

Abstract
Tumor sensitivity to radiation therapy has been known to be dependent on O2 concentrations. However, radiosensitivity of naturally occurring hypoxic tumor cells remains to be well fully investigated in direct comparison to that of their adjacent non-hypoxic tumor cells within the same tumor. We developed a hypoxia-sensing xenograft model using the hypoxia-response element (HRE)-driven enhanced green fluorescence protein (EGFP) as a hypoxia reporter to identify hypoxic tumor cells in situ. Here, we have found that naturally hypoxic tumor cells are moderately radioresistant compared to their neighboring non-hypoxic tumor cells in the same tumor. These naturally hypoxic tumor cells are proficient at repairing DNA damages and resist apoptosis induced by genotoxic stresses, which involves activation of the ATM/CHK1/CHK2 DNA damage-sensing pathway. Inhibition of the checkpoint kinases sensitizes the ex vivo hypoxic tumor cells to ionizing irradiation. Second, the new functional phenotypes acquired by the hypoxic tumor cells in vivo are stable even after they are maintained under non-hypoxic conditions. These new results strongly suggest that the hypoxic tumor microenvironment is capable of selecting stable tumor cell populations with increased resistance to genotoxic stresses and enhanced survival.
Keywords: Checkpoint kinases, DNA Damage Response, Hypoxia, Tumor Microenvironment, Xenograft
1. Introduction
Tumor hypoxia, a hallmark of tumor microenvironment (TME), is an independent prognostic factor for advanced disease progression and poor patient survival [1–5]. Hypoxia negatively impact many modalities of cancer treatment, especially radiotherapy [2, 6, 7]. Molecular oxygen as a critical determinant of cellular response to ionizing irradiation was first reported by Crabtree and Cramer [8]. Subsequently, Gray and his colleagues quantitatively defined the oxygen effects on cellular response to irradiation [9–11]. The first study demonstrating a correlation between hypoxia and radiosensitivity of human tumors was reported by Gatenby et al. who examined 31 fixed lymph node metastases from squamous cell carcinoma of the head and neck and found that tumors containing >26% tumor volume with pO2 ≤ 8 mmHg responded poorly to radiotherapy [12]. However, oxygen effects on ionizing irradiation has so far been extensively studied in cultured cells under defined in vitro hypoxic conditions. The survival of naturally hypoxic tumor cells against ionizing irradiation has only been estimated using the clonogenic survival assay or using clamped tumor models [6]. The radiosensitivity of hypoxic tumor cells that emerge naturally in TME in direct comparison to that of their adjacent non-hypoxic tumor cells within the same tumor remains to be investigated.
In this study, we have developed a hypoxia-sensing xenograft model using human breast cancer cell line and have made several new discoveries with regard to the differential radiosensitivities of the hypoxic and non-hypoxic tumor cells in vivo. First, we have found that the naturally hypoxic tumor cells are only moderately resistant to ionizing irradiation compared to the non-hypoxic tumor cells within the same tumor. Nonetheless, the in vivo irradiated hypoxic tumor cells exhibit enhanced potentials of DNA damage repair. Very interestingly, the therapy-resistant phenotype of the ex vivo hypoxic tumor cells remains stable even after they are maintained under the ambient culture condition. Mechanistically, the canonical DNA damage sensing pathway mediated by ATM/CHK1/CHK2 is preferentially potentiated in ex vivo hypoxic tumor cells. These observations strongly suggest that the hypoxic TME may induce clonal evolution and/or phenotypic changes that leads to the selection of tumor cells with increased DNA damage repair potentials and resistance to genotoxic stresses.
2. Materials and methods
2.1 Chemicals
Etoposide (E1383, Sigma-Aldrich) was dissolved in dimethyl sulfoxide (DMSO) at 50 mM. Bleomycin sulfate (BML-AP302-0010, Enzo Life Science) was dissolved in H2O at 10 mg/ml. AZD7762 (S1532, Selleckchem) was dissolved in DMSO at 10 mM. Stock solutions were diluted in tissue culture media immediately before use to different working concentrations.
2.2 Generation of the hypoxia-sensing tumor cell line
MDA-MB-231 cells were transfected with 5HRE/GFP plasmid [13] and then selected with 500 μg/ml G418. Three rounds of positive (1% O2) and negative selections (normoxia) were done to generate a pool of cells with high hypoxia sensitivity and minimum background EGFP expression.
2.3 Xenografts and detection of tumor hypoxia in situ
MDA-MB-231/HRE-GFP cells were injected either orthotopically in the fourth mammary fat pads or subcutaneously in lower backs of female athymic nude mice (6–8 weeks) at a concentration of 1 × 106 cells per injection. When the tumor sizes reached ~500 mm3, tumor-bearing mice received an intraperitoneal injection of pimonidazole HCl, (60 mg/kg body weight, Hypoxyprobe™-1, Hypoxyprobe, Inc.) at 2 hours before tumor harvest. Tumors were fixed in formalin and cryopreserved in OCT. Tumor cryosections (7 μm) were immunostained with rabbit polyclonal anti-pimonidazole antibody (PAB2627AP, Hypoxyprobe, Inc) followed by Cy5-conjugated goat anti-rabbit IgG antibody (ThermoScientific, A10524). Nuclei were stained with Hoechst 33342 (2 μg/mL).
2.4 Ionizing irradiation
Tumor-bearing mice were irradiated using XRAD 320 (Precision X-RAY) for whole body irradiation or Siemens Stabilipan 250 for tumor-specific irradiation. Tumor cells (60–70% confluency) were irradiated in 6-cm or 10-cm dishes using XRAD 320.
