

Abstract
AMP-activated protein kinase (AMPK) is a crucial metabolic regulator with profound modulatory activities on inflammation. Although the anti-inflammatory benefits of AMPK activators were well documented in experimental studies, the pathological significance of endogenous AMPK in inflammatory disorders largely remains unknown. This study investigated the phosphorylation status of endogenous AMPK and the potential roles of AMPK in mice with lipopolysaccharide (LPS)-induced lethal inflammation. The results indicated that LPS dose-dependently decreased the phosphorylation level of AMPK and its target protein acetyl-CoA carboxylase (ACC). Reactivation of AMPK with the AMPK activator A-769662 suppressed LPS-induced elevation of interleukin 6, alleviated histological abnormalities in lung and improved the survival of LPS-challenged mice. Treatment with A-769662 restored LPS-induced suppression of autophagy, inhibition of autophagy by 3-MA reversed the beneficial effects of A-769662. Treatment with A-769662 suppressed LPS-induced activation of mammalian target of rapamycin (mTOR), co-administration of mTOR activator abolished the beneficial effects of A-769662, and the suppressive effects of A-769662 on uncoordinated-51-like kinase 1 (ULK1) phosphorylation. Inhibition of ULK1 removed the beneficial effects of A-769662. These data indicated that LPS-induced dephosphorylation of AMPK could result in weakened inhibition of mTOR and repression of ULK1-dependent autophagy, which might potentiate the development of LPS-induced inflammatory injury. These data suggest that pharmacological restoration of AMPK activation might be a beneficial approach for the intervention of inflammatory disorders.
Keywords: AMP-activated protein kinase, lipopolysaccharide, autophagy, mammalian target of rapamycin, uncoordinated-51-like kinase 1, inflammation
Introduction
The energy sensitive serine/threonine kinase AMP-activated protein kinase (AMPK) is a crucial metabolic regulator plays central roles in the maintenance energy homeostasis (1). AMPK is a heterotrimer composed of α, β, and γ subunits (2). The α subunit is the catalytic subunit, the phosphorylation of the Thr172 in the α subunit is a hallmark of AMPK activation (3). The regulatory γ subunit binds AMP/ADP under falling energy status and facilitates the phosphorylation of AMPKα and the activation of AMPK (2). AMPK preserves the level of ATP via stimulation of energy-producing pathways and suppression of energy-consuming metabolisms (4).
Inflammation includes a series of highly active molecular responses, which requires intensive energy support (5). Interestingly, there is a growing number of evidence indicates that the energy sensor AMPK is involved in the regulation of inflammation, an energy-intensive response (6). It has been reported that activation of AMPK by pharmacological reagents or molecular approaches suppressed the production of pro-inflammatory mediators and promoted the generation of anti-inflammatory cytokines in lipopolysaccharide (LPS)-stimulated macrophages (7–9). In addition, the anti-inflammatory benefits of the AMPK activators have been observed in animal models with colitis, hepatitis, and myocarditis (10–12). Therefore, AMPK is generally regarded as a negative regulator of inflammation (3, 6).
Severe infection-induced systemic inflammation is one of the most serious inflammatory situations with high mortality (13). The lung is the representative organ suffered from systemic inflammation, which is closely associated with the lethal outcomes (14). Recently, the aberrant activation status of AMPK and its pathological significance have been investigated in animal models with acute hepatitis and chronic obstructive pulmonary disease (15, 16), but the potential roles of endogenous AMPK in LPS-induced lethal inflammation remains unknown. In this study, the phosphorylation status of AMPKα in mice with LPS-induced lethal inflammation was determined. And then, the aberrant status of AMPK was pharmacologically reversed, the degree of inflammatory injury and the downstream molecular mechanisms were investigated.
Materials and Methods
Animals
The Balb/c mice (male, 6–8 weeks old, weighing 18–22 g) were purchased from the Laboratory Animal Center of Chongqing Medical University (Chongqing, China). The animals were maintained under controlled temperature of 20–25°C with a 12-h light/12-h dark schedule and given food and water ad libitum. All experimental procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Chongqing Medical University.
