

Abstract
The glucocorticoid receptor (GR) is a ligand-activated transcription factor that induces expression of many genes. The GR has been useful for understanding how chromatin structure regulates steroid-induced transcription in model systems. However, the effect of glucocorticoids on chromatin structure has been examined on few endogenous mammalian promoters. We investigated the effect of glucocorticoids on the in vivo chromatin structure of the glucocorticoid-responsive IκBα gene promoter, the inhibitor of the ubiquitous transcription factor, nuclear factor kappa B (NFκB). Glucocorticoids inhibit NFκB activity in some tissues by elevating the levels of IκBα. We found that glucocorticoids activated the IκBα promoter in human T47D/A1-2 cells containing the GR. We then investigated the chromatin structure of the IκBα promoter in the absence and presence of glucocorticoids with the use of micrococcal nuclease, restriction enzyme, and deoxyribonuclease (DNaseI) analyses. In untreated cells, the promoter assembles into regularly positioned nucleosomes, and glucocorticoid treatment did not alter nucleosomal position. Restriction enzyme accessiblity studies indicated that the IκBα promoter is assembled as phased nucleosomes that adopt an “open” chromatin architecture in the absence of hormone. However, glucocorticoids may be required for transcription factor binding, because DNaseI footprinting studies suggested that regulatory factors bind to the promoter upon glucocorticoid treatment.
INTRODUCTION
Steroid hormone receptors (SHRs) are ligand-activated transcription factors that regulate the expression of genes involved in development, homeostatic mechanisms, and cellular differentiation (Jenster et al., 1997). A subfamily consisting of the receptors for glucocorticoids, progestins, androgens, and mineralocorticoids share regions of high homology and bind a common hormone response element (HRE) (Amero et al., 1992). SHRs regulate a diverse array of genes in a multitude of cell types. Steroid-induced transcription of eukaryotic genes is carefully controlled, and one critical regulatory mechanism is organization of the gene into chromatin (Collingwood et al., 1999).
In the eukaryotic nucleus, DNA is wrapped around histone proteins, forming chromatin. Highly compact regions of chromatin are associated with low transcriptional activity, whereas less compact regions show higher transcriptional activity (Elgin, 1988). DNA is resistant to nuclease attack when packaged as chromatin, and transcription factors have restricted access to their respective binding sites (Wolffe and Hayes, 1999). Thus, the chromatin structure of promoters is one barrier to transcription that SHRs must overcome (Archer et al., 1997; Wu, 1997). The glucocorticoid receptor (GR) has been a useful model for understanding the effect of chromatin on steroid-induced transcription (Wallberg et al., 2001). In particular, the mouse mammary tumor virus (MMTV) promoter has provided extensive mechanistic information on GR-mediated transcription from a chromatin template (Deroo and Archer, 2001). However, the effect of chromatin structure on glucocorticoid-mediated transcription has been investigated in detail on few endogenous mammalian genes.
To investigate how chromatin structure regulates glucocorticoid activation of genes in vivo, we carried out a detailed analysis of an endogenous, glucocorticoid responsive promoter—the IκBα promoter. IκBα is an inhibitor of the transcription factor, nuclear factor kappa B (NFκB). Members of the NFκB family of transcription factors regulate many immune system genes (for recent reviews, see May and Ghosh, 1997; Ghosh, 1999). Glucocorticoids increase transcription of IκBα in some tissue culture cells (Heck et al., 1997; McKay and Cidlowski, 1999). In vivo studies in humans and mice also demonstrate increased IκBα expression due to glucocorticoid treatment (Auphan et al., 1995; Aljada et al., 1999; Han et al., 1999).
In addition to glucocorticoids, compounds that activate NFκB, such as tumor necrosis factor α (TNF-α), phorbol esters, and lipopolysaccharide also activate the IκBα promoter (Le Bail et al., 1993). The promoter sequences required for activation by NFκB-activating compounds have been characterized by transient transfection assays (Le Bail et al., 1993; Chiao et al., 1994; Ito et al., 1994; Algartéet al., 1999). These studies have been supported by gel shift analysis and footprinting and have identified factors that bind in the presence and absence of stimulation (Le Bail et al., 1993; Chiao et al., 1994; Ito et al., 1994; Algartéet al., 1999). The promoter contains binding sites for NFκB, SP1, AP2, and Ets-1, and occupation of the promoter by these transcription factors was shown to depend on the length of time the promoter was activated (Algartéet al., 1999). However, none of these studies has addressed what role chromatin structure may play in activation of this gene, and the chromatin structure of the native IκBα promoter has not been characterized. What impact stimulation by glucocorticoids or NFκB-activating compounds have on this structure is also not known. Our purpose in this report was twofold: to characterize the chromatin structure of the endogenous IκBα promoter and then to determine the effect of glucocorticoid treatment on this structure.
In T47D/A1-2 breast cancer cells that contain the GR, we have mapped the positions of nucleosomes from nucleotides −900/+100 and investigated the chromatin structure by restriction enzyme hypersensitivity and DNaseI footprinting assays. We found that glucocorticoid activation of the IκBα promoter did not involve a change in position of nucleosomes assembled over the promoter. The chromatin structure was found to be nonrepressed or “open” in the absence of hormone, and hormone treatment did not change accessibility to restriction enzymes. However, steroid treatment appeared to induce transcription factor binding, as suggested by DNaseI footprint analysis. Our results suggest that glucocorticoid activation of this gene proceeds by the recruitment of transcription factors to the IκBα promoter in absence of GR-mediated hypersensitivity at the promoter.
MATERIALS AND METHODS
Cells
T47D/A1-2 cells were derived from T47D breast cancer cells by stable transfection with a pGRneo plasmid as described previously (Nordeen et al., 1989). T47D/A1-2 cells were grown at 37°C with 5% CO2 in modified Eagle's medium containing 10% fetal bovine serum and 0.16 mg/ml Geneticin (Life Technologies, Rockville, MD).