2.5 Tumor cell isolation and cell sorting
A two-step digestion protocol was used to improve dissociation and isolation of tumor cells. First, excised xenograft tumors were minced and dissociated in the 37°C shaker for 2 hours with medium containing 10% Fetal Calf Serum, 0.5 U/ml dispase (#07913, STEMCELL Tech.), 5mg/ml Collagenase Type IV (CLS-4, Worthington Biochem.), and 100 U/ml Penicillin Streptomycin (15-140-122, Gibco) in DMEM (11965-084, ThermoScientific). The digested tumor tissues were pelleted and washed once in PBS before they were resuspended in 0.25% trypsin and briefly digested at room temperature for <5 minutes by repeated gentle pipetting. The dissociated cells were then collected by filtering the digested tissues through a 70-μm cell strainer. Red blood cells were removed using an NH4Cl Solution (07800, STEMCELL Tech.). Host mouse cells were depleted using the Mouse Cell Depletion Kit (130-104-694, Miltenyi Biotec.) EGFP+ (hypoxic) and EGFP− (non-hypoxic) tumor cells were sorted using BD FACSAria™ II.
2.6 Clonogenic assay
Tumor cells were plated at a density of 1,000 and 2,000 cells/well (non-irradiated group), 2,000 and 5,000 cells/well (2Gy group), 5,000 and 10,000 cells/well (7.5 Gy group), 50,000 and 100,000 cells/well (15Gy group) in 6-well plates and incubated for 10 to 14 days. Colonies were stained with Crystal Violet. Plating efficiency = number of colonies (≥50 cells/colony) divided by number of input cells × 100%.
2.7 Immunofluorescence for γH2AX and 53BP1
Cells were seeded in 48-well plates and incubated for 24 hours before treatment with etoposide (6 μM) for another 24 hours. After treatment, cells were washed twice with ice-cold PBS, fixed in a solution containing 2% paraformaldehyde and 1% sucrose for 15 minutes at room temperature, and permeabilized with ice-cold methanol and acetic acid (1:1) for 20 minutes at −20°C. Cells were washed in PBS and then incubated in a blocking buffer (10% horse serum) for 60 minutes. For ionizing irradiation, cells were fixed and processed at different time points after X-irradiation.
Incubation with rabbit polyclonal anti-53BP1 antibody (1:200, Santa Cruz, sc22760) or anti-phospho-Histone H2AX-S139 (1:200, Cell Signaling, 9718) was carried out at 4°C overnight, followed by incubation with Alexa 555–conjugated anti-rabbit IgG (1:500; Invitrogen) for 1 hour at room temperature in the dark. Nuclei were counterstained with DAPI (0.2 μg/mL, Sigma-Aldrich). Immunofluorescence was examined and pictures taken using the EVOS-FL fluorescence microscope (ThermoScientific). Nuclei with >5 foci were counted. Percentage of foci+ cells = number of foci+ cells divided by total number of cells counted x 100%.
2.8 Comet assay
Comet assays were done with CometAssay® Kit (4250-050-K, Trevigen) according to the manufacturer’s instruction. Comet scores were obtained using the Open Comet software.
2.9 Western blots
Western blot analysis were done with the following antibodies: p-CHK1 S345 (#2348, 1:2,000, Cell Signaling), total CHK1 (#04-207, 1:1,000, Millipore), p-CHK2 T68 (#2197, 1:2,000, Cell Signaling), total CHK2 (#6334, 1:1,000, Cell Signaling), p-ATM S1981 (#5883, 1:1,000, Cell Signaling), total ATM (#2873, 1:1,000, Cell Signaling), p-ATR S428 (#2853, 1:1,000, Cell Signaling), total ATR (#2790, 1:1,000, Cell Signaling), p-p53 S15 (#9286, 1:2,000, Cell Signaling), total p53 (#OP03, AB-1, 1:5,000, Oncogene), cleaved Caspase3 (#9661, 1:2,000, Cell Signaling), PARP (#9542, 1:10,000, Cell Signaling), β-actin (#A5316, 1:10,000, Sigma Aldrich), Vinculin (3AB73412, 1:5,000, Abcam). Protein bands were visualized using chemiluminescence substrates (#170-5061, BioRad) and imaged on Kodak X-OMAT 2000A.
2.10 Statistical analysis
Two-group comparison was analyzed by unpaired 2-tailed Student’s t-test (GraphPad Prizm 7). Significant difference was declared if p < 0.05.
3. Results
3.1 Hypoxic tumor cells in vivo are moderately radioresistant compared to non-hypoxic tumor cells in the same tumor
In order to directly examine the radiosensitivity of hypoxic and non-hypoxic tumor cells in the same tumor, we established a hypoxia-sensing tumor model by stably expressing a hypoxia-responsive transcription enhancer element (five tandem repeats of HRE or 5XHRE)-driven destabilized d2EGFP construct [13] in MDA-MB-231 human breast cancer cells. The d2EGFP reporter gene is transcriptionally activated by hypoxia-inducible transcription factor HIF-1 and/or HIF-2, both of which are stabilized by hypoxia [14–16]. Xenografts were generated either orthotopically in mammary fat pads or ectopically in subcutaneous space of lower backs of female athymic nu/nu mice. The hypoxic tumor regions were independently identified using the bioreductive compound pimonidazole HCl (Hypoxyprobe-1) [17, 18]. As shown in Figure 1A, the EGFP+ MDA-MB-231 cells were primarily localized in areas that were positively labeled by pimonidazole. Majority of the EGFP+ tumor cells were located at a distance from the CD31+ blood vessels (right side, Figure 1B). Nonetheless, overlap between EGFP+ tumor cells and blood vessels was also observed (upper left, Figure 1B). Furthermore, EGFP+ tumor cells are found next to necrotic regions (Supplementary Figure 1A) and strongly express the cell surface protein carbonic anhydrase 9 (CA9), another commonly used hypoxia marker (Supplementary Figure 1B). Similar patterns of hypoxia were observed in both orthotopic and subcutaneous xenografts. In addition, we have also confirmed using cDNA microarray analysis that a set of commonly observed hypoxia-inducible genes are significantly upregulated in the EGFP+ cells [19]. These results indicate that chronic or diffusion-limited hypoxia is likely more prevalent than fluctuating hypoxia in these xenografts.