Drugs and Reagents
Lipopolysaccharide (from Escherichia coli, 055:B5) was purchased from Sigma-Aldrich (St. Louis, MO, USA). The AMPK activator 6,7-dihydro-4-hydroxy-3 -(2′-hydroxy[1,1′-biphenyl]-4-yl)-6-oxo-thieno[2,3-b]pyridine-5-carbonitrile (A 769662), the autophagy inhibitor 3-methyl-3H-purin-6-amine (3-MA), the mammalian target of rapamycin (mTOR) activator 3-benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran-2(3H)-one(3-BDO), and the uncoordinated-51-like kinases 1 (ULK1) inhibitor N-[3-[[5-cyclopropyl-2-[(1,2,3,4-tetrahydro-2-methyl-6-isoquinolinyl)amino]-4-pyrimidinyl]amino]propyl]-cyclobutanecarboxamide (MRT68921) were the products from Cayman Chemical (MI, USA). The rabbit anti-mouse α subunit of AMPK, phosphorylated α subunit of AMPK (Thr172), β-actin, acetyl-CoA carboxylase (ACC), phosphorylated ACC (Ser79), ULK1, phosphorylated ULK1 (Ser757), light Chain 3 (LC3), and p62 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The rabbit anti-mouse eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1), phosphorylated 4E-BP1 antibodies were purchased from Abcam (Cambridge, UK). The enzyme-linked immunosorbent assay (ELISA) kit for determination of mouse interleukin 6 (IL-6) was the product from NeoBioscience Technology Company (Shenzhen, China). The BCA protein assay kit, HRP-conjugated goat anti-rabbit antibody, and enhanced chemiluminescence (ECL) reagents were purchased from Thermo Fisher Scientific (Rockford, IL, USA).
Experimental Protocol
Lipopolysaccharide was injected intraperitoneally to induce severe inflammation and acute lung injury. To investigate the phosphorylation status of AMPK, vehicle or various doses of LPS (10 and 20 mg/kg, dissolved in normal saline, n = 8 per group) was injected, the mice were sacrificed by decapitation 3 h post LPS challenge, and lung sample was harvested for determination of the phosphorylation level of AMPK and ACC with immunoblot analysis.
To evaluate the potential roles of AMPK in inflammatory injury, the mice were treated with the AMPK activator A-769662 (30 mg/kg, dissolved in DMSO, i.p.) 30 min before LPS (20 mg/kg) exposure. The mice were sacrificed at 3 or 18 h (n = 8 per group at each time point). Plasma samples and lung sample were collected for further experiments.
To investigate the mechanisms underlying the potential roles of AMPK in inflammatory injury, the autophagy inhibitor 3-MA (15 mg/kg, dissolved in DMSO, i.p.), the mTOR activator 3BDO (100 mg/kg, dissolved in DMSO, i.p.), or the ULK1 inhibitor MRT68921 (50 mg/kg, dissolved in DMSO, i.p.) was co-administered with the AMPK activator A-769662. The mice were sacrificed at 3 or 18 h (n = 8 per group at each time point). Plasma samples and lung sample were collected for further experiments.
Assessment Score
The aberrant physical appearance of LPS-insulted mice was evaluated with the assessment score method as previously described (17). Briefly, the animals were scored according to the appearance of coat, activity, respiration, and posture. The point of coat: 1, smooth; 2, mild ruffling; 3, significant ruffling. The point of activity: 1, normal; 2, moves slowly without stimulation; 3, moves only with stimulation, 4, minimal movement with stimulation. The point of respiration: 1, normal; 2, labored; 3, irregular. The point of posture: 1, moving or resting normally; 2, huddled. The assessment score is the total sum of the points from the above categories.
Histological Analysis
The lung specimens of mice were immersion fixed in 10% formaldehyde at room temperature for 48 h, and then the specimens were embedded in paraffin. Serial paraffin sections (4 µm) were prepared and stained with hematoxylin & eosin (H&E) routinely for conventional morphological evaluation under a light microscope (Olympus, Tokyo, Japan). The histopathological alterations of the lung tissue were blindly scored according to the method described previously (18). Briefly, the histological abnormalities were graded on a scale of 0–4 (0, normal; 1, light; 2, moderate; 3, strong; 4, intense) for the following features: congestion, edema, inflammation, and hemorrhage. A total score for all of the four parameters was calculated.