Preparation of Nuclear Extracts
The protocol for preparing nuclear and cytoplasmic extracts was as described previously (Scheinman et al., 1993). A1-2 cells were plated on 100-mm dishes and grown until 80% confluent before preparation of extracts.
Gel Mobility Shift Assay
Gel shift assays were carried out by preincubating 10 μg of nuclear extract and 1 μl of poly dI/dC (1 μg/ml) in binding buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 3 mM DTT, 10% glycerol, 0.05% NP-40, and 0.1 mM ZnCl2) at room temperature for 10 min (Archer et al., 1990). A double-stranded oligonucleotide for the IL-2 promoter NFκB consensus sequence was end-labeled with γ-32P-ATP and T4 polynucleotide kinase and then incubated with the extract for 20 min at room temperature. The mixture was then electrophoresed on a 5% nondenaturing polyacrylamide gel in 1× Tris-Borate-EDTA buffer. The gels were dried and exposed to film.
Isolation of RNA: Northern Analysis
Cells were left untreated or treated as described in the figure legends. Total cellular RNA was isolated with the use of TRIZOL (Life Technologies) according to the manufacturer's instructions. Ten micrograms of RNA was separated on a 1% agarose gel containing formaldehyde and MOPS (3-(N-morpholino) propane-sulfonic acid) buffer, and the RNA was blotted to Zeta-Probe nylon membrane (Bio-Rad, Hercules, CA) in 10× SSC overnight at room temperature. The membrane was hybridized overnight with a 32P-labeled IκBα cDNA PstI-PstI fragment corresponding to +272/+455 of the IκBα cDNA sequence, which was cut and purified from a CMV–IκBα plasmid (kindly donated by Dr. A. Israel). This fragment was labeled with 32P by random priming (Ready-to-Go beads; Amersham-Pharmacia, Piscataway, NJ), and the membrane was hybridized overnight according to the manufacturer's Standard Protocol instructions. As a loading control, the same membrane was similarly hybridized with either a rat cyclophilin cDNA fragment (kindly donated by Dr. G. DiMattia; London Regional Cancer Center, London, Ontario) or a cyclophilin 40-mer oligonucleotide purchased from Geneka Biotechnologies (Montreal, Canada). The cyclophilin cDNA was labeled by random priming and hybridized as described for the IκBα cDNA fragment. The cyclophilin oligonucleotide was end-labeled with 32P with the use of polynucleotide kinase and incubated according to the Zeta-Probe membrane “oligonucleotide” protocol. After hybridization and washing, the membrane was visualized and quantified by the Molecular Dynamics Phosphorimager (Sunnyvale, CA).
Nucleosome Mapping by Micrococcal Nuclease
Nuclei were isolated as described previously (Archer et al., 1991). Nuclei were resuspended in 100 μL wash buffer containing 1 mM CaCl2 and then digested with 0–200 units/ml micrococcal nuclease (MNase; Worthington Biochemicals, Lakewood, NJ) for 5 min at 30°C. The reaction was stopped by adding 40 μL of 100 mM EDTA, pH 8.0, 10 mM EGTA, pH 7.5. DNA was purified, recut with an appropriate restriction enzyme, and analyzed by Southern blot (see Figure 2, A and B) or reiterative primer extension (see Figure 3, A and B).
Figure 2.

The IκBα promoter is organized into a regular array of nucleosomes. (A) Nuclei (lanes 1 and 2) and genomic DNA (lanes 3 and 4) from A1-2 cells were digested with 0 (lanes 1 and 4), 1 (lane 3), or 200 units/ml (lane 2) micrococcal nuclease (MNase), and the purified DNA recut with HincII. DNA fragments were analyzed by Southern blot, with the use of a radiolabeled HincII (−699)/SgrAI (−536) fragment of the IκBα promoter. (B) Nuclei (lanes 1–5) and genomic DNA (lane 6) were digested with 0 (lane 1), 1 (lane 6), or 20–200 units/ml (lanes 2–5) MNase, and the purified DNA was recut with EcoRI. DNA fragments were analyzed by Southern blot, with the use of a radiolabeled EcoRI (−1229)/AflII (−999) fragment of the IκBα promoter. (C) Schematic diagram of nucleosome positions on the IκBα proximal promoter in A1-2 cells.
Figure 3.

Fine mapping of the −280 to −50 region of the IκBα promoter by micrococcal nuclease. (A) Fine mapping of the IκBα promoter. DNA prepared as in Figure 2 was also analyzed by reiterative primer extension. Lanes 7, 8, and 9: 0, 100, and 200 units/ml MNase digests, respectively. Lane 5: (−623/+11) IκBα-luc plasmid digested with MNase, lanes 1–4, sequencing tracks with the (−623/+11) IκBα-luc plasmid. With the use of the Molecular Dynamics Phosporimager, a line graph representing lane 9 band intensity was created. The adjacent schematic is labeled as follows: ▪, peak locations relative to +1; □, restriction enzyme sites. (B) Digestion of nuclei and reiterative primer extension analysis were carried out as in A except that two different MNase concentrations were used and cells were either untreated or treated with dexamethasone (10−7 M) for 2 h. Lane 2: G sequencing track with (−623/+11) IκBα-luc plasmid. Lane 3: (−623/+11) IκBα-luc plasmid digested with MNase. (C) Line graph comparing lanes 6 and 7.
Southern Blot.
Twenty micrograms of control DNA or DNA isolated from MNase-digested nuclei was separated on a 1.5% agarose gel and transferred to Hybond N+ membrane (Amersham-Pharmacia, Piscataway, NJ; Wolff and Gemmill, 1997). Control genomic DNA was prepared by digesting purified A1-2 genomic DNA with 1 unit/ml MNase for 5 min at 25°C and then redigesting with an appropriate restriction enzyme. Fragments corresponding to HincII (−699)/SgrAI (−536), and EcoRI (−1229)/AflII (−999) were obtained by digesting a 1.4-kB IκBα-CAT plasmid (kindly donated by Dr. R. Scheinman, University of Colorado Health Sciences Center, Denver, CO) and were radiolabeled with 32P by random priming (Amersham-Pharmacia Ready-to-Go beads), and the membrane was hybridized overnight.