Fig. 1. The naturally occurring hypoxic tumor cells have increased clonogenic survival after ionizing irradiation in vivo.

Xenograft tumors were generated in female nu/nu mice from MDA-MB-231 cells stably expressing the HRE-EGFP reporter gene. (A) Hypoxic regions were independently labeled with Hypoxyprobe-1. Tumor cells expressing the endogenous hypoxia reporter gene EGFP were primarily localized in regions labeled with the independent hypoxia marker Hypoxyprobe-1. (B) Localization of the hypoxic (EGFP+) tumor cells relative to blood vessels stained with α-CD31 antibody. (C, D, E) Clonogenic survival of in vivo irradiated hypoxic and non-hypoxic tumor cells. Tumor cells from 3–4 individual tumors were sorted by FACS into hypoxic (EGFP+) and non-hypoxic (EGFP−) populations immediately after γ-irradiation and plated for clonogenic assay in sextuplets using two seeding densities.
We performed the clonogenic assay to examine the relative radiosensitivity of hypoxic tumor cells and their non-hypoxic counterparts after ionizing irradiation in situ. Xenografts were harvested following irradiation at three representative doses of 2, 7.5 and 15 Gy X-rays, respectively. After digestion and depletion of the host mouse cells, individual EGFP+ hypoxic tumor cells and EGFP− non-hypoxic tumor cells were sorted and collected by FACS. As shown in Figure 1C, D, and E, the hypoxic tumor cells showed moderately higher clonogenic survival (approximately 1.7-fold higher) than the non-hypoxic tumor cells at 2 and 7.5 Gy. When irradiated at the high dose of 15 Gy, the classical clonogenic survival of the hypoxic tumor cells was not significantly different from that of the non-hypoxic tumor cells based on the formation of dense colonies. Nevertheless, there were significantly more surviving cells from the in vivo hypoxic tumor cell population than those from the non-hypoxic population (Supplementary Figure 2A). The surviving hypoxic tumor cells after 15-Gy irradiation grew in a scattering pattern and did not form typical densely populated colonies (Supplementary Figure 2A).
3.2 The hypoxic tumor cells repair DNA damages more efficiently than the non-hypoxic tumor cells
To assess the extent of DNA double-strand breaks and repair following ionizing irradiation in vivo, we examined the formation of 53BP1+ and γH2AX+ nuclear foci, as well as neutral comet. Tumor cells were isolated and sorted by FACS immediately after irradiation to tumors. The freshly sorted tumor cells were plated and cultured under the ambient condition. This strategy would allow direct assessment of the DNA damage and repair specifically in individual cells without further complication from the host stromal reactions after irradiation. At 24 hr after irradiation, there were no significant differences in the formation of 53BP1+ (Figure 2A) or γH2AX+ (Figure 2B) nuclear foci in either EGFP+ hypoxic tumor cells or EGFP− non-hypoxic tumor cells, suggesting that both hypoxic and non-hypoxic tumor cells received comparable amounts of DNA damages. At 48 hr after irradiation, the EGFP+ hypoxic tumor cells showed significantly fewer cells with 53BP1+ (Figure 2A) or γH2AX+ (Figure 2B) nuclear foci, suggesting more efficient DNA damage repair potentials of the hypoxic tumor cells. The same observations were made in both orthotopic and ectopic xenograft tumors, suggesting the differential DNA damage repair capacities are primarily determined by tumor hypoxia. Consistent with the foci data, the neutral comet formation was also significantly reduced in the EGFP+ hypoxic tumor cells at 48 hr after isolation from irradiated tumors (Figure 2C).
Fig. 2. The naturally occurring hypoxic tumor cells have proficient DNA damage repair ability in response to ionizing irradiation in vivo.

Xenograft tumors were irradiated with 7 Gy γ-irradiation. Tumor cells were isolated and sorted into hypoxic (EGFP+) and non-hypoxic (EGFP−) populations as described in Figure 1. The freshly isolated tumor cells were plated and cultured under the ambient condition for 24 or 48 hrs before they were fixed for immunofluorescence staining with anti-53BP1 (A) and anti-γH2AX antibodies (B). Cells with >5 nuclear foci were scored in randomly selected microscopic fields (n=16 for the 24 hr samples, n=19 for the 48 hr samples). Double strand DNA damages were assessed by the neutral Comet Assay (C). DNA Tail Moment was calculated using the CometScore software (n=62–123).
3.3 The ex vivo hypoxic tumor cells have acquired an intrinsically radioresistant phenotype that is independent of the hypoxic TME
One of the unresolved questions regarding hypoxic tumor cells in vivo is whether their radioresistance is contingent upon hypoxia in their niche microenvironment. To address this question, we isolated by flow cytometry the EGFP+ hypoxic and EGFP− non-hypoxic tumor cells from the same tumor and cultured them in vitro for 3–5 passages for complete re-acclimation (Figure 3A). When irradiated under the same ambient condition, the ex vivo hypoxic tumor cells still showed moderately higher clonogenic survival than their non-hypoxic counterparts at 2 and 7.5 Gy (Figure 3B). Similar to the in vivo γ-irradiated tumor cells, the percentage of the conventional colony-forming cells were similar between these two ex vivo tumor cell populations at the high dose of 15 Gy. Nonetheless, as with the in vivo irradiated cells, there were significantly more surviving cells from the ex vivo hypoxic tumor cell population than those from the non-hypoxic population and the surviving cells grew as a monolayer instead of forming typical colonies (Supplementary Figure 2B).