Enzyme-Linked Immunosorbent Assay
To determine the level of IL-6 of the harvested blood sample, the ELISA kit was used to according to the manufacturer’s instructions (NeoBioscience, China).
Western Blot Analysis
The total protein was extracted from the lung samples, and the protein concentration was determined using BCA method. The protein extracts were separated using 5–10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then were transferred to nitrocellulose membranes. After incubation with the blocking buffer (containing 5% skim milk, 10 mM Tris–HCl, 150 mM NaCl, and 0.1% Tween-20) for 1 h at room temperature, the membranes were incubated with primary antibody against AMPKα, phosphorylated AMPKα (Thr172), β-actin, ACC, phosphorylated ACC (Ser79), ULK1, phosphorylated ULK1 (Ser757), 4E-BP1, phosphorylated 4E-BP1, LC3, or p62 overnight at 4°C, followed by incubation with the HRP-conjugated secondary antibody. And finally, the blots were visualized with an enhanced chemiluminescence system (Thermo Fisher Scientific) under the ChemiDoc Touch Imaging system (Bio-Rad Laboratories).
Survival Analysis
To evaluate the potential effects of the AMPK activator A769662 on the mortality of LPS-challenged mice, the survival of experimental animals was assessed every 6 h for at least 7 days (n = 20). The cumulative survival rate was expressed by the Kaplan–Meier curve.
Statistical Analyses
Final results from the experiments are presented as the mean ± SD. The quantitative data were compared using the one-way ANOVA, followed by the Tukey’s post hoc test. The assessment score was compared using the Kruskal–Wallis test. The survival rate was compared using a Kaplan–Meier curve and a log-rank test. Results were considered statistically significant when the P value less than 0.05.
Results
LPS-Induced Dephosphorylation of AMPK Potentiated Inflammatory Injury
The phosphorylation of AMPKα at Thr172 is a hallmark of AMPK activation (2). The immunoblot analysis showed that LPS exposure dose-dependently suppressed the phosphorylation of AMPK (Figures 1A–C). Consistently, the phosphorylation of ACC, a representative target of AMPK (19), was also suppressed by LPS (Figures 1A–C). The suppressed phosphorylation of AMPK and ACC could be restored by A-769662 (Figures 1D–F), a widely used AMPK activator (20). In addition, reactivation of AMPK by A-769662 decreased the elevation of assessment score (Figure 2A), suppressed the production of IL-6 (Figure 2B), and improved the survival of LPS-insulted mice (Figure 2C). Meanwhile, LPS-induced histological abnormalities in lung tissue, including alveolar edema, bronchial wall thickening, and leukocyte infiltration, were alleviated by A-769662 (Figures 2D,E). These data suggest that dephosphorylation of AMPK might be involved in the development of LPS-induced inflammation.
Figure 1.

Lipopolysaccharide (LPS) exposure induced dephosphorylation of AMP-activated protein kinase (AMPK). (A–C) Mice were exposed to various doses of LPS (0, 10, and 20 mg/kg), lung samples were harvested 3 h post LPS exposure. The level of phosphorylated AMPK (p-AMPK), total AMPK (AMPK), phosphorylated ACC (p-ACC), and total ACC (ACC) was determined by immunoblot (A), and the blots were semi-quantified (B,C) (n = 4). (D–F) Mice were exposed to LPS (20 mg/kg), vehicle or AMPK activator A-769662 was administered 30 min before LPS exposure. The level of p-AMPK, AMPK, p-ACC, and ACC was determined by immunoblot (D), and the blots were semi-quantified (E,F) (n = 4).
Figure 2.

Restoration of AMP-activated protein kinase (AMPK) phosphorylation alleviated lipopolysaccharide (LPS)-induced lethal inflammation. Mice were exposed to LPS (20 mg/kg) with vehicle or AMPK activator A-769662 administration. The physical appearance of the experimental animals was evaluated 18 h post LPS exposure by the assessment scores (A) (n = 8). The plasma level of interleukin 6 (IL-6) 18 h post LPS exposure was determined by enzyme-linked immunosorbent assay (B) (n = 8). The survival of the experimental animals was monitored every 6 h for 7 days, and the percent survival rate was expressed by Kaplan–Meier curve (C) (n = 20). The histological abnormalities of the lung tissue 18 h post LPS exposure were observed with hematoxylin & eosin staining. The representative lung sections of each group were shown (D), the histopathological alterations were blindly scored (E).
