinical diagnosis of hypertension or two recorded clinic blood pressure readings >140/90 in the previous 12 months.
Treatment with continuous positive airway pressure (CPAP) was allowed, provided that it had been used for a minimum of 3 months prior to participation. Four subjects did not use CPAP. The majority of remaining subjects (69%) were issued auto-titrating CPAP devices (REM Star, Phillips Respironics, Murrysville, PA, USA) for use during the study. The other CPAP users, primarily the ones for whom bi-level positive airway pressure was prescribed, continued to use their own machines during participation. Stable CPAP usage for one month prior to enrollment was documented using the machines’ electronic data cards. The median value for CPAP use duration prior to study enrollment was 11 months.
Potential subjects were excluded if they had overt cardiovascular disease other than hypertension, pulmonary disease with hypoxemia (arterial oxygen saturation [SaO2] <88%), hypertriglyceridemia (triglycerides >300 mg/dL), diabetes (fasting plasma glucose >125 mg/dL), kidney disease (serum creatinine >1.5 mg/dL), hyperkalemia (serum potassium >5.0 mEq/L), or history of adverse reaction to allopurinol or losartan. Also excluded were patients receiving angiotensin converting enzyme inhibitors, alpha-adrenergic and angiotensin receptor antagonists, potassium-sparing diuretics without accompanying loop/thiazide diuretics, allopurinol, oxypurinol, febuxostat, amoxicillin, ampicillin, azathioprine, or mercaptopurine. The first subject was enrolled on January 2, 2013 and the last subject was enrolled March 26, 2015.
Sample size determination
A priori, we estimated the sample size required for 80% power to detect the difference in MSNA:SaO2 slopes we previously observed in a pilot study of 5 patients with untreated OSA and 12 control subjects who were exposed to a progressive hypoxia protocol identical to the one in the current study. This difference was −1.30 vs. −0.54 bursts/min/% SaO2 or 0.76 slope “units”. Based on 10,000 trials of simulated data from similar distributions seen from our previous data, we estimated that 40 subjects per arm would be needed for ANOVA with a two-sided alpha level of 0.05. An updated sample size estimate was made based on the differences and standard deviations in the primary outcome variable existing at the time of a 1-year interim analysis. This calculation revealed that we would have 90% power (vs. 80% in the original sample size calculation) to find a significant between-group difference at a significance level of 0.017 (Bonferroni-adjusted alpha for 3 tests) if the trend continued and we had complete MSNA slope data from 20 subjects per group.
In-laboratory, overnight polysomnography
To ensure consistency in sleep-disordered breathing scoring, all subjects underwent a polysomnogram, without CPAP, in our laboratory. A 16-channel polysomnographic recording system (Tele-factor Heritage digital polysomnography systems, Grass Instruments, Quincy, MA, USA) was used to assess sleep states, and respiratory and cardiac variables. SaO2 was continuously recorded using pulse oximetry (model 3900, Datex-Ohmeda, Louisville, CO, USA). Oral and nasal airflow were detected using a nasal pressure transducer (Pro-Tech, Mukilteo, WA, USA) and Dymedix PVDF (polyvinylidene fluoride film). Rib cage and abdominal excursions were recorded using respiratory inductance plethysmography (Respitrace, Ambulatory Monitoring, Ardsley, NY). Each 30-s interval of the polysomnographic record was inspected visually for episodes of abnormal breathing. Apnoeas, hypopnoeas, and arousals were scored using American Academy of Sleep Medicine guidelines (Iber et al., 2007; Berry et al., 2012). Apnoea-hypopnoea index refers to the number of apnoeas and hypopneas per hour of objectively measured sleep. The respiratory disturbance index was defined as the number of apnoeas, hypopnoeas, and respiratory event-related arousals per hour of sleep. The percentage of time with SaO2 <88% was computed.
Randomization
Eligible subjects were stratified into categories based on their baseline CPAP use (Figure 1). As each subject was assigned to a stratum, a research pharmacist not otherwise involved in the study placed him or her into a treatment group using the next ascending unused randomization number within that stratum. Participants, care providers, investigators, and outcomes assessors were blinded to group assignment. Blinding was maintained through the use of identically appearing capsules in the 3 treatment groups. Figure 2 shows flow of participants through the study.
Figure 1.
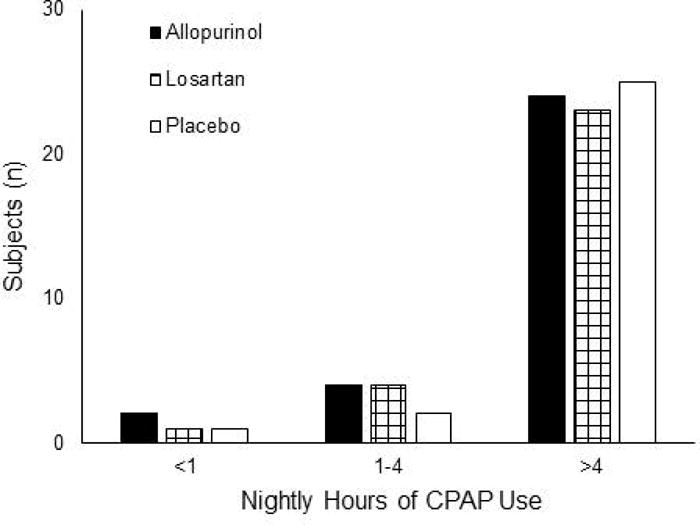
Numbers of subjects allocated to the three treatment groups from the randomization strata. Assignment into a stratum was based on average number of hours of nightly CPAP in the month prior to study enrollment.
Figure 2.
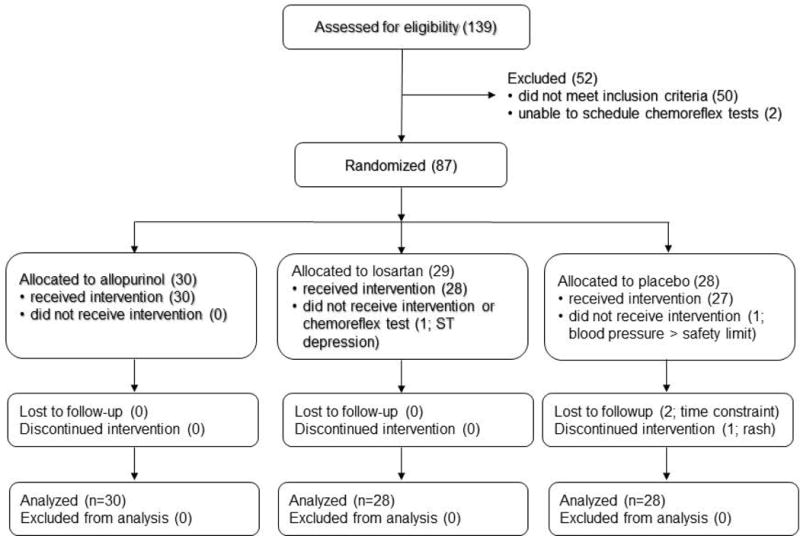
CONSORT diagram showing flow of participants through the study.
Cardiorespiratory measurements
We acquired heart rate from the electrocardiogram, SaO2 by pulse oximetry (Biox 3740; Ohmeda, Madison, WI, USA), MSNA by direct intraneural recordings (see below), and blood pressure by automated sphygmomanometry (Dinamap 1846SX/P; Critikon, Tampa, FL, USA). Mean arterial pressure (MAP) was calculated as 1/3 pulse pressure + diastolic pressure. Forearm blood flow was measured by venous occlusion plethysmography (Greenfield et al., 1963) and forearm vascular conductance (FVC) was calculated as forearm blood flow × 100/MAP, normalized for forearm volume in dL (calculated using the truncated cone formula). Cerebral flow velocity was measured in the M1 segment of the middle cerebral artery by transcranial Doppler ultrasound, as previously described (Reichmuth et al., 2009). Cerebrovascular conductance (CVC) was calculated as velocity-time integral/MAP.
Subjects breathed through a mouthpiece with the nose occluded. Airflow was measured with a heated pneumotachograph (#5719; Hans Rudolph, Kansas City, MO, USA), minute ventilation (VE), tidal volume (VT) and breathing frequency (fB) were calculated, and end-tidal PO2 (PETO2) and PCO2 (PETCO2) were measured by expired air analysis (S-3A/I and CD-3A; Ametek, Pittsburgh, PA, USA). Central respiratory “drive” was estimated by calculating the tidal volume:inspiratory time ratio (VT/Ti).
Muscle sympathetic nerve activity
Postganglionic MSNA was recorded from the fibular nerve using the technique of Vallbo et al. (Vallbo et al., 1979) as previously described (Khayat et al., 2004). Briefly, neural signals were passed to a differential preamplifier, an amplifier (total gain, 100,000), a band-pass filter (700-2,000 Hz), and an integrator (time constant, 100 msec). Placement of the recording electrode within a muscle fascicle was confirmed by: 1) presence of muscle twitches, but not paresthesias, in response to electrical stimulation; 2) pulse-synchronous nature of the nerve activity; 3) appearance of afferent activity in response to tapping or stretching of muscle, but not gentle stroking of skin, in the appropriate receptive fields; and 4) absence of neural activation in response to arousal stimuli. Once an acceptable nerve recording was obtained, the subject was instructed to maintain the leg in a relaxed position for the duration of the study. Acceptable neurograms were obtained in 79% of subjects at the time of baseline studies. If an acceptable recording was not acquired at baseline, we did not attempt to record nerve traffic at the time of follow-up. In 2 subjects, an acceptable recording was obtained at the baseline, but not the follow-up, study.
