

Abstract
Pompe disease is caused by mutations in acid alpha glucosidase (GAA) that causes accumulation of lysosomal glycogen affecting the heart and skeletal muscles, and can be fatal. Enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA) improves muscle function by reducing glycogen accumulation. Limitations of ERT include a short half-life and the formation of antibodies that result in reduced efficacy. By harnessing the immune tolerance induction properties of the liver, liver-targeted gene delivery (with an adeno-associated virus vector containing a liver specific promoter), suppresses immunity against the GAA introduced by gene therapy. This induces immune tolerance to rhGAA by activating regulatory T cells and simultaneously, corrects GAA deficiency. Potentially, liver-targeted gene therapy can be performed once with lasting effects, by administering a relatively low dose of an adeno-associated virus type 8 vector to replace and induce immune tolerance to GAA.
Keywords: Pompe disease, immune tolerance, gene therapy, glycogen storage disease, antibody response, acid alpha-glucosidase, enzyme replacement therapy
Graphical abstract
1.0 Introduction
Pompe disease is an inherited rare disorder (approximately 1 in every 20,000 births) caused by mutations in the gene for the enzyme acid alpha glucosidase (GAA) that disables the heart and skeletal muscles, and is often fatal [1–3]. Enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA) has been shown to decrease heart size, maintain normal heart function, improve muscle function, tone, and strength, and reduce glycogen accumulation. The recommended dose is 20mg/kg every 2 weeks (Myozyme product insert; https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125141s219lbl.pdf), yet the dose used in the clinical setting is often up to 40mg/kg/week [4, 5]. ERT is a lifesaving therapy, yet there are several challenges at this time, including immunogenicity and hypersensitivity such that ERT carries a black-box warning about anaphylaxis in the labeling. Although ERT has prolonged survival in the majority of patients with infantile Pompe disease, many long term sequelae are noted despite high dose ERT. Among the poor responders to ERT are those who formed high, sustained anti-rhGAA IgG antibody titers (HSAT). Patients with HSAT demonstrated greatly increased mortality, in comparison with patients who formed no or low titer antibodies [6]. Cross-reacting immune material (CRIM) negative patients, who lack any residual GAA protein are at highest risk for developing HSAT, yet close to 30% of CRIM positive infantile patients also develop HSAT. Some adult patients with late-onset Pompe disease (LOPD) formed HSAT during ERT, which reduced efficacy [7, 8]. Immune modulation with immune suppressant drugs to prevent anti-rhGAA antibody formation has significantly prolonged the survival in CRIM-negative infants, when it was initiated at the time of ERT. However, there are associated risks (such as increased infection and cancer rate) with the currently used immune tolerance induction (ITI) regimens [9, 10].
2.0 Immunomodulatory Gene Therapy for Pompe Disease
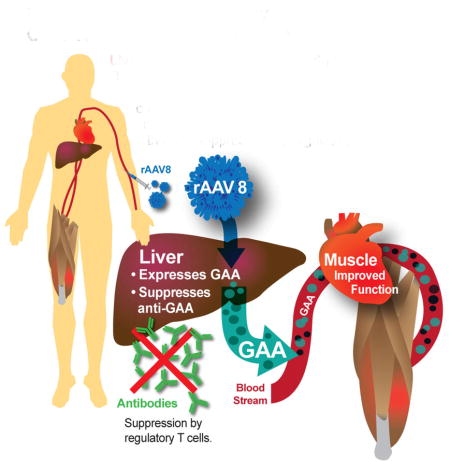
Liver-targeted gene therapy has been proposed to induce immune tolerance to rhGAA and correct GAA deficiency systemically in patients with Pompe disease (Fig. 1). The liver is an attractive target for gene therapies because its contains specialized cells which mediate the tolerogenic response [11]. These tolerogenic properties can be harnessed to prevent the immune system from destroying the foreign, yet beneficial, elements introduced in gene therapy. To that end, an adeno associated virus (AAV) 2 vector has been used for liver-targeted delivery of AAV factor IX vectors to treat hemophilia, but did not achieve stable expression in the plasma of patients due to T cell response to the to the vector capsid [12]. Changing the vector capsid to AAV8 had significant improvement in clinical outcomes [13]. In the latter study, Nathwani and colleagues successfully controlled the anti-capsid T cell responses against AAV8 with transient immune suppression, thereby preserving transgene expression and achieving long-term benefits [13]. AAV8 features enhanced liver tropism, with reduced off-target biodistribution, low seroprevalence and minimal cross-reactivity with other serotypes [14]. Liver depot gene therapy with a recombinant (r) AAV8 vector could treat hemophilia, lysosomal storage disorders, and metabolic diseases involving the liver, which could provide new therapy for genetic diseases on a global scale. The strategy of inducing immune tolerance with an AAV has been termed “immunomodulatory gene therapy” [15]. Liver transduction with a high-expressing AAV vector could effectively cure Pompe disease by creating a stable liver depot for GAA production and tolerizing the immune system to rhGAA.