Dephosphorylation of AMPK Was Associated With Impaired Autophagy
Autophagy was recently suggested to be involved in the progression of inflammation (21). The immunoblot analysis showed that LPS exposure decreased the level of LC3II but increased the level of p62, two molecular markers of autophagy (22), suggesting that LPS suppress autophagy in lung (Figures 3A–C). Reactivation of AMPK by A-769662 reversed the decline of LC3II and the elevation of p62 (Figures 3A–C), suggesting that LPS-induced dephosphorylation of AMPK might result in impaired autophagy. Inhibition of autophagy by 3-MA blocked the suppressive effects of A-769662 on IL-6 induction and histological abnormalities in LPS-exposed mice (Figures 3D–F), these alterations were accompanied with downregulation of LC3II and upregulation of p62 (Figures 3G–I), suggesting that impaired autophagy might contribute to the pathological roles of AMPK dephosphorylation.
Figure 3.

Inhibition of autophagy reversed the beneficial effects of AMP-activated protein kinase (AMPK) activator on inflammatory injury. (A–C) Mice were exposed to lipopolysaccharide (LPS) (20 mg/kg) with vehicle or AMPK activator A-769662 administration. The level of LC3II and p62 was determined by immunoblot (A), and the blots were semi-quantified (B,C) (n = 4). (D–I) Mice were exposed to LPS (20 mg/kg) with vehicle, AMPK activator A-769662, and autophagy inhibitor 3-MA administration. The plasma level of interleukin 6 (IL-6) was determined by enzyme-linked immunosorbent assay (D) (n = 8). The histological abnormalities of the lung tissue were observed with (H,E) staining. The representative lung sections of each group were shown (E), the histopathological alterations were blindly scored (F). The level of LC3II and p62 was determined by immunoblot (G), and the blots were semi-quantified (H,I) (n = 4).
Activation of mTOR Was Relieved by Dephosphorylation of AMPK
The nutrition sensor mTOR is a crucial regulator of autophagy (23). The immunoblot analysis showed that LPS exposure increased the phosphorylation level of 4E-BP1 and S6K1, two protein targets downstreaming mTOR (24), suggesting that mTOR would be activated by LPS (Figures 4A–C). LPS-induced activation of mTOR were suppressed by the AMPK activator A-769662 (Figures 4A–C), suggesting that AMPK dephosphorylation weaken its suppression on mTOR. Co-administration of 3BDO, an activator of mTOR (25), abolished the suppressive effects of A-769662 on LPS-induced mTOR activation (Figures 4A–C). Activation of mTOR by 3BDO also abolished the negative modulation of A-769662 on IL-6 production and histological lesions (Figures 4D–F). In addition, 3BDO abolished A-769662-induced elevation of LC3II and decline of p62 (Figures 4G–I). These data suggest that AMPK stimulate autophagy via inhibition of mTOR.
Figure 4.

Activation of mammalian target of rapamycin (mTOR) reversed the suppressive effects of AMP-activated protein kinase (AMPK) activator on inflammatory injury and autophgay. Mice were exposed to lipopolysaccharide (LPS) (20 mg/kg) with vehicle, AMPK activator A-769662, and mTOR activator 3BDO administration. The level of phosphorylated 4E-BP1 (p-4E-BP1), total 4E-BP1 (4E-BP1), phosphorylated S6K1 (p-S6K1), and total S6K1 (S6K1) was determined by immunoblot (A), and the blots were semi-quantified (B,C) (n = 4). The plasma level of interleukin 6 (IL-6) was determined by enzyme-linked immunosorbent assay (D) (n = 8). The histological abnormalities of the lung tissue were observed with hematoxylin & eosin staining. The representative lung sections of each group were shown (E), the histopathological alterations were blindly scored (F). The level of LC3II and p62 was determined by immunoblot (G), and the blots were semi-quantified (H,I) (n = 4).