Fig. 3. The ex vivo hypoxic tumor cells maintain enhanced clonogenic survival and DNA damage repair potential in response to γ-irradiation under the conventional culture conditions.

The hypoxic (EGFP+) and non-hypoxic (EGFP−) tumor cells were sorted from xenograft tumors and cultured under the ambient condition for <5 passages (approximately 14 days) before γ-irradiation (A). (B) The ex vivo cells were irradiated at indicated doses of γ-irradiation and plated for clonogenic assays in sextuplets using two seeding densities. (C, D) The ex vivo tumor cells were γ-irradiated at 2.5 Gy and then were fixed at the indicated time points for detection of nuclear foci by immunofluorescence with anti-53BP1 (C) or anti-γH2AX antibodies (D). Cells with >5 nuclear foci were scored in three randomly selected microscopic fields with >230 cells counted in each field. **p<0.002, ***p<0.001. (E) Double strand DNA damages were assessed by the neutral Comet Assay. For the control at 0 Gy, n=142 (EGFP−) and n=167 (EGFP+); for the 3 Gy/3hr treatment, n=110 (EGFP−) and n=111 (EGFP+); for the 3 Gy/24hr treatment, n=162 (EGFP−) and n=169 (EGFP+).
We examined the differential responses to ionizing irradiation-induced DNA damages and potential rate of repair between the ex vivo EGFP+ and EGFP− tumor cells in a time-dependent manner. When γ-irradiated at 2.5 Gy under the ambient condition, 53BP1 nuclear foci were formed rapidly in nearly every cell and there were no significant differences between the EGFP+ and EGFP− tumor cell populations within 6 hr post-irradiation (Figure 3C), suggesting that both populations are equally sensitive to γ-irradiation-induced DNA damages. However, at and after 10 hr post-irradiation, significantly fewer 53BP1+ cells were observed in the EGFP+ population than in the EGFP− population (Figure 3C). Similar observations were made when γH2AX nuclear foci were examined (Figure 3D). Even when γ-irradiated at a high dose of 7 Gy, the percentages of 53BP1+ or γH2AX+ cells remained significantly lower in the ex vivo hypoxic tumor cells when examined between 24 and 48 hr (Supplementary Figure 3). Consistent with these observations, the neutral comet assay revealed that the amount of DNA double-strand breaks was significantly lower in the ex vivo hypoxic tumor cells either under the ambient culture condition or after ionizing irradiation (Figure 3D). These findings suggest that the ex vivo EGFP+ tumor cells can more efficiently repair γ-irradiation-induced DNA damages than their EGFP− counterparts do.
To further determine their sensitivity to genotoxic stresses, we treated the ex vivo tumor cells with two commonly used DNA damaging agents: bleomycin and etoposide. Upon bleomycin treatment (middle, Figure 4A), the plating efficiency of the ex vivo hypoxic EGFP+ tumor cells remained significantly higher than that of the EGFP− tumor cells over a dose range of 0.2 to 20 μg/ml although both cell populations appeared to be equally sensitive to bleomycin. Similarly, the ex vivo EGFP+ tumor cells were also more resistant than the EGFP− tumor cells to the clonogenic toxicity of etoposide (0.12 to 12 μg/ml) albeit the EGFP+ cells became highly sensitive to etoposide at ≥1.2 μg/ml (right, Figure 4A).
Fig. 4. The ex vivo hypoxic tumor cells maintain increased survival against DNA damaging agents under the conventional culture conditions.

The sorted hypoxic (EGFP+) and non-hypoxic (EGFP−) tumor cells were maintained under the ambient condition as described in Figure 3. (A) The sorted ex vivo cells were treated with Bleomycin or Etoposide at the indicated doses for 24 hours and then plated for clonogenic assay (n= 6, ****p<0.0001, ***p=0.0003). (B, C, D) The sorted tumor cells were treated with 6 μM Etoposide for 5 hours. 53BP1+ nuclear foci were stained and scored in five (n=5) random microscopic fields (B). Enzymatic cleavage of caspase 3 (C) and PARP (D) were analyzed by Western Blot using specific antibodies.
In response to etoposide, although 53BP1+ nuclear foci were induced in both tumor cell populations, the percentage of the ex vivo hypoxic tumor cells with 53BP1+ nuclear foci remained significantly lower than that of the ex vivo non-hypoxic tumor cells (left, Figure 4B). Furthermore, etoposide strongly induced proteolytic cleavage of caspase 3 (Casp3) and poly-(ADP-ribose) polymerase (PARP) in the ex vivo non-hypoxic tumor cells (Figure 4B), suggesting that the non-hypoxic tumor cells are more sensitive to DNA damage-induced apoptosis than the ex vivo hypoxic tumor cells. Collectively, these results suggest that the hypoxic tumor cells in vivo may undergo clonal evolution in their hypoxic TME to acquire a relatively stable phenotype with enhanced survival against genotoxic stresses.
