

Abstract
INTRODUCTION:
Vici syndrome, a rare autosomal recessive disorder, was first described in 1988 by Vici et al. Only 78 cases have been reported to date. The syndrome is characterised by agenesis of the corpus callosum, hypopigmentation, cardiomyopathy, progressive failure to thrive, dysmorphic features, immunodeficiency and cataracts. Mutations in the gene epg5 have been identified as the cause of Vici syndrome.
CASE DESCRIPTION:
The parents are a consanguineous Saudi couple with two other children diagnosed with Gaucher disease. The patient was born at term and in the first 5 months had many hospital admissions for a recurrent chest infection. Physical examination, investigations and imaging studies revealed that the patient had agenesis of the corpus callosum, cataracts, psychomotor delay, immunodeficiency and hypopigmentation. The initial echocardiogram was normal. At 7 months, genetic testing confirmed the diagnosis of Vici syndrome with a c.3693G>Ap (Gln1231Gln) mutation in the gene EPG5. The patient developed a chest infection and was admitted to the pediatric intensive care unit. An echocardiogram was repeated and showed significant left ventricular dilation with a Z-score of 3.1, moderate mitral and tricuspid regurgitation, and depressed ventricular function with a fractional shortening of 17% and ejection fraction 37%. The patient’s condition deteriorated, and he died aged 8 months.
CONCLUSION:
The symptoms of extensive system involvement in Vici syndrome have been present in the majority of reported cases and should prompt careful evaluation of this syndrome when such symptoms are present in an infant. In confirmed cases, close monitoring of the immune status and cardiac function, the two main causes of death among Vici syndrome patients, is vital to prevent rapid deterioration and improve life expectancy.
Keywords: Vici syndrome, EPG5 gene, Cardiomyopathy
Introduction
Vici syndrome (VICIS) (OMIM 242840) is a rare autosomal recessive disease belonging to the group of congenital disorders of autophagy. The syndrome has a wide range of presentations involving various body systems [1] [2] and is caused by a mutation in epg5, the gene for ectopic P-granules protein 5 (EPG5) on chromosome 18 [2]. The EPG5 protein is responsible for regulating autophagy activity, a pivotal mechanism for the development and proper functioning of body organs. The first two cases were described by Vici and his colleagues in 1988 when they reported on two siblings with a set of clinical features comprising agenesis of the corpus callosum, cutaneous hypopigmentation, bilateral cataract, cleft lip and palate, and combined immunodeficiency [3].
Since that original description of the disorder, an increasing number of cases have been reported, with almost 78 confirmed cases published to date [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22]. The patients have presented, mostly in infancy, with characteristic features of VICIS together with other phenotypic features such as progressive failure to thrive, microcephaly, nystagmus, dysmorphic features, cardiomyopathy, hypotonia, and recurrent pulmonary infection, among others [4]. Features such as hearing loss, lung hypoplasia, and renal tubular necrosis have also been described in isolated cases [6] [7] [8].
In this report, which includes a review of all literature to date, we describe in detail a previously reported case [9] of an 8-month-old male infant diagnosed with VICIS.
Case Description
We report on a boy, the third child of healthy consanguineous first-cousin Saudi parents. His two brothers are confirmed cases of Gaucher disease. The boy is a product of spontaneous vaginal delivery at 36 weeks gestational age with birth weight 3.2 kg. At 10 days of age, he was admitted to the neonatal intensive care unit because of meconium aspiration and group B streptococcus sepsis.
During the first month of life at home, the patient developed poor sucking and choking attacks with each feeding. At the age of 2 months, he was admitted to an outside hospital as a case of aspiration pneumonia and was treated accordingly. Diagnosis of gastroesophageal reflux disease was also made, and he underwent a fundoplication. During that admission, the patient was evaluated for the choking attacks and hypotonia. Chromosomal analysis revealed a normal karyotype (46XY).
At 5 months of age he was admitted to our hospital, for the first time, as a case of pneumonia. On physical examination, he also had profound hypotonia and albinism. Abdominal ultrasound was unremarkable and ruled out any organomegaly for other metabolic syndromes. Visual evoked response (VER) was significant for bilateral P100 prolongation and abnormal NPN configuration. VICIS was suspected as a cause of his multisystem symptoms. Magnetic resonance imaging (MRI) of the brain showed complete agenesis of corpus callosum, hypoplastic pons, and volume loss of white matter (Figure 1). Echocardiographic evaluation showed normal function with only mild left ventricular hypertrophy and fractional shortening of 33.1%. Genetic testing identified a pathogenic variant of epg5 (c.3693G>A p. (Gln1231Gln) and confirmed the diagnosis of VICIS.