Sympathetic bursts were identified by computer-assisted inspection of the mean voltage neurogram using a custom-written program. Briefly, an MSNA delay factor was determined for each subject by interrogating the region of the neurogram 1.3±1 sec after the R wave of the electrocardiogram. Neural signals in this region were divided into twenty 50-msec bins, and the time, relative to the R wave, of the bin with the highest signal content became that subject’s delay factor. Then, a noise level was set by determining, for each cardiac cycle, the mean and standard deviation of the neural signal, excluding the area ±0.15 sec from the computed delay factor. Neural signals occurring within ±0.15 sec of the computed delay factor that were greater than the noise level +1 standard deviation were flagged as bursts. A single human observer (BJM) confirmed the computer-selected bursts in all neurograms in all subjects. For purposes of quantification, MSNA was expressed as burst frequency (bursts/min), burst incidence (bursts/100 heart beats), and total activity (burst frequency × mean burst amplitude).
Physiological assessment protocol
Respiratory and neurocirculatory data were acquired during two tests of chemoreflex function performed during wakefulness with subjects in the supine position. Testing occurred between 1100-1400 hours and room temperature was maintained at 21-24° C. First, chemoreflex-mediated inhibition of MSNA was assessed during four 1-min exposures to hyperoxia (FIO2, 1.0), which were separated by at least 3 min of room air breathing. Fifteen-sec averages of MSNA burst frequency and total activity during the 1-min exposure were computed, and a nadir value determined. The nadir values for the 4 hyperoxic trials in each subject were then averaged. Second, chemoreflex-mediated activation of MSNA and ventilation and hypoxic vasodilation was assessed during a steady-state hypoxia challenge consisting of 3 min of stable room air breathing, followed by three 3-min periods with SaO2 held constant at 90%, 85%, and 80%. Hypoxia was created by supplementation of the inspired air with nitrogen. CO2 was added to the inspired air, as needed, to maintain PETCO2 at the eupneic level. Heart rate and blood pressure responses to hypoxia and hypoxic vasodilation in the forearm and cerebral circulations were measured during the hypoxic challenge. During normoxia and graded hypoxia, 3-min averages were computed for all respiratory and neurocirculatory variables.
To determine whether high levels of ventilation produced by hypoxia per se affected MSNA (Somers et al., 1989; Narkiewicz et al., 1999), heart rate, and blood pressure, subjects completed a 3-min trial of voluntarily controlled breathing in normoxia with tidal volume and breathing frequency maintained at the highest levels reached during hypoxia. PETCO2 was maintained at the eupneic level. Subjects were assisted in maintaining target levels of tidal volume and frequency by visual and auditory feedback.
Both tests of chemoreflex function, assessment of hypoxic vasodilation, controlled breathing, and polysomnograms were repeated after 6 wk of drug treatment.
Drug treatment
Subjects received oral treatment with allopurinol (300 mg/d), losartan (50 or 100 mg/d), or placebo for 6 wk, beginning on the day following baseline assessments of chemoreflex function and hypoxic vasodilation. Losartan was initiated at 50 mg/d and increased to 100 mg/d after 2 wk if blood pressure was >100/60 mmHg. Adherence to study medications was assessed by pill count.
Data analysis
Chemoreflex responses to hypoxia were quantified by computing slopes of the linear relationships between ventilatory and neurocirculatory variables with SaO2. During the initial visit, one subject reported feeling panicky when SaO2 was 90% and could not complete the hypoxia trial. Data points for this subject were deemed outliers, with values up to 9 SDs away from the other subjects’ means. At follow-up, this subject was asked to complete an abbreviated test (normoxia and 90% SaO2 only), and did not report symptoms of panic. This subject’s outlying data were handled statistically by imputing MSNA and ventilatory data based on mean values from the other losartan subjects. A sensitivity analysis was completed based on collected data and imputed data with no major differences in hypothesis testing results.
Hyperoxic sympathoinhibition was quantified by expressing MSNA during exposure as a percentage of the normoxic baseline. Hypoxic vasodilation was quantified by computing slopes of the linear relationships between forearm and cerebral vascular conductance with SaO2.
Statistics
R version 3.3.1 statistical software was used. Between-group comparison of baseline characteristics was conducted with ANOVA or chi-square tests. Between-group comparisons of changes over time were conducted using repeated measures ANOVA (RM-ANOVA) with group, time, and group-by-time interaction as fixed effects, and subject as a random effect. This analysis was performed with and without controlling for CPAP use, measured as average nightly use in minutes, as a covariate. If both methods resulted in similar conclusions, only results obtained with CPAP adherence as a covariate are reported. Post-hoc two-way comparisons were conducted using Tukey’s adjustment for family-wise testing. All analyses were done according to the intent-to-treat principle. “On-protocol” analyses yielded similar conclusions; therefore, only results of intent-to-treat analyses are reported. Because gender differences have been observed in chemoreflex sensitivity (Lozo et al., 2017), hypoxic vasodilation (Casey et al., 2014), and sympathetic regulation of vascular tone (Hinojosa-Laborde et al., 1999), the possibility of differential gender-specific effects was examined using an explorative analysis, adjusted for CPAP adherence.
Results
Participation was terminated after the initial visit in one subject because of excessive blood pressure and in another due to scalp rash. Two other subjects voluntarily withdrew after the initial visit due to time constraints. All four subjects had been randomized to receive placebo. Following the intent-to-treat principle, data collected from these subjects are included in the analyses. One subject who had been allocated to the losartan group did not participate in physiological assessments and did not receive the intervention secondary to ECG abnormalities detected at the initial visit.
Subject characteristics before and after interventions were comparable in the 3 groups (Table 1). No changes were observed in height, weight, body mass index, and the polysomnographic variables. Plasma uric acid levels were reduced by allopurinol (−1.83 [−2.15, −1.50; 95% confidence interval] mg/dL), whereas they remained stable in the losartan (−0.21 [−0.54, 0.12] mg/dL) and placebo (+0.05 [−0.33, 0.42] mg/dL) groups.
Table 1.
Subject characteristics by randomized group at baseline and after interventions. Data shown are mean (SD), median (interquartile range), or n (%). RM-ANOVA revealed no statistically significant differences for group-by-time interaction.
| Allopurinol (n=30) |
Losartan (n=28) |
Placebo (n=28) | P-value* | P-value¶ | |
|---|---|---|---|---|---|
| Sex (female) | 10 (33%) | 15 (54%) | 9 (32%) | 0.180 | ----- |
|
| |||||
| Age (yr) | 49 (8) | 49 (12) | 47 (9) | 0.677 | ----- |
|
| |||||
| Body mass index (kg/m2) | |||||
| Initial | 38.2 (7.2) | 38.6 (7.4) | 34.5 (6.6) | 0.061 | |
| Final | 38.6 (7.1) | 39.1 (7.6) | 34.8 (6.4) | 0.062 | 0.579 |
|
| |||||
| Height (m) | |||||
| Initial | 1.74 (0.10) | 1.74 (0.11) | 1.73 (0.11) | 0.851 | |
| Final | 1.74 (0.10) | 1.74 (0.10) | 1.73 (0.11) | 0.893 | 0.952 |
|
| |||||
| Weight (kg) | |||||
| Initial | 116.6 (26.4) | 118.5 (30.4) | 103.3 (22.5) | 0.071 | |
| Final | 117.2 (25.6) | 119.5 (30.7) | 103.9 (21.8) | 0.078 | 0.504 |
|
| |||||
| Apnoea-hypopnoea index (events/hr) | |||||
| 33 (24-50) | 46 (27-58) | 35 (18-59) | 0.653 | ||
| 45 (22-68) | 44 (25-67) | 26 (15-62) | 0.358 | 0.070 | |
|
| |||||
| Respiratory distress index (events/hr) | |||||
| Initial | 37 (25-51) | 49 (27-60) | 42 (24-61) | 0.734 | |
| Final | 50 (22-69) | 50 (25-67) | 31 (20-64) | 0.445 | 0.074 |
|
| |||||
| Time <88% SaO2 (%) | |||||
| Initial | 5 (1-15) | 10 (4-25) | 5 (1-9) | 0.208 | |
| Final | 8 (2-18) | 12 (2-35) | 3 (1-8) | 0.097 | 0.269 |
|
| |||||
| Fasting blood glucose (mg/dL) | |||||
| Initial | 94 (15) | 96 (10) | 91 (10) | 0.330 | |
| Final | † | † | † | ----- | |
|
| |||||
| Triglycerides (mg/dL) | |||||
| Initial | 145 (56) | 150 (49) | 139 (62) | 0.759 | |
| Final | † | † | † | ----- | |
post-intervention measurements not obtained (these were inclusion criteria)
P-value for between-group comparison at each time point
P-value for group-by-time interaction, adjusted for average daily CPAP use (min)
CPAP pressures, adherence, and residual apnoea-hypopnoea indices did not differ among groups and remained stable throughout the trial (Table 2). Adherence to study medications, assessed by pill counts, did not differ among groups (97±8 [SD], 99±3, and 97±5% in allopurinol-, losartan-, and placebo-treated subjects, respectively). The percentages of subjects taking antihypertensive medications in the allopurinol, losartan, and placebo groups, respectively, were as follows: diuretics, 33, 29, and 4%; beta-adrenergic blockers, 23, 11, and 14%; and calcium channel blockers, 13, 7, and 4%. The types and doses of concomitant antihypertensive agents did not change during the study.
Table 2.