Figure 1. Immunomodulatory Gene Therapy for Pompe Disease.

Liver depot with AAV2/8-LSPhGAA. Treatment with rAAV8 converts the liver to a depot for continuous secretion of GAA, correcting GAA deficiency in the heart and skeletal muscle. Liver expression induces immune tolerance to rhGAA by reducing antibody formation through suppression by regulatory T cells.
3.0 Mechanism for Immunomodulatory Gene Therapy for Pompe Disease
Preclinical experiments have be used demonstrated the ability of gene therapy to modulate the immune response in mice with Pompe disease. In GAA-knock out (KO) mice ERT was efficacious only in the setting of immune tolerance to GAA following AAV vector administration [16]. Preventing HSAT reduced mortality from hypersensitivity that had occurred during ERT in GAA-KO mice. One tolerogenic vector, AAV2/8-LSPhGAA, induced immune tolerance by expressing GAA exclusively in the liver and by activating regulatory T cells (Tregs), which has consistently induced immune tolerance to human GAA in preclinical experiments [15–18].
A model for immunomodulatory gene therapy recognizes the central role of Treg cells and hepatic transgene expression [19]. The precise mechanism of Treg cell stimulation has not been elucidated. It has been proposed that naïve CD4+ T cells interact with liver sinusoidal endothelial cells to differentiate to an anti-inflammatory phenotype, secreting IL-4 and IL-10 [20]. Kuppfer cells and natural killer T (NKT) cells also might be involved in stimulating Treg cells. Previously the depletion of Kupffer cells prevented tolerance induction via portal vein administration of an antigen [21]. Furthermore, stimulation of Kupffer cells was associated with Treg activation in response to hepatic gene expression [22], and this mechanism could play a role in immunomodulatory gene therapy. Additionally, NKT cells may also play a role in stimulating Treg cells, because these cells were activated in liver in the setting of immune suppression [20, 23].
Several factors determine the ability to avoid immune responses against the transgene through liver-specific expression [19]. Liver-specific expression of human coagulation factor IX (hFIX) in mice with hemophilia B prevented antibody formation in response to an immune challenge with hFIX [24]. Furthermore, the adoptive transfer of CD4+CD25+ cells (including Treg cells) to naïve recipient mice, following administration of the AAV vector expressing human hFIX to donor mice, prevented antibody formation in response to an immune challenge with hFIX [24]. An AAV2/8 vector containing a liver-specific regulatory cassette to drive α-galactosidase expression induced immune tolerance to α-galactosidase in Fabry disease mice, and the transfer of splenocytes from vector-treated mice prevented the antibody response against an α-galactosidase challenge in recipient Fabry mice [25]. Finally, anti-CD25 administration prevented the induction of immune tolerance with our LSP-containing vector in GAA-KO mice, presumably be depleting Treg cells [15]. Importantly, immune tolerance induced by AAV2/8-LSPhGAApA prevented antibody responses against ERT with rhGAA, when the AAV vector was administered simultaneously or even shortly before ERT [15, 16, 26]. Taken together, these data strongly support the ability of an AAV vector containing a liver-specific regulatory cassette to induce immune tolerance to an introduced foreign protein in the form of ERT [15, 16].