Figure 1.
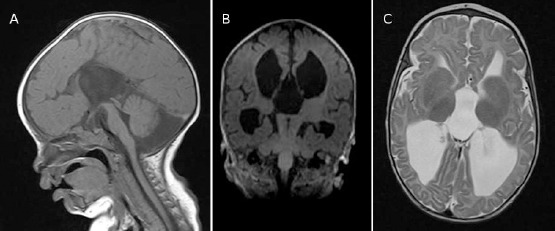
Brain MRI at age 6 months. Medline sagittal T1 weighted sequence (A), showing agenesis of corpus callosum and hypoplastic pons. Coronal T1 (B) and axial T2 (C) showing loss of white matter and dilated ventricular system
Following this admission, the patient presented several times to our hospital with recurrent infections including bronchial pneumonia, urinary tract infection and gastroenteritis. At the age of 7 months he presented to the emergency department following a 3-week episode of bloody diarrhea with severe dehydration. On physical examination, the patient looked pale, cachectic, not oriented, with sunken eyes and anterior fontanelles. His arterial blood gases revealed hypokalemia with metabolic acidosis (K = 2 mmol/L, pH = 7.28, PCO2 = 37 torr, HCO3 = 16.8 mmol/L). His weight had dropped 2.1 kg from the admission a month previously. He was admitted to the pediatric intensive care unit for rehydration and monitoring. During the course of this admission, the immunologic workup revealed reduced absolute B and T cell counts; immunoglobulins A, M and G were within the normal range. A few days later, the patient suddenly became apneic and developed respiratory distress. Chest X-ray revealed cardiomegaly. Repeated echocardiography showed dilated cardiomyopathy. His echocardiogram findings were significant for left ventricular dilation with a Z-score of 3.1, moderate mitral and tricuspid regurgitation, and depressed ventricular function with fractional shortening of 17% and ejection fraction 37%. He deteriorated quickly and developed renal impairment, which worsened his electrolyte imbalance and metabolic acidosis. Eventually, the patient went into cardiopulmonary arrest and died.
Discussion
VICIS is a rare disease that affects multiple systems of the body. Since the condition was first described in 1988, a total of 78 cases —including our patient’s—have been reported (Table 1). The incidence of this disease is yet to be determined. VICIS was first linked to a recessive mutation in EPG5 in 2013 [1]. The EPG5 protein is responsible for regulating autophagy activity, which is a survivor-cell mechanism that selectively removes misfolded proteins and organelles and plays a paramount role in ensuring good development of body systems, especially the nervous system. In our patient, genetic testing showed a c.3693G>A p.(Gln1231Gln) mutation in epg5, confirming the diagnosis of VICIS.
Table 1.
Summary of reported patients with Vici syndrome
| Authors, year | Sex | Age (m) | Clinical outcome |
|---|---|---|---|
| Vici et al., 1988 [3] | M | 24 | Deceased |
| M | 36 | ||
| Del Campo et al., 1999 [4] | M | 24 | Deceased |
| F | 11 | Deceased | |
| M | 36 | Alive | |
| F | 16 | Deceased | |
| Chiyonobu et al., 2002 [5] | F | 19 | Deceased |
| M | 6 | Alive | |
| Miyata et al., 2007 [6] | F | 12 | Deceased |
| M | 11 | Alive | |
| McClelland et al., 2010 [7] | M | 3 | Deceased |
| Al-Owain et al., 2010 [8] | M | 9 | Deceased |
| Rogers et al., 2011 [10] | M | 96 | Deceased |
| F | 96 | Deceased | |
| Said et al., 2012 [11] | F | 15 | Deceased |
| Finocchi et al., 2012 [12] | M | 24 | Alive |
| Özkale et al., 2012 [13] | F | 6 | Deceased |
| Cullup et al., 2013 [1] | F | 15 | Deceased |
| M | 3 | Alive | |
| M | 48 | Alive | |
| M | 36 | Alive | |
| M | 9 | Alive | |
| M | 36 | Alive | |
| M | 24 | Alive | |
| M | 24 | Alive | |
| F | 120 | Alive | |
| Ehmke et al., 2014 [14] | M | 3.5 | Deceased |
| Filloux et al., 2014 [15] | F | 17 | Alive |
| Tasdemir et al., 2015 [16] | M | 9 | Deceased |
| M | 8 | ||
| El-Kersh et al., 2015 [17] | F | 72 | Deceased |
| Byrne et al., 2016 [2] | NS | NS | NS |
| Huenerberg et al., 2016 [18] | F | 11 | Deceased |
| F | 13 | ||
| Maillard et al., 2017 [19] | F | 24 | Alive |
| Hori et al., 2017 [20] | F | 84 | Alive |
| F | 24 | Alive | |
| F | 180 | Alive | |
| F | 12 | Deceased | |
| F | 48 | Alive | |
| F | 24 | Alive | |
| M | 168 | Deceased | |
| M | 60 | Alive | |
| M | 84 | Alive | |
| Hedberg-Oldfors et al., 2017 [21] | M | 8 | Deceased |
| Elsayed et al., 2018 [22] | M | 7 | Deceased |
| This Case | M | 8 | Deceased |
M = male; F = female; NS = not specified; m months.