3.4 The DNA damage-sensing pathway is preferentially potentiated in the ex vivo hypoxic tumor cells
To further understand the mechanisms underlying the observed resistance to genotoxic stresses of the ex vivo hypoxic tumor cells, we examined the key DNA damage-sensing pathway mediated by ATM and ATR following genotoxic stresses [20–22]. In response to bleomycin treatment in vitro (Figure 5A), ATM became quickly phosphorylated at S1981 after 2 hr treatment in the ex vivo hypoxic tumor cells (Figure 5A, lane 3) with much lower ATM-S1981 phosphorylation in the ex vivo non-hypoxic tumor cells (Figure 5A, lane 4). In contrast, phosphorylation of ATR increased moderately in the ex vivo hypoxic tumor cells after 4 hr treatment (Figure 5A, lanes 5 and 6). Phosphorylation of checkpoint kinases CHK1 and CHK2, two direct targets of ATM and ATR, strongly occurred in the ex vivo non-hypoxic tumor cells after 2 hr treatment (Figure 5A, lanes 3 and 4). Further downstream, the tumor suppressor p53, a key target of the ATM/ATR/CHK1/CHK2 pathway, became significantly phosphorylated at S15 in the ex vivo hypoxic tumor cells after 2 hr bleomycin treatment (Figure 5A). Similarly, robust phosphorylation of ATM, CHK1, CHK2, and p53 was observed in ex vivo hypoxic tumor cells after ionizing irradiation (Figure 5B). It is worth noting that the total amounts of these key DNA damage-sensing proteins were by and large equivalent in both EGFP+ and EGFP− tumor cell populations and did not change significantly under the above described experimental conditions.
Fig. 5. The DNA damage-sensing pathway is preferentially activated in the ex vivo hypoxic tumor cells.

The hypoxic (EGFP+) and non-hypoxic (EGFP−) tumor cells were sorted from xenograft tumors and cultured under the ambient condition for <5 passages. (A) Tumor cells were treated with 20 μg/ml Bleomycin for 2 and 4 hrs, respectively. (B) Tumor cells were γ-irradiated at 3 Gy and whole cell lysates were prepared at 1 and 4 hr post-irradiation. Antibodies specific for the phosphor-proteins or the total protein were used for Western blot analysis. Expected protein bands are indicated by an asterisk (*).
Preferential activation of the ATM/CHK1/CHK2 pathway in the ex vivo hypoxic (EGFP+) tumor cells suggests that the in vivo hypoxia-selected cells might rely on this pathway to deal with genetic stresses. Consistent with this notion, phosphorylation of p53-S15 was strongly reduced by the potent checkpoint kinase inhibitor AZD7762 in the ex vivo hypoxic (EGFP+) tumor cells but not in the non-hypoxic (EGFP−) tumor cells in response to the DNA damaging agent bleomycin (Supplementary Figure 4). Importantly, AZD7762, although it alone did not affect clonogenic survival (Figure 6A), significantly reduced the clonogenic survival of the ex vivo hypoxic (EGFP+) tumor cells after 5 Gy X-irradiation (Figure 6B). In contrast, AZD7762 had only minor effects on the survival of the in the non-hypoxic (EGFP−) tumor cells under the same conditions (Figure 6B). On the other hand, AZD7762 had no significant effects in the parental tumor cells strictly under in vitro conditions including in vitro hypoxia (Supplementary Figure 5), suggesting that the differential sensitivity of the ex vivo hypoxic EGFP+ tumor cells to CHK inhibition results from evolution in the hypoxic tumor microenvironment in vivo.
Fig. 6. CHK inhibitors sensitize ex vivo hypoxic tumor cells to ionizing irradiation.
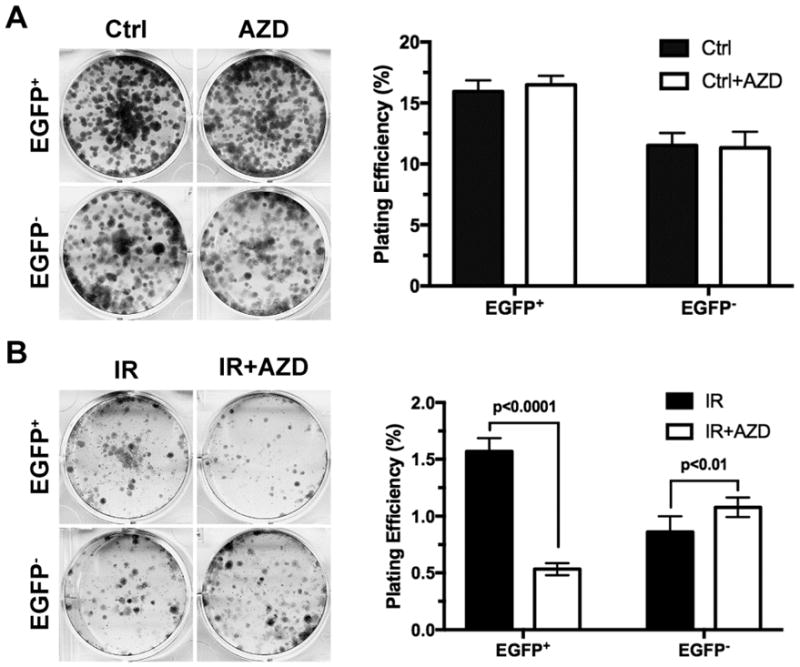
The hypoxic (EGFP+) and non-hypoxic (EGFP−) tumor cells sorted from xenograft tumors and cultured in vitro as described in Figure 5. (A) Tumor cells were treated with the CHK inhibitor AZD7762 at 20 nM for 24 hrs and then plated at 1,000 cells/well (n=6) in 6-well plates for clonogenic assay with vehicle-treated cells as control. (B) Tumor cells were treated with or without AZD7762 at 20 nM for 24 hrs and then irradiated at 5 Gy X-ray. The irradiated cells were then plated at 5,000 cells/well (n=6) in 6-well plates for clonogenic survival assay.