Reiterative Primer Extension.
Twenty micrograms of DNA isolated from MNase-digested nuclei was analyzed with the use of linear Taq polymerase amplification with a 32P-labeled single-strand primer corresponding to the −337/−317 region of the IκBα promoter. Five nanograms of IκBα-luc plasmid was used for sequencing (Mymryk et al., 1997). As a control, IκBα-luc plasmid was digested as follows: 5 μg plasmid was digested with 1 unit/ml MNase in wash buffer containing 1 mM CaCl2. After 5-min digestion at room temperature, the reaction was stopped as described above. The plasmid DNA was purified, and redigested with EcoRI. For all DNA samples, amplified DNA was purified and separated with the use of a 6% polyacrylamide denaturing gel. Statistical significance in Figure 3B was calculated with the use of a paired Student's t test from quadruplicate samples. In Figure 3, the statistical significance was as follows: (A) band at −175, mean = 1.63, p < 0.006, (B) band at −186, mean = 1.85, p < 0.010, (C) Band at −203, mean = 1.38, p < 0.007.
Restriction Enzyme Hypersensitivity Analysis
Cells were either untreated or treated as described in the figure legends. Nuclei were digested in vivo with 5 U DdeI, AvaI, DpnII, PstI, or EcoNI per μg DNA as described previously (Archer et al., 1991). After purification of genomic DNA, samples were recut with DpnII, AvaI, or HindIII. DNA fragments were analyzed with the use of linear Taq polymerase amplification with a 32P-labeled single-strand primer corresponding to the +56 to +73 region of the IκBα coding region or to −629/−610 of the IκBα promoter (Le Bail et al., 1993). Purified extended products were analyzed on 8% polyacrylamide denaturing gels and quantified with the use of the Molecular Dynamics Phosphorimager.
DNaseI Footprinting Analysis
A1-2 cells were untreated or treated with dexamethasone (10−7 M) for 2 h. Nuclei were isolated as above and resuspended in 100 μL wash buffer containing 1 mM CaCl2 and 0.5 mM MgCl2 and then digested with 0, 50, or 100 units/ml deoxyribonuclease I (DNase I; Worthington Biochemicals) for 5 min at 30°C. The reaction was stopped by adding 40 μl of 100 mM EDTA, pH 8.0, 10 mM EGTA, pH 7.5. DNA was purified, recut with an appropriate restriction enzyme, and analyzed by reiterative primer extension (see Figures 5 and 6). Control genomic DNA was prepared by digesting purified A1-2 genomic DNA with 0.01 or 0.04 units/ml DNase for 5 min at 25°C and then redigesting with an appropriate restriction enzyme. Reiterative primer extension was carried out as described for MNase analysis, except that 8 and 10% denaturing gels were used and an additional primer corresponding to the −156/−136 region of the IκBα promoter was also used (see Figure 6).
Figure 5.

DNaseI footprinting of the IκBα promoter (−230 to −60). (A) Nuclei (lanes 3–6) or genomic DNA (lane 2) from A1-2 cells was digested with 0 (lanes 3 and 4), 0.04 (lane 2), or 50 units/ml (lanes 5 and 6) DNaseI, and the purified DNA was recut with EcoRI. DNA fragments were analyzed by reiterative primer extension. Lane 5: G sequencing track with the (−623/+11) IκBα-luc plasmid. (B) A line graph comparing lanes 5 and 6 band intensity with locations of transcription factor binding sites indicated.
Figure 6.

DNaseI footprinting of the IκBα promoter (−100 to + 7). (A) Nuclei (lanes 3–6) or genomic DNA (lane 2) from A1-2 cells was digested with 0 (lanes 3 and 4), 0.01 (lane 2), or 100 units/ml (lanes 5 and 6) DNaseI, and the purified DNA was recut with PstI. DNA fragments were analyzed by reiterative primer extension. Lane 7: C sequencing track with the (−623/+11) IκBα-luc plasmid. (B) A line graph comparing lanes 5 and 6 band intensity with locations of transcription factor binding sites indicated.
RESULTS
Glucocorticoids Activate IκBα Gene Expression and Repress NFκB Activity in T47D/A1-2 Human Breast Cancer Cells
To study glucocorticoid activation of the endogenous IκBα promoter, we used human T47D/A1-2 cells that express high levels of the GR (Nordeen et al., 1989). Glucocorticoids have been shown to increase IκBα RNA levels in some cell types (Auphan et al., 1995; Scheinman et al., 1995). In A1-2 cells, treatment with the synthetic glucocorticoid, dexamethasone (dex) increased IκBα RNA levels three- to fourfold (Figure 1A). The potent NFκB activator, phorbol myristate acetate (PMA), did not increase IκBα RNA levels, as has been observed in other cell lines (Figure 1B; Algartéet al., 1999). To examine the functional consequences of dex induction of IκBα, we determined if glucocorticoid repression of NFκB was observed, because glucocorticoids have been shown to inhibit NFκB activation in several cell types (McKay and Cidlowski, 1999). We found that dex pretreatment completely repressed activation of NFκB by PMA (Figure 1C, cf. lanes 2 and 3). Thus, glucocorticoids increased IκBα RNA levels in A1-2 cells, which correlated with glucocorticoid-induced repression of NFκB activity.
Figure 1.