ULK1 Mediated the Stimulatory Effects of AMPK Activator on Autophagy
ULK1 is a pivotal initiator of autophagy, which is phosphorylated and suppressed by mTOR (26). The immunoblot analysis showed that LPS exposure increased the phosphorylation of ULK1, treatment with the AMPK activator A-769662 suppressed LPS-induced phosphorylation of ULK1, whereas co-administration of the mTOR activator 3BDO removed the suppressive effects of A-769662 on ULK1 phosphorylation (Figures 5A,B). These data suggest that AMPK suppressed ULK1 phosphorylation via repression of mTOR. This study also found that co-administration of MRT68921, an ULK1 inhibitor (27), removed the beneficial effects of A-769662 on IL-6 induction and histological lesions (Figures 5C–E). Co-administration of MRT68921 also reversed the modulatory effects of A-769662 on LC3II and p62 (Figures 5F–H), suggesting that AMPK suppress autophgay and inflammation in an ULK1-dependent manner.
Figure 5.

Inhibition of ULK1 reversed the suppressive effects of AMP-activated protein kinase (AMPK) activator on inflammatory injury and autophgay. (A,B) Mice were exposed to lipopolysaccharide (LPS) (20 mg/kg) with vehicle, AMPK activator A-769662, and mammalian target of rapamycin activator 3BDO administration. The level of phosphorylated ULK1 (p-ULK1) and total ULK 1 (ULK 1) was determined by immunoblot (A), and the blots were semi-quantified (B) (n = 4). (C–G) Mice were exposed to LPS (20 mg/kg) with vehicle, AMPK activator A-769662, and ULK1 inhibitor MRT68921 administration. The plasma level of interleukin 6 (IL-6) was determined by enzyme-linked immunosorbent assay (C) (n = 8). The histological abnormalities of the lung tissue were observed with hematoxylin & eosin staining. The representative lung sections of each group were shown (D), the histopathological alterations were blindly scored (E). The level of LC3II and p62 was determined by immunoblot (F), and the blots were semi-quantified (G,H) (n = 4).
Discussion
The energy sensor AMPK is a crucial regulator in various energy-intensive physiological and pathophysiological processes (28). The profound anti-inflammatory activities of AMPK have been confirmed both in vitro and in vivo (6). In this study, we found that LPS exposure induced the decline of AMPK phosphorylation, while restoration of AMPK phosphorylation by the AMPK activator significantly alleviated LPS-induced lethal inflammation. These results suggest that LPS-induced dephosphorylation of AMPK might be a pathological event involved in the development of LPS-induced lethal inflammation, which provides a molecular basis for the pharmacological intervention with the AMPK activators.
The anti-inflammatory properties of AMPK have been well documented (29), but the underlying mechanisms remains largely unknown. It was reported that SIRT1 was an important target mediating the anti-inflammatory effects of AMPK (30, 31). However, recent studies indicated that SIRT1 might be a detrimental factor in LPS-induced lethal inflammation (32, 33). In addition, the metabolic regulatory effects of AMPK are closely associated with mTOR, and activation of AMPK suppresses the activity of mTOR (34). It has been suggested that mTOR play crucial roles in the development of acute lung injury, because pharmacological inhibition or genetic deletion of mTOR resulted in attenuated airway inflammation and lung injury (35–37). Interestingly, several studies have found that inflammatory stimuli activated mTOR both in vitro and in vivo (36, 38–40). Some studies suggested that LPS-induced activation of PI3K–AKT pathway might be responsible for the activation of mTOR (40, 41). However, this study found that the activation of mTOR might also result from the impaired suppression of mTOR by the dephosphorylation of AMPK in LPS-exposed mice.
There is increasing evidence that mTOR plays pivotal roles in the regulation of inflammation (42, 43), but the underlying mechanisms remain to be investigated. Recently, the crucial roles of autophagy in inflammation regulation have been highlighted (44). In LPS-stimulated human peripheral blood mononuclear cells, inhibition of autophagy resulted in enhanced production of pro-inflammatory cytokines (45). Consistently, macrophages with deletion of autophagy-related 16-like 1 (Atg16L1) also produced higher levels of IL-1β after LPS stimulation and mice lacking Atg16L1 are highly susceptible to dextran sulfate sodium-induced acute colitis (46). The close relationship between AMPK–mTOR and autophagy has been highly concerned (23). Activation of mTOR under amino acid-rich conditions as well as other pathological situations prevents the induction of autophagy (47), whereas inhibition of mTOR by AMPK stimulates autophagy (48). Therefore, suppressed autophagy by dephosphorylation of AMPK and activation of mTOR might be involved in the development of LPS-induced inflammatory injury in this study.