Table 2 summarizes all the important features that have been associated with VICIS to date. The majority of reported cases were associated with recurrent infections (98.7%), agenesis of corpus callosum (97.4%), profound developmental delay (97.4%), skin involvement (96.2%), immunodeficiency (76.9%), cataract (62.8%) and cardiomyopathy (65.4%) with the hypertrophic type being most commonly reported. We suggest, therefore, that VICIS should be considered to head the list of differential diagnoses for an infant who presents with all or most of these features.
Table 2.
Common clinical features of 78 cases of Vici syndrome
| Feature | Positive | Negative | Not reported | n (%) |
|---|---|---|---|---|
| Recurrent infections | 77 | - | 1 | 77/78 (98.7) |
| Corpus callosum agenesis | 76 | - | 2 | 76/78 (97.4) |
| Profound developmental delay | 76 | 1 | 1 | 76/78 (97.4) |
| Cutaneous manifestations | 75 | 3 | - | 75/78 (96.2) |
| Immune system involvement | 60 | 16 | 2 | 60/78 (76.9) |
| Cardiomyopathy | 51 | 18 | 9 | 51/78 (65.4) |
| Cataract | 49 | 25 | 4 | 49/78 (62.8) |
| Microcephaly | 45 | 15 | 18 | 45/78 (57.7) |
| Hypotonia | 37 | - | 41 | 37/78 (47.4) |
| Seizures | 26 | 16 | 36 | 26/78 (33.3) |
| Growth retardation | 24 | - | 54 | 24/78 (30.8) |
Our findings support those published in a review of 38 cases [2] of VICIS where agenesis of corpus callosum, profound developmental delay and immune problems were the most common shared features.
Concurrently with the clinical assessment, there are multiple investigations to help shorten the list of differential diagnoses and assess the extent of organ involvement [23] [24]. Such tests include laboratory investigation, imaging, molecular and other tests. Laboratory investigations will be directed to assess immunodeficiency and the extent of involvement of other organs such as liver and kidneys.
For imaging, a brain MRI is essential for detection of agenesis of corpus callosum and other less specific neuroradiologic abnormalities that have been reported, such as vermis and pons hypoplasia. Chest X-ray and echocardiography are also of benefit for assessing lung and cardiac involvement, respectively. Abdominal ultrasonography helps to confirm laboratory findings, whether abdominal organs are affected or not.
Confirmation of diagnosis requires molecular genetic testing to identify the homogenous or compound heterogenous mutated epg5. Other useful tests for VICIS cases are ophthalmologic tests and electroencephalography (EEG), especially if seizures are present. Our patient underwent all these investigations apart from the EEG, as seizures were absent; this helped to confirm the diagnosis and identify the present complications, thus allowing us to tailor our interventions.
VICIS is a progressive disease with poor prognosis. Survival analysis shows that VICIS patients have a median survival time of 24 months (95% confidence interval, 0–39 months) [2]. Thus, therapeutic interventions for VICIS are merely supportive and directed to relieve the symptoms that result from multi-organ involvement and to improve the survival time.
The most common causes of death among all reported patients with VICIS are recurrent infections and cardiomyopathy. Cardiac functions, in particular, can deteriorate from baseline in a short time, as happened in our patient. Immunity status and cardiac function, therefore, need to be regularly monitored at short intervals for early detection of their VICIS-related manifestations, thus allowing timely intervention for a better outcome and prolonged survival.
In conclusion, in VICIS, the symptoms of extensive system involvement (such as agenesis of corpus callosum, profound developmental delay, recurrent infections and immunodeficiency) that present in the majority of cases should prompt a careful evaluation for this syndrome. When the diagnosis is confirmed, close monitoring of the immune status and cardiac function is vital to prevent rapid deterioration and improve life expectancy, as these have been the two main causes of death among VICIS patients in all known cases to date.