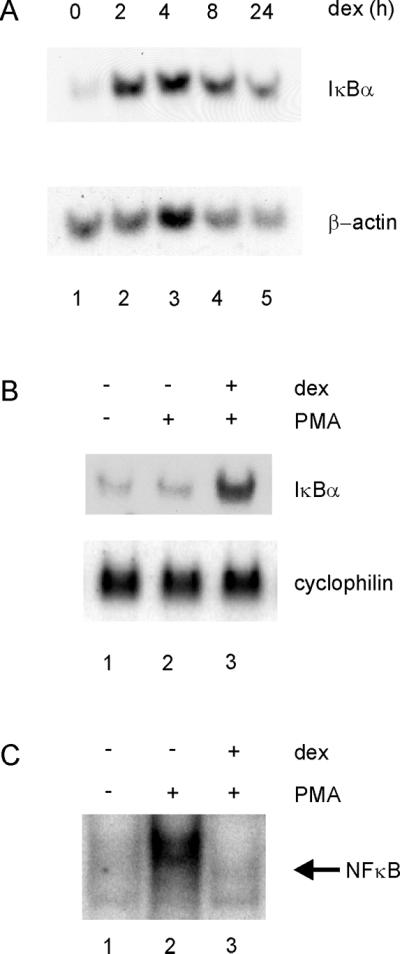
Glucocorticoids increase IκBα transcription in A1-2 cells. (A) Glucocorticoids increase IκBα RNA levels in A1-2 cells. Northern analysis was conducted with the use of A1-2 cells that were either untreated (lane 1) or treated with dexamethasone (10−7 M) for 2, 4, 8, and 24 h (lanes 2–5). Total cellular RNA was prepared and analyzed by Northern blot. Briefly, RNA was separated on a 1% agarose/formaldehyde gel, the RNA (10 μg) transferred to a nylon membrane, and the membrane probed with 32P-labeled IκBα and actin cDNA probes. (B) The NFκB activator, PMA, does not affect glucocorticoid activation of IκBα. Northern analysis was conducted with the use of A1-2 cells that were either untreated (lane 1) or treated with PMA (40 ng/ml) for 45 min (lane 2). In lane 3, cells were pretreated with dexamethasone (10−7 M) for 4 h (lane 3) before treatment with PMA (40 ng/ml) for 45 min. The blot was reprobed for cyclophilin as a control. (C) Glucocorticoids repress NFκB activity in A1-2 cells. Nuclear extracts were prepared from cells treated as in A and analyzed by gel shift, with the use of 10 μg of nuclear extract with a 32P-labeled double-stranded oligonucleotide corresponding to the NFκB consensus sequence of the IL-2 promoter. The binding reactions were analyzed on a 5% nondenaturing polyacrylamide gel, followed by autoradiography.
The IκBα Promoter Is Organized into a Regular Array of Nucleosomes
As a prelude to studying glucocorticoid activation effects, we first analyzed the nucleosomal structure of the human IκBα promoter between −900 and +100 in untreated cells. The region from −225 to +1 has been shown previously to be critical for activation of IκBα by PMA-PHA (phyto-hemagglutinin) or TNF-α (Algartéet al., 1999). To determine the position of nucleosomes on the IκBα promoter, we used MNase digestion in combination with Southern blotting and reiterative primer extension. We isolated nuclei from A1-2 cells and digested them with increasing concentrations of MNase. The promoter region from −900 to +100 was analyzed ±25 bp by Southern blot (Figure 2, A and B). DNA preparations from MNase-digested nuclei were cut with either HincII or EcoRI and probed with a DNA fragment by indirect end-labeling (Figure 2C). Regularly positioned nucleosomes occupied the entire region from −900 to +100 (Figure 2, A–C). Control deproteinized DNA digested with MNase did not produce this ladder of bands, indicating that the in vivo digestion pattern required the presence of nucleosomes. In these in vivo experiments, the average interval between bands was ∼150 bp—smaller than the expected 180–190 bp (Wolffe and Kurumizaka, 1998). This phenomenon has been observed previously for other promoters and may suggest multiple translational positions of nucleosomes (Bortvin and Winston, 1996; Boyes and Felsenfeld, 1996; Bhattacharyya et al., 1997). To confirm the nucleosome positions identified by Southern blot, finer PCR-based mapping was also carried out from −300 to +1 (Figure 3A). This mapping identified two clusters of MNase sensitivity, centering on −135 and −278 (143 bp), suggesting the expected nucleosomal size of ∼146 bp. These two sites, corresponding to the second or “B” nucleosome, were consistent with the sites identified by Southern blot (Figure 2C).
We next wanted to determine if glucocorticoid treatment altered these nucleosomal positions. We focused on the region from −280 to +1 because it was previously shown to be involved in activation by PMA and TNF-α and to contain transcription factor binding sites that were critical for this induction (Algartéet al., 1999). We found that dex treatment had no effect on the band pattern created by MNase (Figure 3B). However, bands representing sites within the nucleosome increased in intensity because of dex treatment (Figure 3C). MNase sensitivity due to dex treatment at −203, −186, and −175 increased 1.63×, 1.85×, and 1.38× (average of quadruplicate samples).
Our MNase analysis indicates that the IκBα promoter is assembled as a phased array of nucleosomes. Glucocorticoid activation did not disrupt the nucleosomal pattern, although increased accessibility at several intranucleosomal sites suggests that steroid activation may alter histone-DNA contacts at these sites.
The IκBα Promoter Is in an Open Chromatin State
One method to detect the position of nucleosomes and/or changes in chromatin structure is the restriction enzyme hypersensitivity assay (Mymryk et al., 1997; Fragoso et al., 1998). DNA over which nucleosomes are positioned is generally resistant to restriction enzyme cleavage, whereas changes in chromatin structure are indicated by changes in promoter hypersensitivity to restriction enzymes. To support the high-resolution nucleosome positioning determined by micrococcal nuclease and to look for steroid-related changes in chromatin structure, we surveyed the sensitivity of the first 500 bp of the IκBα promoter to various restriction enzymes in the absence or presence of dex (Figure 4). We found that percent cleavage (calculated as in vivo band intensity relative to combined in vivo plus in vitro band intensity) correlated to the expected position of the nucleosomes as determined by MNase analysis. For example, cleavage by DdeI or EcoNI in the linker region (as determined in Figure 3A) showed cleavage of 40–50%, whereas cleavage by AvaI, which cuts within a nucleosome, was ∼20%. In addition, DdeI cleavage within a nucleosome was 20%, compared with 40% in the linker. Thus, both MNase and restriction enzyme hypersensitivity data suggest that IκBα nucleosome “B” occupies approximately −135 to −278 of the promoter.
Figure 4.