Several mechanisms underlying the inflammation-regulatory effects of authophgy have recently been revealed. It has been reported that autophagy controlled the production of pro-inflammatory cytokines by targeting pro-IL-1β for degradation and by promoting the degradation of pellino E3 ubiquitin protein ligase family member 3 (PELI3), a scaffold protein implicated in LPS-induced activation of pro-inflammatory signaling cascades (49, 50). In addition, increasing NLRP3 degradation and controlling NLRP3 inflammasome activation were also involved in the anti-inflammatory properties of autophagy (51). In mice with LPS-induced inflammatory lung injury or fulminant hepatitis, inhibition of autophagy exacerbated inflammatory injury but activation of autophagy attenuated injury (35, 52, 53). Therefore, restoration of autophagy might be responsible for the beneficial effects of the AMPK activator in this study.
The induction of autophagy is a tightly organized process; ULK1 is the mammalian homologs of yeast Atg1, which plays crucial roles in the initiation of autophagy (54). mTOR is one of the primary negative regulators of autophagy, it suppresses the activity of ULK1, in part, by phosphorylating ULK1 (23, 55). By contrast, AMPK functions as a suppressor of mTOR and an activator of autophagy. AMPK suppressed the activation of mTOR via phosphorylating TSC2, an inhibitor of mTOR, and thus prevents the inhibitory phosphorylation of ULK1 and promotes the induction of autophagy (56). Therefore, LPS-induced dephosphorylation of AMPK might result in weakened inhibition of mTOR, which led to enhanced mTOR activation, increased ULK1 phosphorylation, suppressed ULK1 activity, and repressed autophagy.
On the other side, LPS-activated pro-inflammatory signaling significantly modulates the induction of autophagy but the experimental data are highly inconsistent (57). Although LPS induced autophagy in macrophages, endothelial cells, and hepatocytes, it suppressed autophagy in pulmonary epithelial cells (35, 53, 58, 59). Therefore, the modulatory effects of LPS on autophagy seem to be cell dependent. In this study, LPS significantly suppressed autophagy in lung tissue. In agree with our findings, mice with cecal ligation and puncture-induced severe peritonitis also exhibited suppressed autophagy (60). In this study as well as the previously reported study in systemic injury models, the significance of autophagy in certain cells, especially pulmonary epithelial cells, is unclear, which is a major limitation of these studies. In addition, although restoration of autophagy by the AMPK activator improved the total survival rate of LPS-challenged mice, it did not prevent the early death of the experimental animals. The inflammatory status dynamically altered post LPS exposure, which orchestrates the outcomes with other pathological processes such as coagulation and apoptosis (61). The status of autophagy at different stages of inflammatory injury and the significance of autophagy in the regulation of inflammation-related coagulation and apoptosis are worthy of further investigation.
Taken together, this study found that LPS exposure induced the dephophorylation of AMPK, which play crucial roles in the development of inflammatory injury. The mechanisms underlying the pathological roles of AMPK dephosphorylation include impaired suppression of mTOR and suppressed ULK1-dependent autophagy (Figure 6). Although the underlying mechanisms remain to be further investigated, this study revealed a novel mechanism contributes to the development of LPS-induced acute lung injury.
Figure 6.