Continuous positive airway pressure (CPAP) treatment parameters in the randomized groups prior to the initial and final visits. Data shown are mean (SD), median (interquartile range), or n (%).
| Allopurinol | Losartan | Placebo | P-value* | P-value¶ | |
|---|---|---|---|---|---|
| PAP (cm/H2O) | |||||
| Initial | 11.0 (3.0) | 10.1 (2.5) | 9.8 (1.6) | 0.217 | |
| Final | 11.2 (3.1) | 9.8 (2.4) | 9.5 (1.2) | 0.066 | 0.449 |
|
| |||||
| CPAP use (hr/night)† | |||||
| Initial | 5.4 (2.6) | 5.6 (2.1) | 5.9 (2.0) | 0.717 | |
| Final | 4.9 (2.8) | 5.5 (2.5) | 5.1(2.7) | 0.690 | 0.579 |
|
| |||||
| Residual AHI (events/hr)† | |||||
| Initial | 2.0 (1.6–3.4) | 2.2 (1.1–3.2) | 2.0 (1.2–3.0) | 0.898 | |
| Final | 2.2 (1.0-3.0) | 2.5 (1.7-4.0) | 2.3 (1.2-3.2) | 0.838 | 0.629 |
|
| |||||
| Poor compliance (n)‡ | |||||
| Initial | 7 (23.3%) | 5 (17.9%) | 3 (10.7%) | 0.466 | |
| Final | 10 (33.3%) | 6 (21.4%) | 7 (25.0%) | 0.619 | 0.449 |
PAP, positive airway pressure; AHI, apnoea-hypopnoea index
recorded during nightly CPAP use
<4 hours of CPAP use per night
P-value for between-group comparison at each time point
P-value for group-by-time interaction
Drug treatment and baseline (normoxic) cardiorespiratory values (Table 3)
Table 3.
Baseline (normoxic) values measured prior to graded hypoxia exposure at initial and final visits. Data shown are mean (SD).
| Allopurinol | Losartan | Placebo | P-value* | P-value¶ | |
|---|---|---|---|---|---|
| MSNA (bursts/min) | |||||
| Initial | 26.7 (12.0) | 20.6 (9.8) | 22.5 (11.1) | 0.189 | |
| Final | 26.4 (11.4) | 27.3 (12.4) | 21.7 (10.8) | 0.267 | 0.130 |
|
| |||||
| MSNA (bursts/100 heart beats) | |||||
| Initial | 39 (17) | 30 (14) | 33 (18) | 0.192 | |
| Final | 39 (17) | 40 (15) | 32 (16) | 0.184 | 0.110 |
|
| |||||
| Heart rate (beats/min) | |||||
| Initial | 68.8 (9.2) | 70.0 (10.1) | 72.5 (11.2) | 0.390 | |
| Final | 69.3 (8.8) | 70.1 (10.1) | 71.8 (11.9) | 0.665 | 0.887 |
|
| |||||
| MAP (mmHg) | |||||
| Initial | 94.1(11.1) | 94.1(8.3) | 97.4(10.4) | 0.371 | |
| Final | 89.5(10.4) | 84.0(8.1) | 97.5(10.1) | < 0.001x,y,z | < 0.001x,y,z |
|
| |||||
| Systolic pressure (mmHg) | |||||
| Initial | 136.9 (18.3) | 136.7 (14.3) | 139.7 (16.4) | 0.744 | |
| Final | 129.5 (15.2) | 122.0 (14.2) | 139.6 (14.1) | < 0.001y,z | < 0.001x,y,z |
|
| |||||
| Diastolic pressure (mmHg) | |||||
| Initial | 72.7(9.1) | 72.8(8.1) | 75.9(9.3) | 0.301 | |
| Final | 69.5(9.7) | 64.7(6.6) | 76.6(9.5) | < 0.001x,y,z | 0.001x,z |
|
| |||||
| FVC (ml/min/dL/mmHg) | |||||
| Initial | 0.43 (0.20) | 0.41 (0.21) | 0.49 (0.31) | 0.420 | |
| Final | 0.55 (0.31) | 0.42 (0.17) | 0.51 (0.29) | 0.185 | 0.522 |
|
| |||||
| CVC (velocity-time integral/mmHg) | |||||
| Initial | 0.27 (0.97) | 0.32 (0.13) | 0.33 (0.11) | 0.150 | |
| Final | 0.31 (0.12) | 0.36 (0.14) | 0.34 (0.12) | 0.479 | 0.139 |
|
| |||||
| VE (liters/min) | |||||
| Initial | 9.3 (2.1) | 8.9 (1.7) | 9.3 (2.1) | 0.692 | |
| Final | 9.8 (2.6) | 8.9 (2.1) | 8.8 (1.9) | 0.181 | 0.095 |
|
| |||||
| VT (liters) | |||||
| Initial | 0.66 (0.27) | 0.64 (0.21) | 0.72 (0.23) | 0.458 | |
| Final | 0.69 (0.21) | 0.66 (0.22) | 0.61 (0.17) | 0.434 | 0.028 |
|
| |||||
| fB (breaths/min) | |||||
| Initial | 15.8 (5.0) | 15.3 (4.1) | 14.5 (4.6) | 0.483 | |
| Final | 16.1 (5.9) | 15.0 (4.0) | 15.6 (4.3) | 0.680 | 0.420 |
|
| |||||
| VT/Ti (l/sec) | |||||
| Initial | 0.38 (0.09) | 0.35 (0.06) | 0.36 (0.08) | 0.443 | |
| Final | 0.40 (0.11) | 0.36 (0.08) | 0.34 (0.07) | 0.041y | 0.090 |
|
| |||||
| SaO2 (%) | |||||
| Initial | 95.6 (1.2) | 95.5 (0.9) | 95.6 (1.0) | 0.895 | |
| Final | 95.7 (1.0) | 95.3 (1.1) | 95.2 (1.0) | 0.120 | 0.183 |
|
| |||||
| PETCO2 (mmHg) | |||||
| Initial | 41.4 (4.0) | 41.8 (3.8) | 41.6 (3.3) | 0.923 | |
| Final | 41.9 (3.7) | 42.3 (3.1) | 42.9 (3.6) | 0.591 | 0.503 |
MSNA, muscle sympathetic nerve activity; MAP, mean arterial pressure; FVC, forearm vascular conductance; CVC, cerebral vascular conductance; VE, minute ventilation; VT, tidal volume; fB, breathing frequency; VT/Ti, tidal volume;inspiratory time ratio; SaO2, arterial oxygen saturation; PETCO2, end-tidal CO2 tension
P-value for between-group comparison at each time point
P-value for group-by-time interaction, adjusted for average daily CPAP use (min)
Pairwise comparisons with Holm adjustment:
p<0.05, allopurinol vs. losartan;
p<0.05, allopurinol vs. placebo;
p<0.05, losartan vs. placebo
Before treatment initiation, all values were comparable in the 3 groups. After 6 wk of treatment, MAP in normoxia prior to chemoreflex testing was significantly reduced in the losartan and allopurinol groups, whereas it remained stable in placebo subjects. The blood pressure lowering effect of losartan was greater than that of allopurinol. The pattern of statistical significance for reductions in systolic pressure mirrored that of MAP; in contrast, diastolic pressure was reduced relative to placebo only by losartan. No other between-group differences were observed.
Within the losartan group, we observed statistically significant increases in baseline MSNA burst frequency (+6.5 [0.4, 12.6] bursts/min) and burst incidence (+0.10 [0.01, 0.18] bursts/100 heart beats); however, these changes were not statistically different from changes in the allopurinol or placebo groups. Within the allopurinol group, baseline FVC increased significantly (+0.12 [0.03, 0.21] units/dL); however, this change was not statistically different from change in the losartan or placebo groups. Within the losartan group, baseline CVC increased significantly (+0.462 [0.006, 0.087]); however, this change was not statistically different from changes in the allopurinol and placebo groups. These within-group changes are apparent in Figure 3, where the 95% confidence intervals for these variables do not contain zero.
Figure 3.
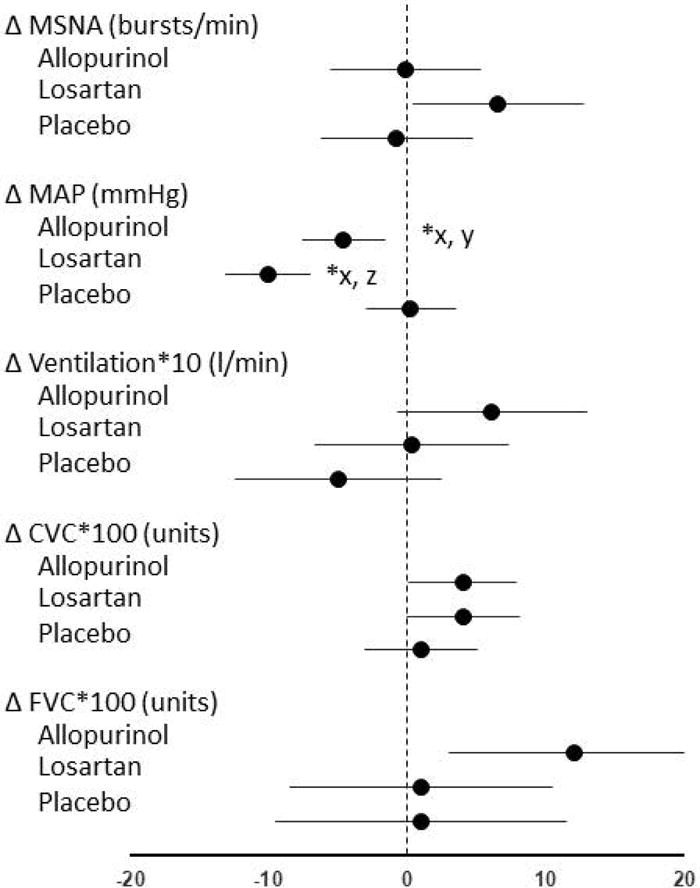
Observed changes in baseline values for primary and secondary outcome measures (mean, 95% CI) in the 3 treatment groups. Mean arterial pressure (MAP) was reduced, relative to placebo, with allopurinol and losartan treatments. The reduction produced by allopurinol was smaller than that caused by losartan. No statistically significant differences in other variables were observed. FVC and CVC values were multiplied by 100 and VE values were multiplied by 10 so that the data points are discernable on these axes.