Restriction enzyme hypersensitivity analysis of the IκBα promoter. (A) A1-2 cells were either untreated (lane 1), or treated for 2 h with dex (10−7 M; lane 2). Nuclei were isolated, digested in vivo with DpnII or PstI and in vitro with AvaI, and analyzed by reiterative primer extension with oligonucleotide TA-80. (B) A1-2 cells were either untreated (lane 1) or treated for 2 h with dex (10−7 M; lane 2). Nuclei were isolated, digested in vivo with AvaI or DdeI and in vitro with DpnII, and analyzed by reiterative primer extension with oligonucleotide TA-53. (C) A1-2 cells were either untreated (lanes 1 and 3) or treated for 2 h with dex (10−7 M; lanes 2 and 4). Nuclei were isolated, digested in vivo with DdeI or EcoNI and in vitro with HindIII, and analyzed by reiterative primer extension with oligonucleotide TA-53. (D) Restriction enzyme hypersensitivity profile of the IκBα promoter in the absence or presence of glucocorticoid. Dotted lines, nucleosome positions determined by low-resolution mapping; solid lines, nucleosomes mapped to base pair resolution.
We also found that enzyme sensitivity at various restriction sites did not change significantly after dex treatment, suggesting that the chromatin configuration of the IκBα is open and accessible to restriction enzyme cleavage (Figure 4). Other promoters, such as the MMTV promoter, show increased sensitivity to restriction enzymes upon hormone treatment (Archer et al., 1992).
DNaseI Footprinting of the IκBα Promoter
DNaseI digestion of DNA organized as rotationally positioned nucleosomes produces a 10-bp ladder, where the enzyme cleaves the minor groove. Transcription factor binding is often indicated by a loss of these bands, where the presence of the factor blocks access of the enzyme to the DNA. We used DNaseI analysis to look for changes in the DNaseI pattern of the IκBα promoter due to glucocorticoid treatment, which suggests binding of factors.
DNaseI digestion of nuclei from untreated A1-2 cells yielded the predicted 10-bp ladder from −230 to −180 and −160 to −120 (marked by arrows), with a gap at −170, where no significant band was present (Figure 5A, lane 5). These results suggest that the DNA around IκBα nuc-B is rotationally phased and that the naïve promoter may be prebound at −170 by a transcription factor. Digestion of deproteinized DNA in vitro with DNaseI yielded a few bands, which did not correspond to those seen in the in vivo digested lanes (Figure 5A, lane 2).
It is not known how glucocorticoids activate transcription of the IκBα promoter, because no consensus HREs have been found in the proximal promoter. However, in a transient transfection assay, −623 bp of promoter is sufficient for glucocorticoid activation, suggesting that the proximal promoter is involved in this activation (Heck et al., 1997). Initial mapping showed that glucocorticoid treatment significantly altered the pattern of DNaseI digestion between −230 and −60 of the IκBα promoter (Figure 5A). Glucocorticoid treatment led to the reduced intensity of several bands and complete disappearance of others (Figure 5A, cf. lanes 5 and 6, and 5B). Bands mapping to −152/−153, −180, −190, −200, and −220 were all affected. We then investigated the effect of dex on chromatin structure closer to the transcription start site by mapping with a different oligo (Figure 6). As seen for the −230/−60 region, several bands were reduced in intensity by glucocorticoid treatment. These bands mapped to −38, −48, −53, −89, and −94 and overlapped factor binding sites for the GR, NFκB, and SP1. The changes we observe in DNaseI pattern are consistent with transcription factor binding to the promoter and suggest that glucocorticoid activation of the IκBα promoter may lead to binding of factors to the proximal promoter region.
DISCUSSION
Steroid hormones mediate gene expression through ligand-activated transcription factors, the SHRs. One barrier to transcription that SHRs must overcome is the assembly of DNA into chromatin. The MMTV promoter has provided extensive information on how glucocorticoids activate promoters assembled as chromatin (Archer et al., 1997). Another glucocorticoid-responsive gene whose chromatin structure has been well defined is the rat tyrosinaminotransferase (TAT) gene (Carr and Richard-Foy, 1990). However, glucocorticoid regulation of chromatin structure has been explored for few other mammalian genes. We sought to expand these studies by investigating steroid activation of an endogenous, glucocorticoid-responsive promoter—the IκBα promoter.
We found that glucocorticoids increased levels of IκBα mRNA in A1-2 cells, and this increase correlated with repression of NFκB activity (Figure 1). We then investigated the impact of transactivation on the chromatin structure of the IκBα promoter with the use of MNase, restriction enzyme hypersensitivity, and DNaseI footprinting assays. The endogenous IκBα promoter was organized into a phased array of nucleosomes, as determined by MNase analysis (Figures 2 and 3). The IκBα nucleosome “B” was rotationally positioned, as indicated by the 10-bp ladder resulting from DNaseI analysis (Figures 5). In addition, the accessibility of the proximal promoter to restriction enzymes correlated with the predicted nucleosome positions (Figure 4). We then investigated whether glucocorticoid activation altered this nucleosomal structure. Glucocorticoid treatment had no effect on the pattern of bands created by MNase, although slight increases in the intensity of several bands, localized around −200/−175 were detected. These hypersensitive bands suggest that some perturbation of chromatin structure by glucocorticoid treatment results in altered sensitivity to enzyme at the site, even though the actual position of the nucleosome does not change. This lack of hormone-dependent change on the IκBα promoter is reminiscent of the MMTV promoter. On this promoter, glucocorticoid treatment did not alter the cleavage pattern by MNase compared with untreated cells (Richard-Foy and Hager, 1987; Fragoso et al., 1995; Mymryk et al., 1995). Retinoic acid activation of the retinoic acid receptor β2 (RARβ2) promoter also has no effect on nucleosomal location (Bhattacharyya et al., 1997).