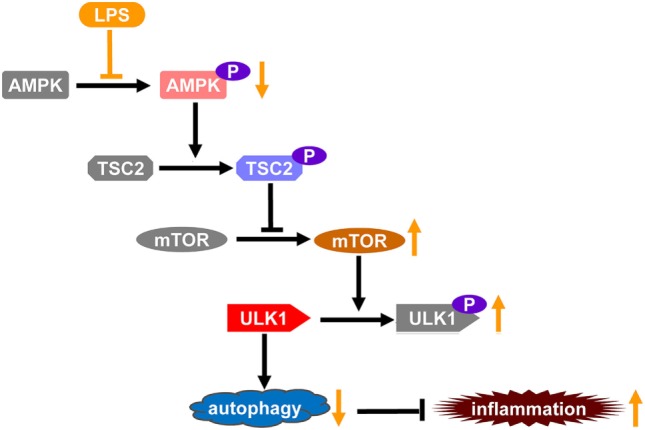
The schematic diagram of the mechanisms through which lipopolysaccharide (LPS)-induced dephosphorylation of AMP-activated protein kinase (AMPK) potentiates inflammatory injury. Autophagy plays crucial roles in the limitation of inflammation, which is a tightly organized process. ULK1 is a key factor promoting the initiation of autophagy, it could be phorphorylated and inhibited by mammalian target of rapamycin (mTOR), a primary negative regulator of autophagy. AMPK functions as a suppressor of mTOR, it suppressed the activation of mTOR via phosphorylating TSC2, an inhibitor of mTOR, and thus prevents the inhibitory phosphorylation of ULK1 and promotes the induction of autophagy. LPS exposure induces the dephosphorylation and suppression of AMPK, which might result in weakened inhibition of mTOR, increased inhibitory phosphorylation of ULK1, and repressed autophagy. Thus, LPS-induced dephosphorylation of AMPK and repression of autophagy might be a novel mechanism underlying the development of inflammatory injury.
Ethics Statement
All experimental procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Chongqing Medical University.
Author Contributions
LZ and JW planned the experiments. KF and RJ performed the experiments. LL and QA analyzed the data. JD prepared the figures. YY and PG drafted the manuscript. GL and LZ proofread the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the grants from the National Nature Science Foundation of China (No. 81370179, 81671953), the grant from Chongqing Research Program of Basic Research and Frontier Technology (No. cstc2017jcyjAX0098), and the grants from Chongqing Yuzhong District Project of Science and Technology (No. 20170114).
References
- 1.Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell (2017) 66(6):789–800. 10.1016/j.molcel.2017.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carling D. AMPK signalling in health and disease. Curr Opin Cell Biol (2017) 45:31–7. 10.1016/j.ceb.2017.01.005 [DOI] [PubMed] [Google Scholar]
- 3.Antonioli L, Colucci R, Pellegrini C, Giustarini G, Sacco D, Tirotta E, et al. The AMPK enzyme-complex: from the regulation of cellular energy homeostasis to a possible new molecular target in the management of chronic inflammatory disorders. Expert Opin Ther Targets (2016) 20(2):179–91. 10.1517/14728222.2016.1086752 [DOI] [PubMed] [Google Scholar]
- 4.Novikova DS, Garabadzhiu AV, Melino G, Barlev NA, Tribulovich VG. AMP-activated protein kinase: structure, function, and role in pathological processes. Biochemistry (Mosc) (2015) 80(2):127–44. 10.1134/S0006297915020017 [DOI] [PubMed] [Google Scholar]
- 5.Gaber T, Strehl C, Buttgereit F. Metabolic regulation of inflammation. Nat Rev Rheumatol (2017) 13(5):267–79. 10.1038/nrrheum.2017.37 [DOI] [PubMed] [Google Scholar]
- 6.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature (2013) 493(7432):346–55. 10.1038/nature11862 [DOI] [PubMed] [Google Scholar]
- 7.Kim J, Kwak HJ, Cha JY, Jeong YS, Rhee SD, Kim KR, et al. Metformin suppresses lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages via activating transcription factor-3 (ATF-3) induction. J Biol Chem (2014) 289(33):23246–55. 10.1074/jbc.M114.577908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu YP, Brown JR, Sag D, Zhang L, Suttles J. Adenosine 5’-monophosphate-activated protein kinase regulates IL-10-mediated anti-inflammatory signaling pathways in macrophages. J Immunol (2015) 194(2):584–94. 10.4049/jimmunol.1401024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem (2010) 285(25):19051–9. 10.1074/jbc.M110.123620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bai A, Ma AG, Yong M, Weiss CR, Ma Y, Guan Q, et al. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem Pharmacol (2010) 80(11):1708–17. 10.1016/j.bcp.2010.08.009 [DOI] [PubMed] [Google Scholar]
- 11.Zhou D, Ai Q, Lin L, Gong X, Ge P, Che Q, et al. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside-attenuates LPS/D-Gal-induced acute hepatitis in mice. Innate Immun (2015) 21(7):698–705. 10.1177/1753425915586231 [DOI] [PubMed] [Google Scholar]