Collectively, these results suggest that potentiated ATM/CHK1/CHK2-mediated DNA damage-sensing pathways in the ex vivo hypoxic tumor cells are likely to be a major mechanism underlying their enhanced clonogenic survival against genotoxic stresses. Checkpoint kinase inhibitors have the potential to sensitized hypoxic tumor cells in vivo to ionizing irradiation or other genotoxic stresses.
4. Discussion
The paradigm for oxygen effects on cellular response to ionizing irradiation is largely based on in vitro studies under well-defined oxygen conditions. It has become widely known that mammalian cells irradiated by X-rays in the absence of oxygen or anoxia are 2.5 to 3 times more resistant to irradiation-induced clonal cell death than those irradiated at or above physiological pO2 [6, 23]. Radiosensitivity of mammalian cells increases sharply from 0 to 10 mmHg with the half-maximum radiosensitivity at approximately pO2 = 3 mmHg [24, 25]. At or above the physiological range of pO2 (>30 mmHg), mammalian cells are close to being fully sensitized by molecular oxygen to ionizing radiation [6, 23]. Therefore, the radiobiological hypoxia where mammalian cells are the most radioresistant occurs primarily in the range of pO2 <3 mmHg or 0.4% O2 [7].
In experimental tumor models, the fractions of radioresistant tumor cells are commonly estimated using the clonogenic survival assay with clamped tumors to mimic maximum radiological hypoxia [6, 17, 26]. Previously, the radiosensitivity of naturally hypoxic tumor cells had been investigated using flow cytometry based on the perfusion and differential uptake of Hoechst 33342 as a marker of tumor hypoxia [27, 28]. However, the potentially deleterious effects on DNA synthesis and genome stability [29], Hoechst 33342 might interfere with cellular response to ionizing radiation and would compromise subsequent culture and growth of tumor cells. In this study, we have developed a hypoxia-sensing tumor model using destabilized d2EGFP (half-life ≅ 2 hrs) as a reporter under the transcriptional control by five tandem HRE repeats [13]. The EGFP+ cells are predominantly located in areas that are also positive for the independent hypoxia marker pimonidazole. However, it should be noted that, despite positive association, the HIF-1α-positive regions do not show complete agreement with other hypoxia markers including carbonic anhydrase IX and nitroimidazoles [30, 31], which is likely due to different mechanisms of activation. Nonetheless, our model has the potential to label majority, if not all, tumor cells in a state of biological hypoxia, either chronically or transiently at the time of irradiation. The advantage of this approach is two-fold. First, both hypoxic (EGFP+) and non-hypoxic (EGFP−) tumor cells are irradiated simultaneously in their native microenvironment within the same tumor. EGFP is non-toxic to cells and will not likely to interfere with cellular response to ionizing irradiation. Second, the hypoxic (EGFP+) and non-hypoxic (EGFP−) tumor cells can be isolated from irradiated tumors and purified by FACS for immediate analysis or for in vitro culture.
Here, we have found that the differences in clonogenic survival after irradiation between hypoxic (EGFP+) and non-hypoxic (EGFP−) tumor cells are quite small, which stands in stark contrast to the textbook example of large differential of radioresistance between anoxic and fully oxic tumor cells [6]. One possible explanation to reconcile these discrepancies is that majority of the hypoxic tumor cells are likely localized in areas of moderate hypoxia. Nitroimidazole molecules are activated and bind to macromolecules at ≤10 mmHg [17, 32, 33] at which HIF-1α and HIF-2α also become stabilized [34]. It is therefore possible that most of the EGFP+ hypoxic tumor cells reside in regions with pO2 close to 10 mmHg or above the range of radiological hypoxia of pO2 = 0–3 mmHg. It is also worth noting that EGFP requires O2 to fluoresce. Although maturation of EGFP can occur at near anoxic conditions [13, 35, 36], it is still possible that some of the extremely hypoxic or anoxic tumor cells might not be effectively identified by this hypoxia reporter and end up in the EGFP− population, which may reduce the differences of radiosensitivity between EGFP+ and EGFP− tumor cells.
Our data also suggest that the truly radiologically hypoxic tumor cells are likely to be a rather small fraction among the entire hypoxic tumor cell population. Nordsmark et al. analyzed a multi-center study of 120 patients with primary cervical cancer but found no significant correlation between pO2 and response to radiotherapy [37]. Although reasons for the lack of correlation are unclear, it is possible that, in light of this study, hypoxic cervical cancer cells in these patients are most likely localized in areas within moderate hypoxia. Consistent with this idea, analysis of an international cohort of 397 patients with head-and-neck cancers found that only deep hypoxia (pO2 ≤ 2.5 mmHg), but not moderate hypoxia (pO2 ≤ 5 mmHg), was significantly associated with poor overall survival [38].
It is worth noting that there are no significant differences in the typical clonogenic survival between hypoxic (EGFP+) and non-hypoxic (EGFP−) tumor cells when they are irradiated in situ with 15 Gy, a high dose at which majority of the non-hypoxic tumor cells are likely to be eliminated [6]. Nonetheless, there are still significantly more surviving cells from the irradiated hypoxic (EGFP+) population than those from their non-hypoxic (EGFP−) counterparts. The hypoxic tumor cells that have survived the 15-Gy irradiation in vivo grow in a scattered pattern rather than as densely populated colonies. Consistent with increased radioresistance, we have found that the in vivo irradiated hypoxic tumor cells are more proficient than their non-hypoxic counterparts in the same tumor at repairing ionizing irradiation-induced DNA damages.