MSNA, muscle sympathetic nerve activity; MAP, mean arterial pressure; CVC, cerebral vascular conductance; FVC, forearm vascular conductance
*p<0.05, group-by-time interaction
Pairwise comparisons with Holm adjustment: xp<0.05, allopurinol vs. losartan; yp<0.05, allopurinol vs. placebo; zp<0.05, losartan vs. placebo
Drug treatment and chemoreflex responsiveness (Table 4)
Table 4.
Slopes (absolute values) of neurocirculatory and ventilatory responses to graded hypoxia at initial and final visits. Data shown are mean (SD).
| Allopurinol | Losartan | Placebo | P-value* | P-value¶ | |
|---|---|---|---|---|---|
| MSNA (bursts/min/% SaO2) | |||||
| Initial | −0.84 (0.47) | −0.54 (0.29) | −0.71 (0.43) | 0.075 | |
| Final | −0.66 (0.38) | −0.69 (0.50) | −0.61 (0.40) | 0.831 | 0.095 |
|
| |||||
| MSNA (bursts/100 heart beats/% SaO2) | |||||
| Initial | −0.006 (0.005) | −0.003 (0.003) | −0.005 (0.005) | 0.293 | |
| Final | −0.004 (0.005) | −0.004 (0.006) | −0.004 (0.005) | 0.951 | 0.376 |
|
| |||||
| Heart rate (beats/min/% SaO2) | |||||
| Initial | −0.85 (0.31) | −0.88 (0.30) | −0.84 (0.38) | 0.902 | |
| Final | −0.82 (0.34) | −0.80 (0.31) | −0.83 (0.33) | 0.966 | 0.493 |
|
| |||||
| MAP (mmHg/% SaO2) | |||||
| Initial | −0.17 (0.45) | −0.29 (0.42) | −0.26 (0.40) | 0.540 | |
| Final | −0.22 (0.42) | −0.38 (0.31) | −0.19 (0.29) | 0.121 | 0.382 |
|
| |||||
| FVC (ml/min/dL/mmHg/% SaO2) | |||||
| Initial | −0.009 (0.008) | −0.003 (0.006) | −0.009 (0.014) | 0.058 | |
| Final | −0.008 (0.010) | −0.005 (0.007) | −0.009 (0.018) | 0.481 | 0.769 |
|
| |||||
| CVC (velocity-time integral/mmHg/% SaO2) | |||||
| Initial | −0.40 (0.29) | −0.31 (0.19) | −0.39 (0.31) | 0.472 | |
| Final | −0.27 (0.27) | −0.12 (0.32) | −0.30 (0.17) | 0.080 | 0.765 |
|
| |||||
| VE (liters/min/% SaO2) | |||||
| Initial | −0.53 (0.41) | −0.41 (0.28) | −0.44 (0.29) | 0.390 | |
| Final | −0.43 (0.37) | −0.37 (0.25) | −0.48 (0.32) | 0.471 | 0.298 |
|
| |||||
| VT (liters/% SaO2) | |||||
| Initial | −0.03 (0.03) | −0.02 (0.02) | −0.03 (0.03) | 0.521 | |
| Final | −0.03 (0.03) | −0.02 (0.02) | −0.03 (0.02) | 0.266 | 0.322 |
|
| |||||
| fB (breaths/min/% SaO2) | |||||
| Initial | −0.06 (0.22) | −0.07 (0.20) | −0.03 (0.16) | 0.687 | |
| Final | −0.03 (0.23) | −0.07 (0.18) | −0.05 (0.18) | 0.768 | 0.609 |
|
| |||||
| VT/Ti (l/sec/% SaO2) | |||||
| Initial | −0.02 (0.01) | −0.02 (0.01) | −0.02 (0.01) | 0.453 | |
| Final | −0.02 (0.01) | −0.01 (0.01) | −0.02 (0.01) | 0.549 | 0.273 |
MSNA, muscle sympathetic nerve activity; MAP, mean arterial pressure; FVC, forearm vascular conductance; CVC, cerebral vascular conductance; VE, minute ventilation; VT, tidal volume; fB, breathing frequency; VT/Ti, tidal volume;inspiratory time ratio
P-value for between-group comparison at each time point
P-value for group-by-time interaction, adjusted for average daily CPAP use (min)
Acute exposure to hypoxia caused brisk, progressive increases in VE, caused mainly by increases in VT. There were no between-group differences in the relationships between VE or its components and SaO2 at either time point. Treatment-related changes in VE:SaO2 relationships did not differ among groups.
Acute exposure to graded hypoxia produced progressive sympathetic activation. We observed no between-group differences in the slopes of MSNA:SaO2 relationships, either at the initial visit or after drug treatment. Treatment-related changes in MSNA slopes did not differ among groups.
Exposure to hyperoxia resulted in a rapid rise in PETO2 to >250 mmHg within 2.3 [0.7] breaths or 12.4 [4.5] sec. Within the losartan group, hyperoxic inhibition of total MSNA was significantly reduced after treatment (−11.2 [0.2, −22.7] %; Figure 4), but this change was not statistically different from change in the allopurinol (−0.7 [8.9, −10.2] %) or placebo (−2.3 [7.9, −12.4]) groups.
Figure 4.
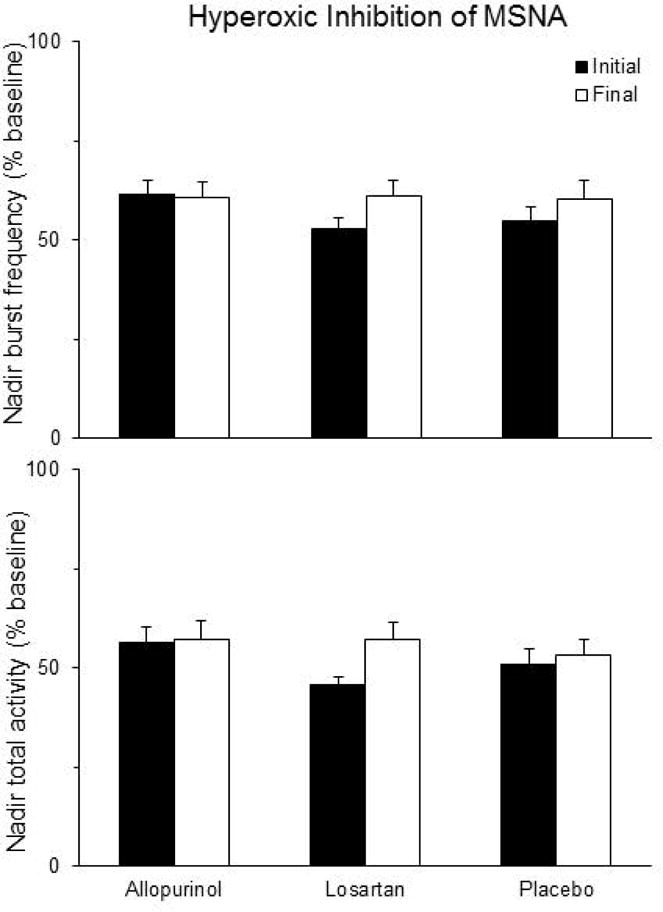
Brief hyperoxic exposure decreased muscle sympathetic activity (MSNA) in all treatment groups. Within the losartan group, the amount of hyperoxic inhibition of total MSNA activity was diminished significantly after 6 weeks of treatment, but this change wasn’t statistically different from changes in the allopurinol or placebo group (RM-ANOVA p>0.05). Data shown are mean±SEM.
Haemodynamic responses to acute hypoxia (Table 4)
Hypoxia elicited modest increases in MAP and heart rate. We observed no between-group differences in the slopes of the MAP or heart rate responses to hypoxia, either at the initial visit or after drug treatment. In the forearm and cerebral circulations, acute hypoxic exposure produced vasodilation. Treatment-related changes in hypoxic vasodilation did not differ among groups.
Influence of hyperpnoea on MSNA
When subjects voluntarily reproduced, in normoxia, the same elevated levels of ventilation elicited by SaO2 reduction to 80%, MSNA was the same as during eupnea (RM-ANOVA p>0.05; Figure 5). Likewise, neither heart rate nor MAP differed in voluntary hyperpnoea vs. eupnea (data not shown).
Figure 5.
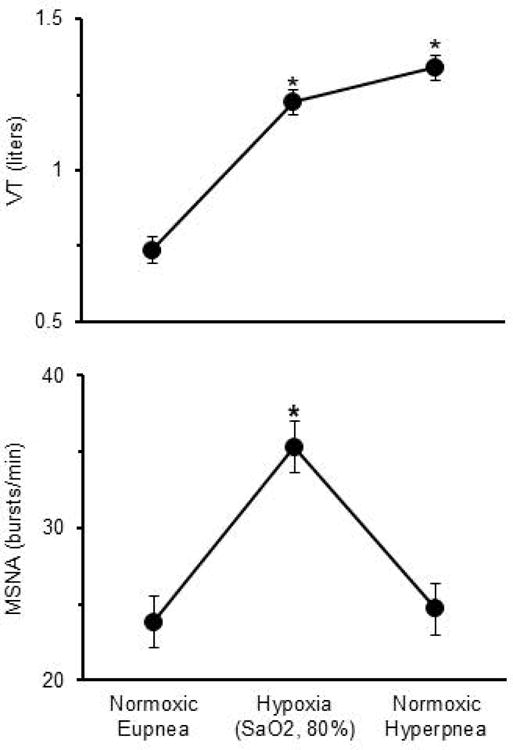
Tidal volume (VT) and muscle sympathetic nerve activity (MSNA) during normoxia, hypoxia (SaO2, 80%), and when subjects voluntarily mimicked, in normoxia, the same levels of VT and breathing frequency elicited by the hypoxic exposure. MSNA was comparable during normoxic eupnea and normoxic hyperpnoea, indicating that increased ventilatory volumes and rates (not shown), per se, have no effect on MSNA. Data shown are mean±SEM.