Footnotes
Funding: This research did not receive any financial support
Competing Interests: The authors have declared that no competing interests exist
References
- 1.Cullup T, Kho A, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nature Genetics. 2012;45(1):83–87. doi: 10.1038/ng.2497. https://doi.org/10.1038/ng.2497 PMid:23222957 PMCid: PMC4012842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byrne S, Jansen L, U-King-Im J, Siddiqui A, Lidov H, Bodi I, et al. EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain. 2016;139(3):765–781. doi: 10.1093/brain/awv393. https://doi.org/10.1093/brain/awv393 PMid:26917586 PMCid: PMC4766378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, Boldrini R, et al. Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. American Journal of Medical Genetics. 1988;29(1):1–8. doi: 10.1002/ajmg.1320290102. https://doi.org/10.1002/ajmg.1320290102 PMid:3344762. [DOI] [PubMed] [Google Scholar]
- 4.del Campo M, Hall B, Aeby A, Nassogne M, Verloes A, Roche C, et al. Albinism and agenesis of the corpus callosum with profound developmental delay: Vici syndrome, evidence for autosomal recessive inheritance. American Journal of Medical Genetics. 1999;85(5):479–485. doi: 10.1002/(sici)1096-8628(19990827)85:5<479::aid-ajmg9>3.3.co;2-4. https://doi.org/10.1002/(SICI)1096-8628(19990827)85:5<479:: AID-AJMG9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 5.Chiyonobu T, Yoshihara T, Fukushima Y, Yamamoto Y, Tsunamoto K, Nishimura Y, et al. Sister and brother with Vici syndrome: Agenesis of the corpus callosum, albinism, and recurrent infections. American Journal of Medical Genetics. 2002;109(1):61–66. doi: 10.1002/ajmg.10298. https://doi.org/10.1002/ajmg.10298 PMid:11932994. [DOI] [PubMed] [Google Scholar]
- 6.Miyata R, Hayashi M, Sato H, Sugawara Y, Yui T, Araki S, et al. Sibling cases of Vici syndrome: Sleep abnormalities and complications of renal tubular acidosis. American Journal of Medical Genetics Part A. 2007;143A(2):189–194. doi: 10.1002/ajmg.a.31584. https://doi.org/10.1002/ajmg.a.31584 PMid:17163544. [DOI] [PubMed] [Google Scholar]
- 7.McClelland V, Cullup T, Bodi I, Ruddy D, Buj-Bello A, Biancalana V, et al. Vici syndrome associated with sensorineural hearing loss and evidence of neuromuscular involvement on muscle biopsy. American Journal of Medical Genetics Part A. 2010;152A(3):741–747. doi: 10.1002/ajmg.a.33296. https://doi.org/10.1002/ajmg.a.33296 PMid:20186778. [DOI] [PubMed] [Google Scholar]
- 8.Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, Al-Homoud I, et al. Vici syndrome associated with unilateral lung hypoplasia and myopathy. American Journal of Medical Genetics Part A. 2010;152A(7):1849–1853. doi: 10.1002/ajmg.a.33421. https://doi.org/10.1002/ajmg.a.33421 PMid:20583151. [DOI] [PubMed] [Google Scholar]
- 9.Waggass R. Dilated cardiomyopathy in a Saudi male infant with Vici syndrome. Heart, Lung and Circulation. 2015;24:S428. https://doi.org/10.1016/j.hlc.2015.06.734. [Google Scholar]
- 10.Rogers R, Aufmuth B, Monesson S. Vici Syndrome: A Rare Autosomal Recessive Syndrome with Brain Anomalies, Cardiomyopathy, and Severe Intellectual Disability. Case Reports in Genetics. 2011;2011:1–4. doi: 10.1155/2011/421582. https://doi.org/10.1155/2011/421582 PMid:23091746 PMCid: PMC3447215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Said E, Soler D, Sewry C. Vici syndrome-A rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. American Journal of Medical Genetics Part A. 2011;158A(2):440–444. doi: 10.1002/ajmg.a.34273. https://doi.org/10.1002/ajmg.a.34273 PMid:21964879. [DOI] [PubMed] [Google Scholar]