Surprisingly, after the sorted cells are maintained under the ambient culture conditions, the ex vivo hypoxic tumor cells continue to show higher clonogenic survival and resistance to apoptosis against genotoxic stresses that the ex vivo non-hypoxic tumor cells from the same tumors. These ex vivo hypoxic tumor cells also maintain their proficient DNA damage repair abilities. The canonical DNA damage-signaling pathway [20–22], especially the ATM/CHK1/CHK2 pathway, is preferentially activated in the hypoxic tumor cells in response to ionizing irradiation or DNA damage agents. We have recently found that cancer stem cell-like populations are significantly enriched in the in vivo hypoxic tumor cells [19]. Our results are consistent with the recent view that cancer stem cells exhibit increased activation of the DNA damage checkpoint [39].
5. Conclusions
This study strongly suggests that the hypoxic TME may induce adaptive responses, either by clonal evolution or selection, such that hypoxic tumor cells in vivo acquire new functional traits that confer resistance to genotoxic stresses and enhance survival. The relatively stable phenotype exhibited by the ex vivo hypoxic tumor cells implies that the tumor hypoxia in vivo has the potential to exert long range ectopic effects after the hypoxic tumor cells have migrated into non-hypoxic microenvironment or after a hypoxic niche restores normoxia. These observations further underscore the importance of targeting hypoxic tumor cells to improve tumor control and patient survival. Our data also suggest a combination between checkpoint kinase inhibitors and ionizing irradiation or other DNA-damaging agents have the potential to effectively eliminate the clonogenic tumor cells selected by the hypoxic tumor microenvironment.
Supplementary Material
Highlights.
Radiosensitivity of naturally hypoxic tumor cells remains to be fully understood.
We developed a hypoxia-sensing tumor model to identify hypoxic tumor cells in situ.
Tumor hypoxia in vivo selects a stable population of radioresistant tumor cells.
ATM/CHK1/CHK2 DNA damage-sensing pathway is preferentially activated in hypoxic cells.
Acknowledgments
Funding:
This work was supported by a grant from the National Institutes of Health to ZY (R01CA178254).
We thank Dr. Peter Glazer of the Department of Therapeutic Radiology, Yale School of Medicine for reagents and helpful discussions.
Footnotes
Authors’ Contributions
Conception and Design: Z. Yun
Development of Methodology: H. Kim, Q. Lin and Z. Yun
Data Acquisition, Analysis and Interpretation: H. Kim, Q. Lin and Z. Yun
Writing, Review, and/or Revision of the Manuscript: H. Kim, Q. Lin and Z. Yun
Study Supervision: Z. Yun
Conflicts of Interests:
The authors claim no potential conflicts of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Hoon Kim, Department of Therapeutic Radiology, Yale School of Medicine, New Haven, CT 06510, USA.
Qun Lin, Department of Therapeutic Radiology, Yale School of Medicine, New Haven, CT 06510, USA.
Zhong Yun, Department of Therapeutic Radiology, Yale School of Medicine, New Haven, CT 06510, USA.
References
- 1.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol. 2004;14:198–206. doi: 10.1016/j.semradonc.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Dhani N, Fyles A, Hedley D, Milosevic M. The clinical significance of hypoxia in human cancers. Semin Nucl Med. 2015;45:110–121. doi: 10.1053/j.semnuclmed.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 5.Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56:4509–4515. [PubMed] [Google Scholar]
- 6.Rockwell S, Dobrucki IT, Kim EY, Marrison ST, Vu VT. Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med. 2009;9:442–458. doi: 10.2174/156652409788167087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu C, Lin Q, Yun Z. Cellular and molecular mechanisms underlying oxygen-dependent radiosensitivity. Radiat Res. 2015;183:487–496. doi: 10.1667/RR13959.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crabtree HG, Cramer W. The Action of Radium on Cancer Cells. II.--Some Factors Determining the Susceptibility of Cancer Cells to Radium. Proc R Soc B-Biol Sci. 1933;113:238–250. [Google Scholar]
- 9.Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9:539–549. doi: 10.1038/bjc.1955.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol. 1953;26:638–648. doi: 10.1259/0007-1285-26-312-638. [DOI] [PubMed] [Google Scholar]
- 11.Gray LH. Oxygenation in radiotherapy. I. Radiobiological considerations. Br J Radiol. 1957;30:403–406. doi: 10.1259/0007-1285-30-356-403. [DOI] [PubMed] [Google Scholar]
- 12.Gatenby RA, Kessler HB, Rosenblum JS, Coia LR, Moldofsky PJ, Hartz WH, Broder GJ. Oxygen distribution in squamous cell carcinoma metastases and its relationship to outcome of radiation therapy. Int J Radiat Oncol Biol Phys. 1988;14:831–838. doi: 10.1016/0360-3016(88)90002-8. [DOI] [PubMed] [Google Scholar]
- 13.Vordermark D, Shibata T, Brown JM. Green fluorescent protein is a suitable reporter of tumor hypoxia despite an oxygen requirement for chromophore formation. Neoplasia. 2001;3:527–534. doi: 10.1038/sj.neo.7900192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata T, Giaccia AJ, Brown JM. Development of a hypoxia-responsive vector for tumor-specific gene therapy. Gene Ther. 2000;7:493–498. doi: 10.1038/sj.gt.3301124. [DOI] [PubMed] [Google Scholar]