*p<0.05 vs. normoxic eupnea
Gender subgroup analysis
Allopurinol- and losartan-induced reductions in baseline MAP were present in both male (RM-ANOVA p=0.001) and female (RM-ANOVA p=0.050) subjects. Absence of treatment effects on baseline ventilation, heart rate, and MSNA was observed in both males and females (data not shown). In females, baseline FVC increased significantly with allopurinol, whereas it remained stable in losartan- and placebo-treated females (RM-ANOVA p=0.009; Figure 6). Baseline FVC in males was not affected by any treatment (RM-ANOVA p=0.248). Baseline CVC did not differ among groups, either in males or females (Figure 6). No gender effects were noted in chemoreflex sensitivity or hypoxic vasodilation (data not shown).
Figure 6.
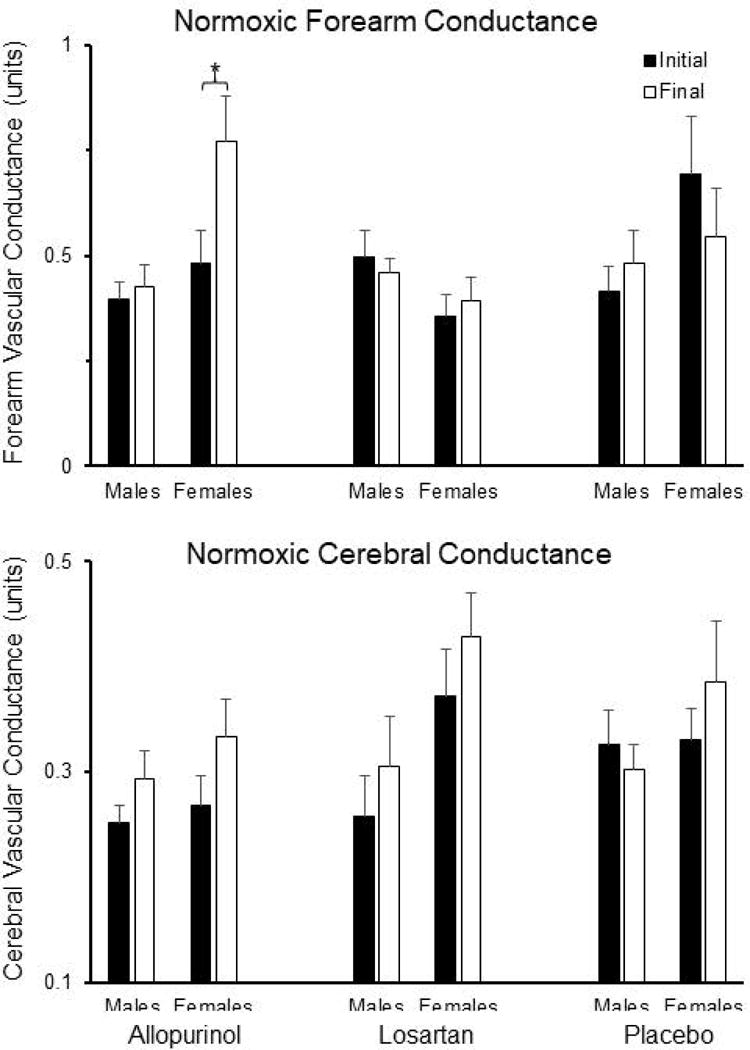
Upper panel shows initial and final values for normoxic forearm vascular conductance (FVC), separated by sex, in the three treatment groups. In female subjects, baseline FVC was significantly increased by allopurinol, but was unaffected by losartan and placebo treatments (RM-ANOVA p=0.009). Baseline FVC did not change with any treatment in male subjects. Lower panel shows initial and final values for normoxic cerebral vascular conductance (CVC), separated by sex, in the three treatment groups. CVC did not change with any treatment and subgroup analysis revealed no sex differences. Data shown are mean±SEM.
Discussion
We evaluated the effects of xanthine oxidase inhibition with allopurinol and angiotensin II receptor antagonism with losartan on respiratory and neurocirculatory function in hypertensive patients with moderate-to-severe OSA. The major finding is that both drugs significantly lowered blood pressure during normoxic, eupneic breathing and during acute exposure to hypoxia without reducing sympathetic vasoconstrictor activity. In contrast to our hypotheses, based on previous findings in rats exposed to CIH (Marcus et al., 2010; Dopp et al., 2011; Marcus et al., 2012; Morgan et al., 2016a; Morgan et al., 2016b), the slopes of the chemoreflex and vasodilatory responses to acute hypoxia were not altered by losartan or allopurinol.
Insufficient statistical power is an unlikely explanation for our negative findings regarding chemoreflex responsivity. A priori, we established a “clinically relevant difference” for the MSNA:SaO2 relationship (0.76 slope “units”) by calculating the difference in slope between patients with untreated OSA and healthy control subjects in a pilot study. In the present study, 95% confidence intervals surrounding change in MSNA slope were sufficiently narrow (~±0.31) to detect a difference of this magnitude, had one existed.
We considered the possibility that augmented hypoxic ventilatory drive obscured treatment-related changes in the sympathoexcitatory response, via the sympatholytic effect proposed by previous investigators (Somers et al., 1989; Narkiewicz et al., 1999); however, none of the treatments altered hypoxic ventilatory responses. Moreover, we found no evidence of sympathetic inhibition when subjects voluntarily mimicked, in normoxia, the same high levels of ventilation elicited by acute hypoxia, either before or after drug treatment (Figure 5).
In the current subjects, sympathetic burst frequency and incidence were lower under control conditions than in untreated OSA patients previously described, and in fact, were comparable in magnitude to values reported for control subjects without OSA (Carlson et al., 1993; Waradekar et al., 1996; Narkiewicz et al., 1999; Donadio et al., 2007; Imadojemu et al., 2007; Grassi et al., 2014; Goya et al., 2016; Henderson et al., 2016). Excessive daytime sleepiness, which is associated with sympathetic hyperactivity (Donadio et al., 2007) and cardiovascular morbidity (Feng et al., 2012) in patients with OSA, was absent in the majority of our subjects. We suspect that the stringent exclusion criteria used to minimize effects of co-morbid conditions, along with concurrent CPAP usage, resulted in a relatively healthy cohort with normal chemoreflex responsiveness.
For ethical reasons, we did not ask our subjects to delay or discontinue CPAP treatment for the duration of the study. Instead, we requested that subjects maintain customary CPAP use patterns during participation. CPAP adherence was relatively high (5.6 hr/night) and was indeed stable over the study period. Continued effectiveness of clinically recommended pressure settings was demonstrated by low and stable residual apnoea-hypopnoea indices (Table 2). Concurrent CPAP usage resulted in nightly intermittent hypoxia exposures substantially lower than those suggested by the subjects’ pre-CPAP apnoea-hypopnoea indices. Therefore, concurrent CPAP usage allowed by our experimental design may have obscured the beneficial effects we were seeking. Nevertheless, our design allowed us to evaluate the cardiorespiratory effects of the drugs in a common clinical scenario, i.e. partial-night CPAP use, where some intermittent hypoxia episodes remain untreated (Weaver & Grunstein, 2008).
Chronic intermittent hypoxia in rodents as a model for human sleep apnea
Perhaps the positive effects of losartan and allopurinol on chemosensitivity and hypoxic vasodilation we previously demonstrated in CIH-exposed rats were not observed in this group of human subjects simply because CIH is a poor model for OSA. It is certainly true that the model fails to mimic all aspects of OSA (e.g. no upper airway occlusion, no concurrent hypercapnia, no sleep disruption); nevertheless, our CIH paradigm features a temporal pattern of cyclical decreases in SaO2 (i.e. slow desaturations followed by rapid resaturations) that is “human-like” (Lim et al., 2014; Morgan et al., 2016a; Morgan et al., 2016b). Moreover, our CIH paradigm is a good model for the cardiorespiratory consequences OSA, because both produce diurnal increases in arterial pressure (Fletcher et al., 1992b; Becker et al., 2003; Marcus et al., 2009), sympathetic activation (Carlson et al., 1993; Marcus et al., 2010), impaired endothelium-dependent vasodilation (Carlson et al., 1996; Phillips et al., 2004), increased arterial stiffness (Phillips et al., 2005; Phillips et al., 2006), and evidence of oxidative stress (Lavie, 2015; Morgan et al., 2016b). Accordingly, we believe that hypotheses based on our rat data were not upheld in humans, not because of a failure of the model per se, but rather because the total daily “dose” of intermittent hypoxia in our rat model was more severe than in our human subjects, leading to more pronounced ventilatory and neurocirculatory impairments. Our rats were exposed to CIH for 10-12 hours/day (15 events/hour) whereas the majority of our human subjects were adequately treated with CPAP (5-6 hours of CPAP use/night; residual apnoea-hypopnoea index, 2-3 events/hour). In our previous rat studies, allopurinol and losartan, which were administered prior to and during exposure to CIH, were observed to prevent chemoreflex sensitization and impaired vascular function. In the present human study, we investigated the ability of these agents to reverse OSA-induced abnormalities in cardiorespiratory regulation. The outcomes may have been different if allopurinol and losartan had been administered at the time of OSA diagnosis.