4.0 Improving Treatment through Liver-Targeted Immunomodulation
ERT does not target skeletal muscle efficiently resulting in the need for repeated dosing and a number of long term sequelae [27–29]. The development of liver depot gene therapy for Pompe disease might advance a new paradigm for the treatment of genetic diseases with protein replacement (Table 1). Rather than frequent infusions of a recombinant protein, as in ERT, gene therapy with a rAAV8 vector will be performed once with long-lasting effects. In the initial clinical application of immunomodulatory gene therapy, liver-specific expression of the therapeutic protein will prevent neutralizing antibody responses against the therapeutic protein. This strategy will induce specific immune tolerance to GAA in Pompe disease with a low dosage of an rAAV8 vector containing a liver-specific promoter (LSP) that expressed GAA only in liver and induced immune tolerance to GAA [15, 16], thereby providing a safe immune tolerance induction (ITI) without the risk for immune suppression or other toxic effects of current drug regimens [9]. Direct comparisons of ERT with AAV vector-mediated production of GAA have shown advantages for gene therapy [30, 31]
Table 1.
Comparison of ERT with immunomodulatory liver depot gene therapy.
| ERT | Potential benefits of immunomodulatory gene therapy* | |
|---|---|---|
| Stability | Short half-life in blood, requiring repeated administration (every week to 2 weeks) | Continuous GAA secretion in bloodstream |
| GAA delivery to muscle | Lack of efficient uptake in skeletal muscle, especially type 2 fibers | Increased delivery to muscle due to increased exposure |
| Immune responses | High titer antibody response in CRIM negative, ~30 %CRIM positive and 10 LOPD | Immune tolerance induction |
| Population | Some patients fail to respond | Larger patient population |
| Efficacy | Partial | More complete correction |
Based upon preclinical research
4.1 Increased Transduction Efficiency for Liver
Groups have proposed direct transduction of muscle with an AAV vector [32] however the efficacy of muscle-specific GAA expression was blunted by antibody responses [33, 34]. In preclinical experiments, gene therapy that transduced the liver with an AAV2/8 vector continuously secreted GAA into the blood, accompanied by receptor-mediated uptake in the heart and skeletal muscle, and to a lesser extent the central nervous system (CNS) with high efficacy [17, 18]. This strategy converted the liver into a depot organ for GAA production that essentially performed continuous ERT, which markedly improved efficacy in preclinical experiments that demonstrated the widespread correction of GAA deficiency in association with clearance of accumulated glycogen and improved muscle function [17, 26, 35]. Liver-targeted gene therapy has been much more efficient than the direct transduction of muscle, either vectors containing a muscle-specific or ubiquitous regulatory cassette (Table 2). For example, direct transduction of striated muscle with the vector containing a muscle-specific promoter featured 5–10 fold higher dosage requirements than the liver depot strategy [17, 26, 34]. Universal expression of GAA with the vector containing a constitutive promoter provoked neutralizing immune responses [17, 26], which emphasized the unique advantages of a liver depot strategy for gene therapy. If similar results are observed in clinical trials, the liver-targeted gene therapy approach will surpass the benefits associated with ERT or muscle directed gene therapy for Pompe disease.
Table 2.
Regulatory Cassette for AAV2/8 Vectors Evaluated in Adult GAA-KO Mice
| Vector | Regulatory Cassette | Minimum Effective Dose | Highly Effective Dose | IgG Antibody Responses to GAA | Reference |
|---|---|---|---|---|---|
| AAV2/8-LSPhGAA | Liver specific | 8×1011 vg/kg | 4×1012 vg/kg | None detected | [17, 26] |
| AAV2/8-MHCK7hGAA | Muscle-specific | 4×1012 vg/kg | 4×1013 vg/kg | Antibody responses | [34] |
| AAV2/8-CBhGAA | Constitutive | Not applicable* | Not applicable* | Antibody and T cell responses | [17, 26] |
AAV2/8-CBhGAA expressed GAA highly for two weeks, prior to onset of neutralizing immune responses.