As a measure of chromatin change during promoter activation, we determined sensitivity to restriction enzyme cleavage before and after steroid treatment (Figure 4). We found that the accessibility of the DNA to restriction enzyme cleavage reflected the expected position of the nucleosomes, as has been demonstrated for other promoters, including MMTV (Archer et al., 1991; Fragoso et al., 1998; Gregory et al., 1999; Polach and Widom, 1999). However, we found that restriction enzyme sensitivity of the IκBα promoter did not significantly change after treatment with glucocorticoids (Figure 4). These results suggest that the chromatin structure of the IκBα promoter in untreated cells is already hypersensitive or open and does not require hormone-dependant chromatin disruption to initiate transcription.
In contrast to many other glucocorticoid-responsive genes, the IκBα promoter contains no known full HREs up to approximately −1200 bp of the promoter, although several half-HREs are present. However, a reporter plasmid containing up to −623 bp of the promoter is sufficient for glucocorticoid activation when the promoter is transiently transfected into tissue culture cells (Heck et al., 1997). On the basis of this data, we wanted to determine if glucocorticoid treatment altered the pattern of bands created by DNaseI. In this assay, the footprint, or areas lacking bands, often indicate bound transcription factors. Bands that increase in intensity may flank these footprints and indicate perturbations in chromatin structure that render the DNA more accessible to cleavage. Therefore, we looked for glucocorticoid-mediated alterations in the 10-bp ladder obtained by DNaseI analysis of untreated cells. We found that this ladder was interrupted at −170, where cleavage by DNaseI did not appear to occur, suggesting that a transcription factor such as CP2 may be prebound to the promoter. Interestingly, we also found that several sites were protected from digestion in the glucocorticoid-treated samples, compared with untreated controls (Figures 5 and 6). Several of these footprints overlapped with putative transcription factor binding sites. These sites include a GR half-site at −91/−86, an NFκB-like site at −152/−153, and the NFκB sites at −225/−216 and −63/−53. The Ets-1 site at −103/−96 and the SP1 site at −44/−36 may also be protected. These changes in DNaseI suggest that glucocorticoid activation of IκBα transcription may involve factor binding to the proximal region of the promoter. Indeed, previous in vivo footprinting assays have suggested that Ets-1, AP-2, NFκB, and SP-1 factors may bind constitutively to the IκBα promoter in Jurkat cells (Algartéet al., 1999). In contrast, in A1-2 breast cancer cells, our data suggest that factors bind only after glucocorticoid activation. It will be important to further evaluate if these potential differences represent tissue specific regulation of the IκBα promoter in Jurkat and breast cancer cells.
Transcription factor binding can occur even when changes in nucleosome position do not occur. For example, binding of NF1 and OTFs upon hormone induction of the MMTV promoter does not alter nucleosome position (Lee and Archer, 1994; Mymryk et al., 1995). Similarly, on the RARβ2 promoter, DNA binding of the RXR-RAR heterodimer did not alter the nucleosomal organization (Bhattacharyya et al., 1997). Thus, in A1-2 cells, although glucocorticoids were required to induce IκBα transcription, they do not result in chromatin remodeling. These results could place IκBα into the category of “preset” promoters, which have open chromatin structures before activation. These promoters may be prebound with transcription factors, but usually require other factors for activation. On preset promoters, transcription is independent of chromatin disruption but dependent on binding of new transcription factors or on modification of prebound factors. There are many examples of preset promoters in the literature. The IL-6 promoter in MDA-MB-231 cells, which is extensively occupied by prebound factors both before and after activation by TNF-α 20 (Armenante et al., 1999). The Xenopus hsp70 promoter is preset by the transcription factor NF-Y, but requires the acetyltransferase activity of p300 for activation (Li et al., 1998). The gadd45 gene is activated by ionizing radiation and may be prebound by octamer transcription factors, and AP-1 and p53 (Graunke et al., 1999). Drosophila hsp26 and hsp70 are examples of other preset promoters (Cartwright and Elgin, 1986; Thomas and Elgin, 1988). As with these promoters, IκBα appears preset for transcription, requiring glucocorticoids to activate transcription, but not to remodel chromatin.
This open structure via bound factors is analagous to binding of NF1 on the MMTV promoter after transient transfection, where this binding is coincident with a constitutive open architecture of the transfected DNA (Archer et al., 1992). The MMTV promoter acquires a similar architecture when stably integrated into T47D/2963.1 cells, which contain the progesterone receptor (PR), but not the GR. In these cells, MMTV is in an open configuration, and the PR is consititutively bound to nuc-B of the promoter (Mymryk et al., 1995). Progestin is required to activate transcription but not to remodel the chromatin structure of MMTV. Similarly, in the T47D/M10 cell line, which contains the GR but not the PR, the stably integrated MMTV promoter is constitutively open but requires glucocorticoid for activation (Kinyamu et al., 2000).
The low- and high-resolution analysis of the IκBα promoter reported in this investigation strongly indicate that the unstimulated IκBα promoter in A1-2 cells is organized into a phased array of nucleosomes. Glucocorticoid treatment leads to an increase in IκBα mRNA and specific changes in the MNase sensitivity of the proximal nucleosomes, but does not alter sensitivity to restriction enzymes (Figure 7). In contrast, analysis with DNase I revealed a limited but significant alteration in the chromatin architecture of the promoter upon hormone treatment. Rather than the induction of hypersensitive sites, there was a reduction of cleavage that was consistent with the stable binding of transcription factors at the promoter. This hormone-dependent “hyposensitivity” may reflect the lack of canonical GREs within the proximal promoter and/or the function of the various other transcription factors that appear to be recruited to the promoter by the GR. Consequently, the GR-mediated activation of the IκBα promoter may represent a novel mechanism by which SHRs stimulate gene expression from single copy genes within chromosomes. In the future, the characterization of additional glucocorticoid responsive genes will allow us to determine if this mechanism is used at other promoters.
Figure 7.

Schematic representation of the IκBα promoter. The region of IκBα that was mapped to bp resolution is shown. ▵, sites of glucocorticoid-induced MNase hypersensitivity; ▾, sites where glucocorticoid treatment reduced DNaseI cleavage.