Mechanisms of chemoreflex hypersensitivity caused by intermittent hypoxia
We previously demonstrated that chemoreflex hypersensitivity caused by CIH in rats is dependent on AT1R signaling in the carotid body (Marcus et al., 2010). In CIH-exposed pheochromocytoma cells, xanthine oxidase is a critical component of reactive oxygen species-generating pathways that activate hypoxia inducible factor-1α and degrade hypoxia inducible factor-2α (Nanduri et al., 2013; Nanduri et al., 2015). If xanthine oxidase were to play an analogous role in carotid body, it could be another important determinant of chemoreflex sensitivity. The present observations in humans with OSA do not provide support for these notions, however, because neither treatment (AT1R blockade nor xanthine oxidase inhibition), altered chemoreflex control of ventilation or sympathetic vasoconstrictor outflow. We would like to emphasize, however, that marked chemoreflex hyperresponsiveness was not present in our hypertensive, but otherwise-healthy, OSA patients.
Allopurinol and losartan contribute to blood pressure control in patients with OSA
Despite the absence of treatment effects on chemoreflex and vascular responses to hypoxia, our treatments significantly reduced blood pressure. Average values for systolic pressure in both groups were reduced from the “Stage 1 Hypertension” to the “Elevated” category, as defined in recent guidelines (Whelton et al., 2017), with greater reductions in the losartan than in the allopurinol group. Average diastolic pressure, which was not elevated at baseline in either group, was reduced relative to placebo only by losartan.
Antihypertensive effects of losartan (Thunström et al., 2016) and valsartan (Pépin et al., 2010) in patients with OSA have been reported previously. In the present losartan-treated subjects, 68% of whom received 100 mg/day, changes in blood pressure are comparable to those produced by 160 mg/day valsartan (Pépin et al., 2010) and slightly larger than those seen with 50 mg/day losartan (Thunström et al., 2016). Our study extends the previous findings via inclusion of a placebo-treated control group and by demonstrating a potent antihypertensive effect of allopurinol.
Allopurinol decreases blood pressure in patients with hyperuricemia, essential hypertension, and chronic kidney disease (Feig et al., 2008; Satirapoj et al., 2015; Qu et al., 2017); however, an antihypertensive effect of allopurinol has not been demonstrated in OSA. The mechanism of this effect in our subjects is unlikely to be a reduction in sympathetic tone, because MSNA was not affected by allopurinol. Heart rate did not change with allopurinol; however, we cannot speak to possible cardiac output effects. In a rat model of hypertension, allopurinol lowered blood pressure, reduced cardiac function (dp/dtmax), and enhanced vasodilation in isolated arteries (El-Bassossy et al., 2018). Our observation of increased forearm vascular conductance in allopurinol-treated subjects, at least in females, is consistent with this notion and with the observation by previous investigators that allopurinol improves endothelium-dependent vasodilation in OSA patients (El Solh et al., 2006).
Losartan-induced blood pressure lowering and increased MSNA
In losartan-treated subjects, baseline MSNA burst frequency increased by ~33%, which is more than twice the size of the previously reported day-to-day variability in this measurement (Vallbo et al., 1979). An analogous effect of eprosartan was observed in young healthy males (Heusser et al., 2003); however, losartan either decreased (Béchir et al., 2005) or did not affect MSNA and whole-body norepinephrine spillover (Krum et al., 2006) in patients with essential hypertension. This discrepancy may be explained by the smaller daily dose of losartan and smaller blood pressure reductions in the previous study (Krum et al., 2006). We attribute sympathetic activation in our subjects to baroreflex unloading elicited by losartan-induced decreases in blood pressure. Changes in MSNA and MAP were moderately correlated (r=-0.67) in losartan-treated, but not allopurinol- or placebo-treated subjects (r=−0.11 and −0.07). Although MSNA increased significantly in losartan-treated subjects, between-group differences were insignificant, perhaps due to lack of statistical power. We cannot rule out this possibility; nevertheless, losartan-induced increases in baseline MSNA may have clinical importance.
Methodologic considerations
Our blood pressure measurements were made immediately prior to laboratory tests of chemoreflex and vascular function. They are averages of 3-5 measurements made at 1-min intervals after at least 45 min of supine rest. Further studies are required to determine the effects of allopurinol and losartan on 24-h blood pressure profiles.
We did not have the scheduling flexibility to test female subjects at a standardized phase of the menstrual cycle. Ventilation (Macnutt et al., 2012), MSNA (Minson et al., 2000; Carter et al., 2013), and ventilatory (Schoene et al., 1981; Macnutt et al., 2012) and sympathoexcitatory (Usselman et al., 2015) responses to hypoxia vary across the menstrual cycle, whereas vascular responses to hypoxia remain stable (Minson et al., 2000). Thus, hormone fluctuations may have obscured or amplified treatment effects on ventilation and MSNA in our pre-menopausal subjects (47% of female participants), but it is unlikely that they affected vascular conductance. Allopurinol-induced increases in forearm vascular conductance were comparable in pre- and post-menopausal females, as were the effects of drug treatment on all other ventilatory and neurocirculatory variables.
Changes in Doppler velocity reflect changes in volume flow only when cross-sectional area of the insonated vessel remains constant. We did not measure middle cerebral artery diameter; however, diameter changes minimally during fluctuations in arterial pressure, fraction of inspired CO2, and gravitational stress (Giller et al., 1993; Serrador et al., 2000) and a strong correlation between middle cerebral artery velocity and volume flow has been demonstrated (Kirkham et al., 1986).
We recorded sympathetic outflow to vascular structures in the leg. This neural discharge is representative of sympathetic outflow to skeletal muscle vascular beds throughout the body (Rea & Wallin, 1989); however, our findings cannot be extrapolated to other organs or vascular beds.
Conclusions
Six weeks of treatment with losartan and allopurinol lowered arterial pressure in hypertensive patients with moderate-to-severe OSA who were, on average, adherent to CPAP treatment. The effects of CPAP alone on arterial pressure are variable (Becker et al., 2003; Campos-Rodriguez et al., 2006; Robinson et al., 2006) and, at best, modest (Hu et al., 2015; Sun et al., 2016). Optimal blood pressure control in OSA often requires additional pharmacotherapy; however, no specific class of antihypertensive agents has been judged to be superior (Ahmad et al., 2017). We propose that both allopurinol and losartan are viable therapeutic options for patients with OSA, even those who are adequately treated with CPAP.
New Findings.
What is the central question of this study? In sleep apnoea, a putative link between intermittent hypoxia and hypertension is generation of oxygen radicals by angiotensin II and xanthine oxidase within the chemoreflex arc and vasculature. We tested whether chemoreflex control of sympathetic outflow, hypoxic vasodilation, and blood pressure are altered by angiotensin blockade (losartan) and/or xanthine oxidase inhibition (allopurinol).
What is the main finding and its importance? Both drugs lowered blood pressure without altering sympathetic outflow, reducing chemoreflex sensitivity, or enhancing hypoxic vasodilation. Losartan and allopurinol are effective therapies for achieving blood pressure control in sleep apnoea.
Acknowledgments
The authors would like to thank: Jacqueline K. Limberg, PhD of the University of Missouri for her insightful comments on the manuscript; Jaime A. Boero, MD of Marshfield Clinic, Daren G. Tobert, MD of Gundersen Lutheran Clinic, and Taylor McGinnis of Aurora BayCare Medical Center for help with subject recruitment; Robert J. Kotloski, MD and Marianne Hines, NP-C of UW Health for performing medical histories and physical exams; Christine Sorkness, PharmD of the UW Institute for Clinical and Translational Research for executive oversight; and Howard H. Bailey, MD of the Wisconsin Network for Health Research for clinical and administrative support. We are appreciative of the skillful assistance of Rich Severin, Tricia Denman, Debbie Friedland, Amanda Rasmuson, and Meghan Hackbarth in data collection and analysis. We thank Philips Respironics for the generous loan of positive airway pressure equipment.
Funding
National Heart, Lung, and Blood Institute (UO-1 HL 105365) and the Clinical and Translational Science Award (CTSA) program, through the NIH National Center for Advancing Translational Sciences (NCATS), grant UL1TR000427. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing interests
None of the authors have competing interests to report.
Author contributions
These experiments were performed in the James B. Skatrud Pulmonary/Sleep Laboratory at the William S. Middleton Veterans Administration Hospital. Author contributions are as follows: 1) concept/design (BJM, MT, SJH, JMD); 2) acquisition, analysis, interpretation (BJM, MT, DFP, ERJ, DLS, DTP, JPG, SJH, JMD); and 3) drafting or revision of manuscript (BJM, MT, DFP, ERJ, DLS, DTP, JPG, SJH, JMD). All authors have approved the final version of the manuscript and all agree to be accountable for all aspects of the work. All authors qualify for authorship and all who qualify are listed.