4.2. Minimum Effective Dose
Liver-target gene therapy will more effectively treat Pompe disease as determined by reducing the amount of GAA required to show functional outcomes due to the immune tolerance induced – which in turn will reduce the AAV dose required. Therefore, we evaluated the biochemical efficacy of 3 lower dosages of AAV2/8-LSPhGAA in GAA-KO mice, reduced to as low as 2×1010 vg/kg, either alone or in combination with ERT [30]. The minimum effective dose (MED) of AAV2/8-LSPhGAA was at least 10-fold lower than previously estimated [16, 26], because 8×1010 vg/kg significantly reduced glycogen content in the striated muscle of GAA-KO mice [30]. Thus, the MED was considered to be the vector dose that significantly decreased the glycogen content of heart and diaphragm, without affecting the glycogen content of muscle. Importantly, the MED of rAAV8 without ERT significantly reduced the glycogen content of heart (p<0.01), and diaphragm (p<0.01), which demonstrated that the glycogen storage in muscle associated with Pompe disease was substantially cross-corrected by GAA secretion from liver accompanied by receptor-mediated uptake in striated muscle [30]. The efficacy of AAV2/8-LSPhGAA [26] at the low dose (2×1010 vg, equivalent to 8×1011 vg/kg body weight) was comparable to long-term ERT [36, 37] with regard to biochemical correction. Furthermore, administering ERT by itself had no significant effect on the glycogen content of quadriceps, but ERT following administration of the MED of AAV2/8-LSPhGAA significantly reduced glycogen content of quadriceps by 38% (p<0.05) indicating that gene therapy with AAV2/8-LSPhGAA made ERT effective. In summary, the MED for AAV2/8-LSPhGAA (8×1010 vg/kg) has been estimated at least 10-fold lower [30], in comparison with previous data [15, 16].
A pharmacology/toxicology study with AAV2/8-LSPhGAApA was completed under good laboratory practice (GLP) by the Gene Therapy Resource Program (GTRP) Toxicology Laboratory at Lovelace Respiratory Research Institute [38]. The experimental groups were designed to detect early and late toxicity [38]. The data revealed no early or late toxicity, and demonstrated biochemical correction [38]. Briefly, intravenous administration of the AAV2/8LSPhGAA vector at 1.6 × 1013vg/kg (8-fold higher than the proposed higher dose in the Phase 1 trial) did not cause significant short- or long-term toxicity. The vector genome was sustained in all tissues through 16-week post dosing, except for in blood with a similar tissue tropism between males and females [38]. Administration of the vector alone, or combined with the ERT, was effective in producing significantly increased GAA activity and consequently decreased glycogen accumulation in multiple tissues, in comparison with administration of vehicle. The urinary Glc4 was reduced in association with the correction of glycogen accumulations by 16 weeks following vector administration either with or without concurrent ERT, in comparison with vehicle [38].
4.3 Decreased Immunogenicity of rhGAA-ERT and GAA
To overcome the limitations in rhGAA-ERT, liver-targeted gene therapy can be used for ITI to rhGGA. The potential role of immune tolerance in mediating the response to ERT among CRIM-negative Pompe subjects was evaluated in GAA-KO mice. A low, sub-therapeutic dose of AAV2/8 vector particles was administered to 3 month-old GAA-KO mice, 6 weeks prior to an immune challenge with rhGAA [16]. An immune challenge consisting of rhGAA (20 mg/kg, the standard dose for humans [39] was administered with modified Freund’s adjuvant to 4.5 month-old GAA-KO mice, either vector-treated or mock-treated with PBS. Anti-hGAA antibodies were detected only in the mock-treated GAA-KO mice at 6 and 7.5 months of age [16]. The anti-rhGAA antibody titer for mock-treated GAA-KO mice was significantly elevated at 6 months of age, in comparison with AAV vector-treated GAA-KO mice. The absence of anti-rhGAA antibody formation suggested immune tolerance to rhGAA following AAV2/8 vector administration at dosages suitable for a Phase I clinical trial (<2×1012 vector genomes/kg) [30]. Immune tolerance to rhGAA could reduce serious adverse events, improve tolerability to ERT and reduce frequency and dosing requirements of ERT.