ACKNOWLEDGMENTS
We thank Dr. A. Israel for the CMV-IκBα and (−623/+11) IκBα-luc constructs, Dr. R. Scheinman for the 1.4-kB IκBα–CAT plasmid, and Dr. G. DiMattia for the cyclophilin cDNA. We thank Dr. C. Weinberger, Dr. D. Swope, and Dr. L. McKay and members of the Archer laboratory for critical review of the manuscript. B.J.D. was supported by a Medical Research Council of Canada Doctoral Scholarship.
REFERENCES
- Algarté M, Kwon H, Genin P, Hiscott J. Identification by in vivo genomic footprinting of a transcriptional switch containing NF-kappaB and Sp1 that regulates the IkappaBalpha promoter. Mol Cell Biol. 1999;19:6140–6153. doi: 10.1128/mcb.19.9.6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljada A, Ghanim H, Assian E, Mohanty P, Hamouda W, Garg R, Dandona P. Increased IkappaB expression and diminished nuclear NF-kappaB in human mononuclear cells following hydrocortisone injection. J Clin Endocrinol Metab. 1999;84:3386–3389. doi: 10.1210/jcem.84.9.6104. [DOI] [PubMed] [Google Scholar]
- Amero SA, Kretsinger RH, Moncrief ND, Yamamoto KR, Pearson WR. The origin of nuclear receptor proteins: a single precursor distinct from other transcription factors. Mol Endocrinol. 1992;6:3–7. doi: 10.1210/mend.6.1.1738368. [DOI] [PubMed] [Google Scholar]
- Archer TK, Cordingley MG, Wolford RG, Hager GL. Transcription factor access is mediated by accurately positioned nucleosomes on the mouse mammary tumor virus promoter. Mol Cell Biol. 1991;11:688–698. doi: 10.1128/mcb.11.2.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer TK, Deroo BJ, Fryer CJ. Chromatin modulation of glucocorticoid and progesterone receptor activity. Trends Endocrinol Metab. 1997;8:384–390. doi: 10.1016/s1043-2760(97)00159-8. [DOI] [PubMed] [Google Scholar]
- Archer TK, Hager GL, Omichinski JG. Sequence-specific DNA binding by glucocorticoid receptor “zinc finger peptides.”. Proc Natl Acad Sci USA. 1990;87:7560–7564. doi: 10.1073/pnas.87.19.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer TK, Lefebvre P, Wolford RG, Hager GL. Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation [published erratum appears in Science 1992 Apr 10;256(5054):161] Science. 1992;255:1573–1576. doi: 10.1126/science.1347958. [DOI] [PubMed] [Google Scholar]
- Armenante F, Merola M, Furia A, Tovey M, Palmieri M. Interleukin-6 repression is associated with a distinctive chromatin structure of the gene. Nucleic Acids Res. 1999;27:4483–4490. doi: 10.1093/nar/27.22.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis [see comments] Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya N, Dey A, Minucci S, Zimmer A, John S, Hager G, Ozato K. Retinoid-induced chromatin structure alterations in the retinoic acid receptor beta2 promoter. Mol Cell Biol. 1997;17:6481–6490. doi: 10.1128/mcb.17.11.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortvin A, Winston F. Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science. 1996;272:1473–1476. doi: 10.1126/science.272.5267.1473. [DOI] [PubMed] [Google Scholar]
- Boyes J, Felsenfeld G. Tissue-specific factors additively increase the probability of the all- or-none formation of a hypersensitive site. EMBO J. 1996;15:2496–2507. [PMC free article] [PubMed] [Google Scholar]
- Carr KD, Richard-Foy H. Glucocorticoids locally disrupt an array of positioned nucleosomes on the rat tyrosine aminotransferase promoter in hepatoma cells. Proc Natl Acad Sci USA. 1990;87:9300–9304. doi: 10.1073/pnas.87.23.9300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright IL, Elgin SC. Nucleosomal instability and induction of new upstream protein-DNA associations accompany activation of four small heat shock protein genes in Drosophila melanogaster. Mol Cell Biol. 1986;6:779–791. doi: 10.1128/mcb.6.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiao PJ, Miyamoto S, Verma IM. Autoregulation of I kappa B alpha activity. Proc Natl Acad Sci USA. 1994;91:28–32. doi: 10.1073/pnas.91.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingwood TN, Urnov FD, Wolffe AP. Nuclear receptors: coactivators, corepressors and chromatin remodeling in the control of transcription. J Mol Endocrinol. 1999;23:255–275. doi: 10.1677/jme.0.0230255. [DOI] [PubMed] [Google Scholar]
- Deroo BJ, Archer TK. Glucocorticoid receptor-mediated chromatin remodeling in vivo. Oncogene Rev. 2001;20:3039–3046. doi: 10.1038/sj.onc.1204328. [DOI] [PubMed] [Google Scholar]
- Elgin SC. The formation and function of DNase I hypersensitive sites in the process of gene activation. J Biol Chem. 1988;263:19259–19262. [PubMed] [Google Scholar]
- Fragoso G, John S, Roberts MS, Hager GL. Nucleosome positioning on the MMTV LTR results from the frequency-biased occupancy of multiple frames. Genes Dev. 1995;9:1933–1947. doi: 10.1101/gad.9.15.1933. [DOI] [PubMed] [Google Scholar]
- Fragoso G, Pennie WD, John S, Hager GL. The position and length of the steroid-dependent hypersensitive region in the mouse mammary tumor virus long terminal repeat are invariant despite multiple nucleosome B frames. Mol Cell Biol. 1998;18:3633–3644. doi: 10.1128/mcb.18.6.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S. Regulation of inducible gene expression by the transcription factor NF- kappaB. Immunol Res. 1999;19:183–189. doi: 10.1007/BF02786486. [DOI] [PubMed] [Google Scholar]
- Graunke DM, Fornace AJ, Jr, Pieper RO. Presetting of chromatin structure and transcription factor binding poise the human GADD45 gene for rapid transcriptional up-regulation. Nucleic Acids Res, 1999;27:3881–3890. doi: 10.1093/nar/27.19.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PD, Barbaric S, Horz W. Restriction nucleases as probes for chromatin structure. Methods Mol Biol. 1999;119:417–425. doi: 10.1385/1-59259-681-9:417. [DOI] [PubMed] [Google Scholar]