- 15.Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, Calvo V, Cheung JF, Bravo-Cordero JJ, Entenberg D, Castracane J, Verkhusha V, Keely PJ, Condeelis J, Aguirre-Ghiso JA. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. 2017;19:120–132. doi: 10.1038/ncb3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le A, Stine ZE, Nguyen C, Afzal J, Sun P, Hamaker M, Siegel NM, Gouw AM, Kang BH, Yu SH, Cochran RL, Sailor KA, Song H, Dang CV. Tumorigenicity of hypoxic respiring cancer cells revealed by a hypoxia-cell cycle dual reporter. Proc Natl Acad Sci U S A. 2014;111:12486–12491. doi: 10.1073/pnas.1402012111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raleigh JA, Chou SC, Arteel GE, Horsman MR. Comparisons among pimonidazole binding, oxygen electrode measurements, and radiation response in C3H mouse tumors. Radiat Res. 1999;151:580–589. [PubMed] [Google Scholar]
- 18.Pogue BW, Paulsen KD, O’Hara JA, Wilmot CM, Swartz HM. Estimation of oxygen distribution in RIF-1 tumors by diffusion model-based interpretation of pimonidazole hypoxia and eppendorf measurements. Radiat Res. 2001;155:15–25. doi: 10.1667/0033-7587(2001)155[0015:eoodir]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 19.Kim H, Lin Q, Glazer PM, Yun Z. The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res. 2018;20:16. doi: 10.1186/s13058-018-0944-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashwell S, Zabludoff S. DNA damage detection and repair pathways--recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin Cancer Res. 2008;14:4032–4037. doi: 10.1158/1078-0432.CCR-07-5138. [DOI] [PubMed] [Google Scholar]
- 21.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 23.Kirkpatrick JP, Cardenas-Navia LI, Dewhirst MW. Predicting the effect of temporal variations in PO2 on tumor radiosensitivity. Int J Radiat Oncol Biol Phys. 2004;59:822–833. doi: 10.1016/j.ijrobp.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 24.Hall EJ, Giacia AJ. Radiobiology fo the Radiologist. 7. Lippincott Williams & Wilkins; Philadelphia: 2012. [Google Scholar]
- 25.Hockel M, Schlenger K, Knoop C, Vaupel P. Oxygenation of carcinomas of the uterine cervix: evaluation by computerized O2 tension measurements. Cancer Res. 1991;51:6098–6102. [PubMed] [Google Scholar]
- 26.Rockwell S, Moulder JE. Hypoxic fractions of human tumors xenografted into mice: a review. Int J Radiat Oncol Biol Phys. 1990;19:197–202. doi: 10.1016/0360-3016(90)90154-c. [DOI] [PubMed] [Google Scholar]
- 27.Durand RE, Raleigh JA. Identification of nonproliferating but viable hypoxic tumor cells in vivo. Cancer Res. 1998;58:3547–3550. [PubMed] [Google Scholar]
- 28.Olive PL, Banath JP. Phosphorylation of histone H2AX as a measure of radiosensitivity. Int J Radiat Oncol Biol Phys. 2004;58:331–335. doi: 10.1016/j.ijrobp.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 29.Durand RE, Olive PL. Cytotoxicity, Mutagenicity and DNA damage by Hoechst 33342. J Histochem Cytochem. 1982;30:111–116. doi: 10.1177/30.2.7061816. [DOI] [PubMed] [Google Scholar]
- 30.Jankovic B, Aquino-Parsons C, Raleigh JA, Stanbridge EJ, Durand RE, Banath JP, MacPhail SH, Olive PL. Comparison between pimonidazole binding, oxygen electrode measurements, and expression of endogenous hypoxia markers in cancer of the uterine cervix. Cytometry B Clin Cytom. 2006;70:45–55. doi: 10.1002/cyto.b.20086. [DOI] [PubMed] [Google Scholar]
- 31.Vukovic V, Haugland HK, Nicklee T, Morrison AJ, Hedley DW. Hypoxia-inducible factor-1α is an intrinsic marker for hypoxia in cervical cancer xenografts. Cancer Res. 2001;61:7394–7398. [PubMed] [Google Scholar]
- 32.Gross MW, Karbach U, Groebe K, Franko AJ, Mueller-Klieser W. Calibration of misonidazole labeling by simultaneous measurement of oxygen tension and labeling density in multicellular spheroids. Int J Cancer. 1995;61:567–573. doi: 10.1002/ijc.2910610422. [DOI] [PubMed] [Google Scholar]
- 33.Koch CJ. Measurement of absolute oxygen levels in cells and tissues using oxygen sensors and 2-nitroimidazole EF5. Methods Enzymol. 2002;352:3–31. doi: 10.1016/s0076-6879(02)52003-6. [DOI] [PubMed] [Google Scholar]
- 34.Lin Q, Cong X, Yun Z. Differential hypoxic regulation of hypoxia-inducible factors 1α and 2α. Mol Cancer Res. 2011;9:757–765. doi: 10.1158/1541-7786.MCR-11-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansen MC, Palmer RJ, Jr, Udsen C, White DC, Molin S. Assessment of GFP fluorescence in cells of Streptococcus gordonii under conditions of low pH and low oxygen concentration. Microbiology. 2001;147:1383–1391. doi: 10.1099/00221287-147-5-1383. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi E, Takano T, Nomura Y, Okano S, Nakajima O, Sato M. In vivo oxygen imaging using green fluorescent protein. Am J Physiol Cell Physiol. 2006;291:C781–787. doi: 10.1152/ajpcell.00067.2006. [DOI] [PubMed] [Google Scholar]
- 37.Nordsmark M, Loncaster J, Aquino-Parsons C, Chou SC, Gebski V, West C, Lindegaard JC, Havsteen H, Davidson SE, Hunter R, Raleigh JA, Overgaard J. The prognostic value of pimonidazole and tumour pO2 in human cervix carcinomas after radiation therapy: a prospective international multi-center study. Radiother Oncol. 2006;80:123–131. doi: 10.1016/j.radonc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 38.Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, Becker A, Adam M, Molls M, Dunst J, Terris DJ, Overgaard J. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol. 2005;77:18–24. doi: 10.1016/j.radonc.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 39.Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007;67:8980–8984. doi: 10.1158/0008-5472.CAN-07-0895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.