The impact of immune tolerance upon long-term ERT was evaluated in vector-treated and mock-treated GAA- KO mice [16]. ERT was administered every other week for 12 weeks starting at 4.5 months of age (20 mg/kg/dose), consistent with recommended clinical doses [39]. Mock-treated GAA-KO mice died within hours following the second or third dose of rhGAA consistent with anaphylaxis, as reported for non-tolerant GAA-KO mice [36]. Endurance was significantly improved for the 10.5 month-old vector-treated GAA-KO mice following 12 weeks of sustained ERT, in comparison with vector-treated GAA-KO mice that received no ERT, as demonstrated by prolonged Rotarod times [16] The efficacy of 12 weeks sustained ERT was further evaluated in vector-treated GAA-KO mice through the evaluation of biochemical correction of GAA deficiency and glycogen accumulations in muscle [16]. In contrast to single infusion of high-dose rhGAA, sustained ERT significantly increased the GAA activity in heart and skeletal muscle in vector-treated GAA-KO mice two weeks following the last injection of rhGAA. Glycogen content was reduced significantly in the heart and diaphragm only for vector-treated mice following ERT [16]. These data indicated that efficacy from ERT was achieved only in GAA-KO mice rendered tolerant to rhGAA through gene therapy.
The timing of AAV vector administration relative to the immune challenge with rhGAA was further evaluated in adult GAA-KO mice [15]. AAV2/8-LSPhGAApA administration (2×1010 vg intravenously) significantly prolonged survival, if administered either prior to or three weeks following initial rhGAA injection. Furthermore, other experiments showed that prior administration of an immunogenic AAV vector, AAV2/8-CBhGAApA (2×1010 vg) that contained a ubiquitously active regulatory cassette [17], caused increased mortality in response to ERT [15]. AAV2/8-LSPhGAApA administration enhanced the efficacy of ERT, as reflected by the increased time that vector-treated GAA-KO mice could run on the Rotarod apparatus [15]. Decreased Rotarod time indicates progressive loss of muscle function in GAA-KO mice, which can be prevented by reducing the glycogen content of striated muscle [17]. Urinary hexose tetrasaccharide (Hex4), a biomarker that was decreased in correlation with biochemical correction in Pompe disease mice, was reduced following AAV2/8-LSPhGAApA administration, in comparison with GAA-KO mice that received rhGAA injections only [15]. The negative impact of preexisting anti-rhGAA antibodies was demonstrated by the persistent elevations of urinary Hex4 following antibody formation. GAA-KO mice that were immunized with two injections of rhGAA demonstrated elevated urinary Hex4 at week 10, in comparison with a group of PBS-injected GAA-KO mice that had not yet formed anti-rhGAA antibodies [15]. The formation of anti-rhGAA antibodies occurred uniformly in response to ERT, if mice were not treated with AAV-LSPhGAApA [15]. In contrast, tolerogenic AAV vector administration suppressed IgG titers, even when vector administration followed the initial rhGAA injection by three weeks [15]. These data predicted that administration of AAV2/8-LSPhGAApA either prior to, simultaneously with, or three weeks after initiation of ERT would prevent antibody formation and increase the efficacy from ERT in Pompe disease.
The importance of liver-specific GAA expression to induce immune tolerance during gene therapy was corroborated by a study in which the vector plasmid containing the liver-specific promoter was mixed with a vector plasmid containing a ubiquitously active promoter during vector production [40]. Co-packaging of the two vectors demonstrated expression in cardiac muscle, skeletal muscle, peripheral nerve, and the spinal cord. Furthermore, high-level expression in the liver led to the expansion of GAA-specific Tregs and induction of immune tolerance. Transfer of Tregs into naïve recipients allowed repeated ERT challenges. This study was preceded by another study that co-administered vector containing the liver-specific promoter mixed with a vector containing a ubiquitously active promoter, and demonstrated the immunodominant effect of liver-specific expression over constitutive expression of GAA [26].