- 12.Finocchi A, Angelino G, Cantarutti N, Corbari M, Bevivino E, Cascioli S, et al. Immunodeficiency in Vici syndrome: A heterogeneous phenotype. American Journal of Medical Genetics Part A. 2011;158A(2):434–439. doi: 10.1002/ajmg.a.34244. https://doi.org/10.1002/ajmg.a.34244 PMid:21965116. [DOI] [PubMed] [Google Scholar]
- 13.Özkale M, Erol I, Gümüş A, Özkale Y, Alehan F. Vici Syndrome Associated With Sensorineural Hearing Loss and Laryngomalacia. Pediatric Neurology. 2012;47(5):375–378. doi: 10.1016/j.pediatrneurol.2012.07.007. https://doi.org/10.1016/j.pediatrneurol.2012.07.007 PMid:23044023. [DOI] [PubMed] [Google Scholar]
- 14.Ehmke N, Parvaneh N, Krawitz P, Ashrafi M, Karimi P, Mehdizadeh M, et al. First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon ofEPG5and review of the literature. American Journal of Medical Genetics Part A. 2014;164(12):3170–3175. doi: 10.1002/ajmg.a.36772. https://doi.org/10.1002/ajmg.a.36772 PMid:25331754. [DOI] [PubMed] [Google Scholar]
- 15.Filloux F, Hoffman R, Viskochil D, Jungbluth H, Creel D. Ophthalmologic Features of Vici Syndrome. Journal of Pediatric Ophthalmology & Strabismus. 2014;51(4):214–220. doi: 10.3928/01913913-20140423-02. https://doi.org/10.3928/01913913-20140423-02 PMid:24779424. [DOI] [PubMed] [Google Scholar]
- 16.Tasdemir S, Sahin I, Cayır A, Yuce I, Ceylaner S, Tatar A. Vici syndrome in siblings born to consanguineous parents. American Journal of Medical Genetics Part A. 2015;170(1):220–225. doi: 10.1002/ajmg.a.37398. https://doi.org/10.1002/ajmg.a.37398 PMid:26395118. [DOI] [PubMed] [Google Scholar]
- 17.El-Kersh K, Jungbluth H, Gringras P, Senthilvel E. Severe Central Sleep Apnea in Vici Syndrome. Pediatrics. 2015;136(5):e1390–e1394. doi: 10.1542/peds.2015-0297. https://doi.org/10.1542/peds.2015-0297 PMid:26482670. [DOI] [PubMed] [Google Scholar]
- 18.Huenerberg K, Hudspeth M, Bergmann S, Pai S, Singh B, Duong A. Two cases of Vici syndrome associated with Idiopathic Thrombocytopenic Purpura (ITP) with a review of the literature. American Journal of Medical Genetics Part A. 2016;170(5):1343–1346. doi: 10.1002/ajmg.a.37589. https://doi.org/10.1002/ajmg.a.37589 PMid:26854214. [DOI] [PubMed] [Google Scholar]
- 19.Maillard C, Cavallin M, Piquand K, Philbert M, Bault J, Millischer A, et al. Prenatal and postnatal presentations of corpus callosum agenesis with polymicrogyria caused byEGP5mutation. American Journal of Medical Genetics Part A. 2017;173(3):706–711. doi: 10.1002/ajmg.a.38061. https://doi.org/10.1002/ajmg.a.38061 PMid:28168853. [DOI] [PubMed] [Google Scholar]
- 20.Hori I, Otomo T, Nakashima M, Miya F, Negishi Y, Shiraishi H, et al. Defects in autophagosome-lysosome fusion underlie Vici syndrome, a neurodevelopmental disorder with multisystem involvement. Scientific Reports. 2017;7(1) doi: 10.1038/s41598-017-02840-8. https://doi.org/10.1038/s41598-017-02840-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hedberg-Oldfors C, Darin N, Oldfors A. Muscle pathology in Vici syndrome–A case study with a novel mutation in EPG5 and a summary of the literature. Neuromuscular Disorders. 2017;27(8):771–776. doi: 10.1016/j.nmd.2017.05.005. https://doi.org/10.1016/j.nmd.2017.05.005 PMid:28624465. [DOI] [PubMed] [Google Scholar]
- 22.Elsayed S, Gamal R. Cardiomyopathy in Vici syndrome. Egyptian Journal of Medical Human Genetics. 2018;19(1):49–50. https://doi.org/10.1016/j.ejmhg.2017.12.003. [Google Scholar]
- 23.Cullup T, Dionisi-Vici C, Kho A, Yau S, Mohammed S, Gautel M, et al. Clinical utility gene card for: Vici Syndrome. European Journal of Human Genetics. 2013;22(3) doi: 10.1038/ejhg.2013.142. PMid 23838600; PMCid: PMC3925270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H. Vici syndrome: a review. Orphanet Journal of Rare Diseases. 2016;11(1) doi: 10.1186/s13023-016-0399-x. https://doi.org/10.1186/s13023-016-0399-x PMid:26927810 PMCid: PMC4772338. [DOI] [PMC free article] [PubMed] [Google Scholar]