References
- Ahmad M, Makati D, Akbar S. Review of and Updates on Hypertension in Obstructive Sleep Apnea. Int J Hypertens. 2017;2017:1848375. doi: 10.1155/2017/1848375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker HF, Jerrentrup A, Ploch T, Grote L, Penzel T, Sullivan CE, Peter JH. Effect of nasal continuous positive airway pressure treatment on blood pressure in patients with obstructive sleep apnea. Circulation. 2003;107:68–73. doi: 10.1161/01.cir.0000042706.47107.7a. [DOI] [PubMed] [Google Scholar]
- Berry RB, Budhiraja R, Gottlieb DJ, Gozal D, Iber C, Kapur VK, Marcus CL, Mehra R, Parthasarathy S, Quan SF, Redline S, Strohl KP, Davidson Ward SL, Tangredi MM, Medicine AAoS Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8:597–619. doi: 10.5664/jcsm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béchir M, Enseleit F, Chenevard R, Lüscher TF, Noll G. Effect of losartan on muscle sympathetic activity and baroreceptor function in systemic hypertension. Am J Cardiol. 2005;95:129–131. doi: 10.1016/j.amjcard.2004.08.079. [DOI] [PubMed] [Google Scholar]
- Campos-Rodriguez F, Grilo-Reina A, Perez-Ronchel J, Merino-Sanchez M, Gonzalez-Benitez MA, Beltran-Robles M, Almeida-Gonzalez C. Effect of continuous positive airway pressure on ambulatory BP in patients with sleep apnea and hypertension: a placebo-controlled trial. Chest. 2006;129:1459–1467. doi: 10.1378/chest.129.6.1459. [DOI] [PubMed] [Google Scholar]
- Carlson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest. 1993;103:1763–1768. doi: 10.1378/chest.103.6.1763. [DOI] [PubMed] [Google Scholar]
- Carlson JT, Rångemark C, Hedner JA. Attenuated endothelium-dependent vascular relaxation in patients with sleep apnoea. J Hypertens. 1996;14:577–584. doi: 10.1097/00004872-199605000-00006. [DOI] [PubMed] [Google Scholar]
- Carter JR, Fu Q, Minson CT, Joyner MJ. Ovarian cycle and sympathoexcitation in premenopausal women. Hypertension. 2013;61:395–399. doi: 10.1161/HYPERTENSIONAHA.112.202598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Shepherd JR, Joyner MJ. Sex and vasodilator responses to hypoxia at rest and during exercise. J Appl Physiol (1985) 2014;116:927–936. doi: 10.1152/japplphysiol.00409.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio R, Andrade DC, Lucero C, Arias P, Iturriaga R. Carotid Body Ablation Abrogates Hypertension and Autonomic Alterations Induced by Intermittent Hypoxia in Rats. Hypertension. 2016;68:436–445. doi: 10.1161/HYPERTENSIONAHA.116.07255. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Iturriaga R. Carotid body and cardiorespiratory alterations in intermittent hypoxia: the oxidative link. Eur Respir J. 2010;36:143–150. doi: 10.1183/09031936.00158109. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Iturriaga R. Carotid body potentiation during chronic intermittent hypoxia: implication for hypertension. Front Physiol. 2014;5:434. doi: 10.3389/fphys.2014.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadio V, Liguori R, Vetrugno R, Contin M, Elam M, Wallin BG, Karlsson T, Bugiardini E, Baruzzi A, Montagna P. Daytime sympathetic hyperactivity in OSAS is related to excessive daytime sleepiness. J Sleep Res. 2007;16:327–332. doi: 10.1111/j.1365-2869.2007.00602.x. [DOI] [PubMed] [Google Scholar]
- Dopp JM, Philippi NR, Marcus NJ, Olson EB, Bird CE, Moran JJ, Mueller SW, Morgan BJ. Xanthine oxidase inhibition attenuates endothelial dysfunction caused by chronic intermittent hypoxia in rats. Respiration. 2011;82:458–467. doi: 10.1159/000329341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Solh AA, Saliba R, Bosinski T, Grant BJ, Berbary E, Miller N. Allopurinol improves endothelial function in sleep apnoea: a randomised controlled study. Eur Respir J. 2006;27:997–1002. doi: 10.1183/09031936.06.00101005. [DOI] [PubMed] [Google Scholar]
- El-Bassossy HM, Mahmoud MF, Eid BG. The vasodilatory effect of allopurinol mediates its antihypertensive effect: Effects on calcium movement and cardiac hemodynamics. Biomed Pharmacother. 2018;100:381–387. doi: 10.1016/j.biopha.2018.02.033. [DOI] [PubMed] [Google Scholar]
- Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA. 2008;300:924–932. doi: 10.1001/jama.300.8.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, He QY, Zhang XL, Chen BY, Sleep Breath Disorder Group ScoRM Epworth Sleepiness Scale may be an indicator for blood pressure profile and prevalence of coronary artery disease and cerebrovascular disease in patients with obstructive sleep apnea. Sleep Breath. 2012;16:31–40. doi: 10.1007/s11325-011-0481-5. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol (1985) 1992a;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Qian W, Miller CC, Unger T. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension. 1992b;19:555–561. doi: 10.1161/01.hyp.19.6.555. [DOI] [PubMed] [Google Scholar]
- Giller CA, Bowman G, Dyer H, Mootz L, Krippner W. Cerebral arterial diameters during changes in blood pressure and carbon dioxide during craniotomy. Neurosurgery. 1993;32:737–741. discussion 741-732. [PubMed] [Google Scholar]
- Goya TT, Silva RF, Guerra RS, Lima MF, Barbosa ER, Cunha PJ, Lobo DM, Buchpiguel CA, Busatto-Filho G, Negrão CE, Lorenzi-Filho G, Ueno-Pardi LM. Increased Muscle Sympathetic Nerve Activity and Impaired Executive Performance Capacity in Obstructive Sleep Apnea. Sleep. 2016;39:25–33. doi: 10.5665/sleep.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Brambilla G, Buzzi S, Volpe M, Cesana F, Dell’oro R, Mancia G. Regional differences in sympathetic activation in lean and obese normotensive individuals with obstructive sleep apnoea. J Hypertens. 2014;32:383–388. doi: 10.1097/HJH.0000000000000034. [DOI] [PubMed] [Google Scholar]
- Greenfield AD, Whitney RJ, Mowbray JF. Methods for the investigation of peripheral blood flow. Br Med Bull. 1963;19:101–109. doi: 10.1093/oxfordjournals.bmb.a070026. [DOI] [PubMed] [Google Scholar]
- Henderson LA, Fatouleh RH, Lundblad LC, McKenzie DK, Macefield VG. Effects of 12 Months Continuous Positive Airway Pressure on Sympathetic Activity Related Brainstem Function and Structure in Obstructive Sleep Apnea. Front Neurosci. 2016;10:90. doi: 10.3389/fnins.2016.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusser K, Vitkovsky J, Raasch W, Schmieder RE, Schobel HP. Elevation of sympathetic activity by eprosartan in young male subjects. Am J Hypertens. 2003;16:658–664. doi: 10.1016/s0895-7061(03)00917-8. [DOI] [PubMed] [Google Scholar]
- Hinojosa-Laborde C, Chapa I, Lange D, Haywood JR. Gender differences in sympathetic nervous system regulation. Clin Exp Pharmacol Physiol. 1999;26:122–126. doi: 10.1046/j.1440-1681.1999.02995.x. [DOI] [PubMed] [Google Scholar]
- Hu X, Fan J, Chen S, Yin Y, Zrenner B. The role of continuous positive airway pressure in blood pressure control for patients with obstructive sleep apnea and hypertension: a meta-analysis of randomized controlled trials. J Clin Hypertens (Greenwich) 2015;17:215–222. doi: 10.1111/jch.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iber C, Ancoli-Israel S, Chesson A, SF Q. The AASM manual for the scoring of sleep and associated events: rules, terminology, and technical specifications. American Academy of Sleep Medicine; Westchester, IL: 2007. [Google Scholar]
- Imadojemu VA, Mawji Z, Kunselman A, Gray KS, Hogeman CS, Leuenberger UA. Sympathetic chemoreflex responses in obstructive sleep apnea and effects of continuous positive airway pressure therapy. Chest. 2007;131:1406–1413. doi: 10.1378/chest.06-2580. [DOI] [PubMed] [Google Scholar]
- Khayat RN, Przybylowski T, Meyer KC, Skatrud JB, Morgan BJ. Role of sensory input from the lungs in control of muscle sympathetic nerve activity during and after apnea in humans. J Appl Physiol. 2004;97:635–640. doi: 10.1152/japplphysiol.00241.2004. [DOI] [PubMed] [Google Scholar]
- Khayat RN, Varadharaj S, Porter K, Sow A, Jarjoura D, Gavrilin MA, Zweier JL. Angiotensin Receptor Expression and Vascular Endothelial Dysfunction in Obstructive Sleep Apnea. Am J Hypertens. 2018;31:355–361. doi: 10.1093/ajh/hpx174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkham FJ, Padayachee TS, Parsons S, Seargeant LS, House FR, Gosling RG. Transcranial measurement of blood velocities in the basal cerebral arteries using pulsed Doppler ultrasound: velocity as an index of flow. Ultrasound Med Biol. 1986;12:15–21. doi: 10.1016/0301-5629(86)90139-0. [DOI] [PubMed] [Google Scholar]
- Krum H, Lambert E, Windebank E, Campbell DJ, Esler M. Effect of angiotensin II receptor blockade on autonomic nervous system function in patients with essential hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H1706–1712. doi: 10.1152/ajpheart.00885.2005. [DOI] [PubMed] [Google Scholar]