5.0 Conclusion
Treatment of GAA-KO mice with AAV2/8-LSPhGAApA achieved stable GAA secretion from the liver into the bloodstream [17, 41] [15, 16, 30]. An analogous gene therapy approach resulted in successful treatment of dogs with hemophilia B for up to 8 years [42]. If similarly, effective in patients with Pompe disease, gene therapy will address the inherent limitations of long term ERT – i.e. the need for frequent repeated infusions and associated high costs of treatment [43]. If the preclinical data translates to the clinic, by preventing anti-rhGAA formation, liver-targeted gene therapy with AAV2/8-LSPhGAA will increase GAA activity in muscle and subsequently will have beneficial effects upon muscle strength and function. In addition, the immune tolerance induced by liver targeted gene therapy to GAA and rhGAA will permit ERT to be effective with reduced with reduced immune responses [10]. The ability to treat with lower AAV dose or ERT dose will reduce the cost associated with manufacturing and treatment increasing patient and doctor acceptance. Potentially, AAV vectors can be re-administered to maintain efficacy lasting several decades, through prophylactic immunosuppression before administration or by the development of different serotypes, which will be critically important in the setting of a pediatric population [44–46]. Liver targeted rAAV8 vector-mediated gene therapy has the potential to revolutionize the treatment of genetic diseases and other disorders currently treated by protein replacement, where antibody formation has complicated available therapy.
Highlights.
ERT has been ineffective for Pompe disease patients who form high, sustained antibody titers
AAV-mediated liver-specific expression induced tolerance to ERT through activation of Tregs
The liver depot of GAA is more efficient than the direct transduction of muscle
Acknowledgments
Thank you to Laura Hughes for preparing figure 1.
Funding: This work was supported by the National Institutes of Health R01AR065873 from the National Institute of Arthritis and Musculoskeletal and Skin Disorders, and by the Duke Translational Medicine Institute/Duke CTSA, the Alice and Y.-T. Chen Pediatric Genetics and Genomics Center
Abbreviations
- AAV
Adeno-associated virus
- CNS
central nervous system
- FIX
coagulation factor IX
- CRIM
cross-reacting immune material
- ERT
enzyme replacement therapy
- GAA
acid alpha-glucosidase
- Glc4
glucose tetrasaccharide
- Hex4
hexose tetrasaccharide
- HSAT
high, sustained (anti-rhGAA IgG) antibody titers
- ITI
immune tolerance induction
- KO
knock-out
- LOPD
late-onset Pompe disease
- LSP
liver specific promoter
- MED
minimum effective dose
- rhGAA
recombinant human GAA
- Tregs
regulatory T-cells
- vg
vector genome
Footnotes
Disclosure/Conflict of interest: DDK has developed the technology that is being used in the study. If the technology is commercially successful in the future, the developers and Duke University may benefit financially. DDK has received research/grant support from Sanofi Genzyme Corporation in the past, and rhGAA for these studies was supplied by Sanofi Genzyme.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elliott S, Buroker N, Cournoyer JJ, et al. Pilot study of newborn screening for six lysosomal storage diseases using Tandem Mass Spectrometry. Mol Genet Metab. 2016;118:304–9. doi: 10.1016/j.ymgme.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hopkins PV, Campbell C, Klug T, et al. Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J Pediatr. 2015;166:172–177. doi: 10.1016/j.jpeds.2014.09.023. [DOI] [PubMed] [Google Scholar]
- 3.Kishnani PS, Hwu WL, Mandel H, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 4.van Gelder CM, Poelman E, Plug I, et al. Effects of a higher dose of alglucosidase alfa on ventilator-free survival and motor outcome in classic infantile Pompe disease: an open-label single-center study. J Inherit Metab Dis. 2016;39:383–390. doi: 10.1007/s10545-015-9912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Case LE, Bjartmar C, Morgan C, et al. Safety and efficacy of alternative alglucosidase alfa regimens in Pompe disease. Neuromuscul Disord. 2015;25:321–332. doi: 10.1016/j.nmd.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. 2011;13:729–736. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- 8.Patel TT, Banugaria SG, Case LE, et al. The impact of antibodies in late-onset Pompe disease: A case series and literature review. Mol Genet Metab. 2012;106:301–309. doi: 10.1016/j.ymgme.2012.04.027. [DOI] [PubMed] [Google Scholar]