- Han SJ, Choi JH, Ko HM, Yang HW, Choi IW, Lee HK, Lee OH, Im SY. Glucocorticoids prevent NF-kappaB activation by inhibiting the early release of platelet-activating factor in response to lipopolysaccharide. Eur J Immunol. 1999;29:1334–1341. doi: 10.1002/(SICI)1521-4141(199904)29:04<1334::AID-IMMU1334>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Heck S, Bender K, Kullmann M, Gottlicher M, Herrlich P, Cato AC. I kappaB alpha-independent downregulation of NF-kappaB activity by glucocorticoid receptor. EMBO J. 1997;16:4698–4707. doi: 10.1093/emboj/16.15.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito CY, Kazantsev AG, Baldwin AS., Jr Three NF-kappa B sites in the I kappa B-alpha promoter are required for induction of gene expression by TNF alpha. Nucleic Acids Res. 1994;22:3787–3792. doi: 10.1093/nar/22.18.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenster G, Spencer TE, Burcin MM, Tsai SY, Tsai MJ, O'Malley BW. Steroid receptor induction of gene transcription: a two-step model. Proc Natl Acad Sci USA. 1997;94:7879–7884. doi: 10.1073/pnas.94.15.7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinyamu HK, Fryer CJ, Horwitz KB, Archer TK. The MMTV promoter adopts distinct chromatin structures in human breast cancer cells with and without glucocorticoid receptor. J Biol Chem. 2000;275:20061–20068. doi: 10.1074/jbc.M001142200. [DOI] [PubMed] [Google Scholar]
- Le Bail O, Schmidt-Ullrich R, Israel A. Promoter analysis of the gene encoding the I kappa B-alpha/MAD3 inhibitor of NF-kappa B: positive regulation by members of the rel/NF- kappa B family. EMBO J. 1993;12:5043–5049. doi: 10.1002/j.1460-2075.1993.tb06197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HL, Archer TK. Nucleosome-mediated disruption of transcription factor-chromatin initiation complexes at the mouse mammary tumor virus long terminal repeat in vivo. Mol Cell Biol. 1994;14:32–41. doi: 10.1128/mcb.14.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Herrler M, Landsberger N, Kaludov N, Ogryzko VV, Nakatani Y, Wolffe AP. Xenopus NF-Y pre-sets chromatin to potentiate p300 and acetylation- responsive transcription from the Xenopus hsp70 promoter in vivo. EMBO J. 1998;17:6300–6315. doi: 10.1093/emboj/17.21.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May MJ, Ghosh S. Rel/NF-kappa B and I kappa B proteins: an overview. Semin Cancer Biol. 1997;8:63–73. doi: 10.1006/scbi.1997.0057. [DOI] [PubMed] [Google Scholar]
- McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev. 1999;20:435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- Mymryk JS, Berard D, Hager GL, Archer TK. Mouse mammary tumor virus chromatin in human breast cancer cells is constitutively hypersensitive and exhibits steroid hormone-independent loading of transcription factors in vivo. Mol Cell Biol. 1995;15:26–34. doi: 10.1128/mcb.15.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mymryk JS, Fryer CJ, Jung LA, Archer TK. Analysis of chromatin structure in vivo. Methods. 1997;12:105–114. doi: 10.1006/meth.1997.0452. [DOI] [PubMed] [Google Scholar]
- Nordeen SK, Kuhnel B, Lawler-Heavner J, Barber DA, Edwards DP. A quantitative comparison of dual control of a hormone response element by progestins and glucocorticoids in the same cell line. Mol Endocrinol. 1989;3:1270–1278. doi: 10.1210/mend-3-8-1270. [DOI] [PubMed] [Google Scholar]
- Polach KJ, Widom J. Restriction enzymes as probes of nucleosome stability and dynamics. Methods Enzymol, 1999;304:278–298. doi: 10.1016/s0076-6879(99)04017-3. [DOI] [PubMed] [Google Scholar]
- Richard-Foy H, Hager GL. Sequence-specific positioning of nucleosomes over the steroid-inducible MMTV promoter. EMBO J. 1987;6:2321–2328. doi: 10.1002/j.1460-2075.1987.tb02507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinman RI, Beg AA, Baldwin AS., Jr NF-kappa B p100 (Lyt-10) is a component of H2TF1 and can function as an I kappa B-like molecule. Mol Cell Biol. 1993;13:6089–6101. doi: 10.1128/mcb.13.10.6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS., Jr Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids [see comments] Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- Thomas GH, Elgin SC. Protein/DNA architecture of the DNase I hypersensitive region of the Drosophila hsp26 promoter [published erratum appears in EMBO J 1988 Oct;7(10):3300] EMBO J. 1988;7:2191–2201. doi: 10.1002/j.1460-2075.1988.tb03058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallberg AE, Wright A, Gustafsson JA. Chromatin-remodeling complexes involved in gene activation by the glucocorticoid receptor. Vit Horm. 2001;60:75–122. doi: 10.1016/s0083-6729(00)60017-1. [DOI] [PubMed] [Google Scholar]
- Wolff R, Gemmill R. Purifying and analyzing genomic DNA. In: Birren B, editor. Genome Analysis: A Laboratory Manual. Vol. 1. Plainview, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 43–77. [Google Scholar]
- Wolffe AP, Hayes JJ. Chromatin disruption and modification. Nucleic Acids Res. 1999;27:711–720. doi: 10.1093/nar/27.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe AP, Kurumizaka H. The nucleosome: a powerful regulator of transcription. Prog Nucleic Acid Res Mol Biol. 1998;61:379–422. doi: 10.1016/s0079-6603(08)60832-6. [DOI] [PubMed] [Google Scholar]
- Wu C. Chromatin remodeling and the control of gene expression. J Biol Chem. 1997;272:28171–28174. doi: 10.1074/jbc.272.45.28171. [DOI] [PubMed] [Google Scholar]