- Lavie L. Oxidative stress in obstructive sleep apnea and intermittent hypoxia–revisited–the bad ugly and good: implications to the heart and brain. Sleep Med Rev. 2015;20:27–45. doi: 10.1016/j.smrv.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Lim DC, Brady DC, Po P, Chuang LP, Marcondes L, Kim EY, Keenan BT, Guo X, Maislin G, Galante RJ, Pack AI. Simulating obstructive sleep apnea patients’ oxygenation characteristics into a mouse model of cyclical intermittent hypoxia. J Appl Physiol (1985) 2014 doi: 10.1152/japplphysiol.00629.2014. jap.00629.02014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozo T, Komnenov D, Badr MS, Mateika JH. Sex differences in sleep disordered breathing in adults. Respir Physiol Neurobiol. 2017;245:65–75. doi: 10.1016/j.resp.2016.11.001. [DOI] [PubMed] [Google Scholar]
- Macnutt MJ, De Souza MJ, Tomczak SE, Homer JL, Sheel AW. Resting and exercise ventilatory chemosensitivity across the menstrual cycle. J Appl Physiol (1985) 2012;112:737–747. doi: 10.1152/japplphysiol.00727.2011. [DOI] [PubMed] [Google Scholar]
- Mansukhani MP, Wang S, Somers VK. Chemoreflex physiology and implications for sleep apnoea: insights from studies in humans. Exp Physiol. 2015;100:130–135. doi: 10.1113/expphysiol.2014.082826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Li YL, Bird CE, Schultz HD, Morgan BJ. Chronic intermittent hypoxia augments chemoreflex control of sympathetic activity: role of the angiotensin II type 1 receptor. Respir Physiol Neurobiol. 2010;171:36–45. doi: 10.1016/j.resp.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Olson EB, Bird CE, Philippi NR, Morgan BJ. Time-dependent adaptation in the hemodynamic response to hypoxia. Respir Physiol Neurobiol. 2009;165:90–96. doi: 10.1016/j.resp.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Philippi NR, Bird CE, Li YL, Schultz HD, Morgan BJ. Effect of AT1 receptor blockade on intermittent hypoxia-induced endothelial dysfunction. Respir Physiol Neurobiol. 2012;183:67–74. doi: 10.1016/j.resp.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minson CT, Halliwill JR, Young TM, Joyner MJ. Influence of the menstrual cycle on sympathetic activity, baroreflex sensitivity, and vascular transduction in young women. Circulation. 2000;101:862–868. doi: 10.1161/01.cir.101.8.862. [DOI] [PubMed] [Google Scholar]
- Morgan BJ, Adrian R, Wang ZY, Bates ML, Dopp JM. Chronic intermittent hypoxia alters ventilatory and metabolic responses to acute hypoxia in rats. J Appl Physiol (1985) 2016a doi: 10.1152/japplphysiol.00015.2016. jap.00015.02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BJ, Bates ML, Rio RD, Wang Z, Dopp JM. Oxidative stress augments chemoreflex sensitivity in rats exposed to chronic intermittent hypoxia. Respir Physiol Neurobiol. 2016b;234:47–59. doi: 10.1016/j.resp.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Vaddi DR, Khan SA, Wang N, Makarenko V, Semenza GL, Prabhakar NR. HIF-1α activation by intermittent hypoxia requires NADPH oxidase stimulation by xanthine oxidase. PLoS One. 2015;10:e0119762. doi: 10.1371/journal.pone.0119762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Vaddi DR, Khan SA, Wang N, Makerenko V, Prabhakar NR. Xanthine oxidase mediates hypoxia-inducible factor-2α degradation by intermittent hypoxia. PLoS One. 2013;8:e75838. doi: 10.1371/journal.pone.0075838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Phillips C, Hedner J, Berend N, Grunstein R. Diurnal and obstructive sleep apnea influences on arterial stiffness and central blood pressure in men. Sleep. 2005;28:604–609. doi: 10.1093/sleep/28.5.604. [DOI] [PubMed] [Google Scholar]
- Phillips SA, Olson EB, Lombard JH, Morgan BJ. Chronic intermittent hypoxia alters NE reactivity and mechanics of skeletal muscle resistance arteries. J Appl Physiol (1985) 2006;100:1117–1123. doi: 10.1152/japplphysiol.00994.2005. [DOI] [PubMed] [Google Scholar]
- Phillips SA, Olson EB, Morgan BJ, Lombard JH. Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol. 2004;286:H388–393. doi: 10.1152/ajpheart.00683.2003. [DOI] [PubMed] [Google Scholar]
- Pialoux V, Hanly PJ, Foster GE, Brugniaux JV, Beaudin AE, Hartmann SE, Pun M, Duggan CT, Poulin MJ. Effects of exposure to intermittent hypoxia on oxidative stress and acute hypoxic ventilatory response in humans. Am J Respir Crit Care Med. 2009;180:1002–1009. doi: 10.1164/rccm.200905-0671OC. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Carotid body chemoreflex: a driver of autonomic abnormalities in sleep apnoea. Exp Physiol. 2016;101:975–985. doi: 10.1113/EP085624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Kumar GK, Nanduri J. Peripheral chemoreception and arterial pressure responses to intermittent hypoxia. Compr Physiol. 2015;5:561–577. doi: 10.1002/cphy.c140039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pépin JL, Tamisier R, Barone-Rochette G, Launois SH, Lévy P, Baguet JP. Comparison of continuous positive airway pressure and valsartan in hypertensive patients with sleep apnea. Am J Respir Crit Care Med. 2010;182:954–960. doi: 10.1164/rccm.200912-1803OC. [DOI] [PubMed] [Google Scholar]
- Qu LH, Jiang H, Chen JH. Effect of uric acid-lowering therapy on blood pressure: systematic review and meta-analysis. Ann Med. 2017;49:142–156. doi: 10.1080/07853890.2016.1243803. [DOI] [PubMed] [Google Scholar]
- Rea RF, Wallin BG. Sympathetic nerve activity in arm and leg muscles during lower body negative pressure in humans. J Appl Physiol (1985) 1989;66:2778–2781. doi: 10.1152/jappl.1989.66.6.2778. [DOI] [PubMed] [Google Scholar]
- Reichmuth KJ, Dopp JM, Barczi SR, Skatrud JB, Wojdyla P, Hayes D, Morgan BJ. Impaired vascular regulation in patients with obstructive sleep apnea: effects of continuous positive airway pressure treatment. Am J Respir Crit Care Med. 2009;180:1143–1150. doi: 10.1164/rccm.200903-0393OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GV, Smith DM, Langford BA, Davies RJ, Stradling JR. Continuous positive airway pressure does not reduce blood pressure in nonsleepy hypertensive OSA patients. Eur Respir J. 2006;27:1229–1235. doi: 10.1183/09031936.06.00062805. [DOI] [PubMed] [Google Scholar]
- Satirapoj B, Wirajit O, Burata A, Supasyndh O, Ruangkanchanasetr P. Benefits of Allopurinol Treatment on Blood Pressure and Renal Function in Patients with Early Stage of Chronic Kidney Disease. J Med Assoc Thai. 2015;98:1155–1161. [PubMed] [Google Scholar]
- Schoene RB, Robertson HT, Pierson DJ, Peterson AP. Respiratory drives and exercise in menstrual cycles of athletic and nonathletic women. J Appl Physiol Respir Environ Exerc Physiol. 1981;50:1300–1305. doi: 10.1152/jappl.1981.50.6.1300. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Prabhakar NR. Neural Regulation of Hypoxia-Inducible Factors and Redox State Drives the Pathogenesis of Hypertension in a Rodent Model of Sleep Apnea. J Appl Physiol (1985) 2015 doi: 10.1152/japplphysiol.00162.2015. jap.00162.02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke. 2000;31:1672–1678. doi: 10.1161/01.str.31.7.1672. [DOI] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Zavala DC, Abboud FM. Influence of ventilation and hypocapnia on sympathetic nerve responses to hypoxia in normal humans. J Appl Physiol (1985) 1989;67:2095–2100. doi: 10.1152/jappl.1989.67.5.2095. [DOI] [PubMed] [Google Scholar]
- Sun Y, Huang ZY, Sun QR, Qiu LP, Zhou TT, Zhou GH. CPAP therapy reduces blood pressure for patients with obstructive sleep apnoea: an update meta-analysis of randomized clinical trials. Acta Cardiol. 2016;71:275–280. doi: 10.2143/AC.71.3.3152087. [DOI] [PubMed] [Google Scholar]
- Tamisier R, Pépin JL, Rémy J, Baguet JP, Taylor JA, Weiss JW, Lévy P. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J. 2011;37:119–128. doi: 10.1183/09031936.00204209. [DOI] [PubMed] [Google Scholar]
- Thunström E, Manhem K, Rosengren A, Peker Y. Blood Pressure Response to Losartan and Continuous Positive Airway Pressure in Hypertension and Obstructive Sleep Apnea. Am J Respir Crit Care Med. 2016;193:310–320. doi: 10.1164/rccm.201505-0998OC. [DOI] [PubMed] [Google Scholar]
- Usselman CW, Gimon TI, Nielson CA, Luchyshyn TA, Coverdale NS, Van Uum SH, Shoemaker JK. Menstrual cycle and sex effects on sympathetic responses to acute chemoreflex stress. Am J Physiol Heart Circ Physiol. 2015;308:H664–671. doi: 10.1152/ajpheart.00345.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallbo AB, Hagbarth KE, Torebjörk HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev. 1979;59:919–957. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- Waradekar NV, Sinoway LI, Zwillich CW, Leuenberger UA. Influence of treatment on muscle sympathetic nerve activity in sleep apnea. Am J Respir Crit Care Med. 1996;153:1333–1338. doi: 10.1164/ajrccm.153.4.8616563. [DOI] [PubMed] [Google Scholar]
- Weaver TE, Grunstein RR. Adherence to continuous positive airway pressure therapy: the challenge to effective treatment. Proc Am Thorac Soc. 2008;5:173–178. doi: 10.1513/pats.200708-119MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA, Williamson JD, Wright JT. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2017 doi: 10.1016/j.jacc.2017.11.006. [DOI] [PubMed] [Google Scholar]