- 9.Messinger YH, Mendelsohn NJ, Rhead W, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. 2012;14:135–142. doi: 10.1038/gim.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banugaria SG, Prater SN, Patel TT, et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile pompe disease: a step towards improving the efficacy of ERT. PLoS One. 2013;8:e67052. doi: 10.1371/journal.pone.0067052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karimi MH, Geramizadeh B, Malek-Hosseini SA. Tolerance Induction in Liver. Int J Organ Transplant Med. 2015;6:45–54. [PMC free article] [PubMed] [Google Scholar]
- 12.Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 13.Nathwani AC, Reiss UM, Tuddenham EG, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sands MS. AAV-mediated liver-directed gene therapy. Methods Mol Biol. 2011;807:141–157. doi: 10.1007/978-1-61779-370-7_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun B, Kulis MD, Young SP, et al. Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine pompe disease. Mol Ther. 2010;18:353–360. doi: 10.1038/mt.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun B, Bird A, Young SP, et al. Enhanced response to enzyme replacement therapy in pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franco LM, Sun B, Yang X, et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther. 2005;12:876–884. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 18.Ziegler RJ, Bercury SD, Fidler J, et al. Ability of adeno-associated virus serotype 8-mediated hepatic expression of acid alpha-glucosidase to correct the biochemical and motor function deficits of presymptomatic and symptomatic Pompe mice. Hum Gene Ther. 2008;19:609–621. doi: 10.1089/hum.2008.010. [DOI] [PubMed] [Google Scholar]
- 19.Koeberl DD, Kishnani PS. Immunomodulatory gene therapy in lysosomal storage disorders. Curr Gene Ther. 2009;9:503–510. doi: 10.2174/156652309790031094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crispe IN. Hepatic T cells and liver tolerance. Nat Rev Immunol. 2003;3:51–62. doi: 10.1038/nri981. [DOI] [PubMed] [Google Scholar]
- 21.Roland CR, Mangino MJ, Duffy BF, Flye MW. Lymphocyte suppression by Kupffer cells prevents portal venous tolerance induction: a study of macrophage function after intravenous gadolinium. Transplantation. 1993;55:1151–1158. doi: 10.1097/00007890-199305000-00041. [DOI] [PubMed] [Google Scholar]
- 22.You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology. 2008;48:978–990. doi: 10.1002/hep.22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YG, Choisy-Rossi CM, Holl TM, et al. Activated NKT cells inhibit autoimmune diabetes through tolerogenic recruitment of dendritic cells to pancreatic lymph nodes. J Immunol. 2005;174:1196–1204. doi: 10.4049/jimmunol.174.3.1196. [DOI] [PubMed] [Google Scholar]
- 24.Cao O, Dobrzynski E, Wang L, et al. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110:1132–1140. doi: 10.1182/blood-2007-02-073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziegler RJ, Cherry M, Barbon CM, et al. Correction of the Biochemical and Functional Deficits in Fabry Mice Following AAV8-mediated Hepatic Expression of alpha-galactosidase A. Mol Ther. 2007;15:492–500. doi: 10.1038/sj.mt.6300066. [DOI] [PubMed] [Google Scholar]
- 26.Zhang P, Sun B, Osada T, et al. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum Gene Ther. 2012;23:460–472. doi: 10.1089/hum.2011.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prater SN, Banugaria SG, Dearmey SM, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med. 2012;14:800–810. doi: 10.1038/gim.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- 29.Chan J, Desai AK, Kazi ZB, et al. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol Genet Metab. 2017;120:163–172. doi: 10.1016/j.ymgme.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 30.Han SO, Ronzitti G, Arnson B, et al. Low-Dose Liver-Targeted Gene Therapy for Pompe Disease Enhances Therapeutic Efficacy of ERT via Immune Tolerance Induction. Mol Ther Methods Clin Dev. 2017;4:126–136. doi: 10.1016/j.omtm.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falk DJ, Soustek MS, Todd AG, et al. Comparative impact of AAV and enzyme replacement therapy on respiratory and cardiac function in adult Pompe mice. Mol Ther Methods Clin Dev. 2015;2:15007. doi: 10.1038/mtm.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith BK, Collins SW, Conlon TJ, et al. Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther. 2013;24:630–640. doi: 10.1089/hum.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun B, Zhang H, Franco LM, et al. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol Ther. 2005;11:889–898. doi: 10.1016/j.ymthe.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 34.Sun B, Young SP, Li P, et al. Correction of multiple striated muscles in murine Pompe disease through adeno-associated virus-mediated gene therapy. Mol Ther. 2008;16:1366–1371. doi: 10.1038/mt.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li S, Sun B, Nilsson MI, et al. Adjunctive beta2-agonists reverse neuromuscular involvement in murine Pompe disease. FASEB J. 2013;27:34–44. doi: 10.1096/fj.12-207472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raben N, Danon M, Gilbert AL, et al. Enzyme replacement therapy in the mouse model of Pompe disease. Molecular Genetics and Metabolism. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 37.Raben N, Fukuda T, Gilbert AL, et al. Replacing acid alpha-glucosidase in Pompe disease: Recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Molecular Therapy. 2005;11:48–56. doi: 10.1016/j.ymthe.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 38.Wang G, Young SP, Bali D, et al. Assessment of toxicity and biodistribution of recombinant AAV8 vector-mediated immunomodulatory gene therapy in mice with Pompe disease. Mol Ther Methods Clin Dev. 2014;1:14018. doi: 10.1038/mtm.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid {alpha}-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 40.Doerfler PA, Todd AG, Clement N, et al. Copackaged AAV9 Vectors Promote Simultaneous Immune Tolerance and Phenotypic Correction of Pompe Disease. Hum Gene Ther. 2016;27:43–59. doi: 10.1089/hum.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun B, Zhang H, Benjamin DK, Jr, et al. Enhanced Efficacy of an AAV Vector Encoding Chimeric, Highly Secreted Acid alpha-Glucosidase in Glycogen Storage Disease Type II. Mol Ther. 2006;14:822–830. doi: 10.1016/j.ymthe.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niemeyer GP, Herzog RW, Mount J, et al. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood. 2009;113:797–806. doi: 10.1182/blood-2008-10-181479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Desnick RJ. Enzyme replacement and enhancement therapies for lysosomal diseases. Inher Metab Dis. 2004;27:385–410. doi: 10.1023/B:BOLI.0000031101.12838.c6. [DOI] [PubMed] [Google Scholar]
- 44.Halbert CL, Standaert TA, Aitken ML, et al. Transduction by adeno-associated virus vectors in the rabbit airway: efficiency, persistence, and readministration. J Virol. 1997;71:5932–5941. doi: 10.1128/jvi.71.8.5932-5941.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang LL, Calcedo R, Nichols TC, et al. Sustained correction of disease in naive and AAV2-pretreated hemophilia B dogs: AAV2/8-mediated, liver-directed gene therapy. Blood. 2005;105:3079–3086. doi: 10.1182/blood-2004-10-3867. [DOI] [PubMed] [Google Scholar]
- 46.Demaster A, Luo X, Curtis S, et al. Long-term efficacy following readministration of an adeno-associated virus vector in dogs with glycogen storage disease type Ia. Hum Gene Ther. 2012;23:407–418. doi: 10.1089/hum.2011.106. [DOI] [PMC free article] [PubMed] [Google Scholar]