- 12.Liu G, Wu K, Zhang L, Dai J, Huang W, Lin L, et al. Metformin attenuated endotoxin-induced acute myocarditis via activating AMPK. Int Immunopharmacol (2017) 47:166–72. 10.1016/j.intimp.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 13.Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers (2016) 2:16045. 10.1038/nrdp.2016.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadowitz B, Roy S, Gatto LA, Habashi N, Nieman G. Lung injury induced by sepsis: lessons learned from large animal models and future directions for treatment. Expert Rev Anti Infect Ther (2011) 9(12):1169–78. 10.1586/eri.11.141 [DOI] [PubMed] [Google Scholar]
- 15.Hu K, Gong X, Ai Q, Lin L, Dai J, Cai L, et al. Endogenous AMPK acts as a detrimental factor in fulminant hepatitis via potentiating JNK-dependent hepatocyte apoptosis. Cell Death Dis (2017) 8(3):e2637. 10.1038/cddis.2017.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JS, Park SJ, Cho YS, Huh JW, Oh YM, Lee SD. Role of AMP-activated protein kinase (AMPK) in smoking-induced lung inflammation and emphysema. Tuberc Respir Dis (Seoul) (2015) 78(1):8–17. 10.4046/trd.2015.78.1.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Konter JM, Parker JL, Baez E, Li SZ, Ranscht B, Denzel M, et al. Adiponectin attenuates lipopolysaccharide-induced acute lung injury through suppression of endothelial cell activation. J Immunol (2012) 188(2):854–63. 10.4049/jimmunol.1100426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu K, Yang Y, Lin L, Ai Q, Dai J, Fan K, et al. Caloric restriction mimetic 2-deoxyglucose alleviated inflammatory lung injury via suppressing nuclear pyruvate kinase M2-signal transducer and activator of transcription 3 pathway. Front Immunol (2018) 9:426. 10.3389/fimmu.2018.00426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saha AK, Ruderman NB. Malonyl-CoA and AMP-activated protein kinase: an expanding partnership. Mol Cell Biochem (2003) 253(1–2):65–70. 10.1023/A:1026053302036 [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med (2016) 48:e224. 10.1038/emm.2016.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med (2017) 6(1):24. 10.1186/s40169-017-0154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munz C. Autophagy proteins in phagocyte endocytosis and exocytosis. Front Immunol (2017) 8:1183. 10.3389/fimmu.2017.01183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol (2014) 36:121–9. 10.1016/j.semcdb.2014.08.006 [DOI] [PubMed] [Google Scholar]
- 24.Linke M, Fritsch SD, Sukhbaatar N, Hengstschlager M, Weichhart T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett (2017) 591(19):3089–103. 10.1002/1873-3468.12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge D, Han L, Huang S, Peng N, Wang P, Jiang Z, et al. Identification of a novel mTOR activator and discovery of a competing endogenous RNA regulating autophagy in vascular endothelial cells. Autophagy (2014) 10(6):957–71. 10.4161/auto.28363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang B, Kundu M. Canonical and noncanonical functions of ULK/Atg1. Curr Opin Cell Biol (2017) 45:47–54. 10.1016/j.ceb.2017.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petherick KJ, Conway OJ, Mpamhanga C, Osborne SA, Kamal A, Saxty B, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem (2015) 290(18):11376–83. 10.1074/jbc.C114.627778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol (2016) 26(3):190–201. 10.1016/j.tcb.2015.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J, Yang G, Ha J. Targeting of AMP-activated protein kinase: prospects for computer-aided drug design. Expert Opin Drug Discov (2017) 12(1):47–59. 10.1080/17460441.2017.1255194 [DOI] [PubMed] [Google Scholar]
- 30.Guo JM, Shu H, Wang L, Xu JJ, Niu XC, Zhang L. SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci Ther (2017) 23(4):360–9. 10.1111/cns.12686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal (2013) 25(10):1939–48. 10.1016/j.cellsig.2013.06.007 [DOI] [PubMed] [Google Scholar]
- 32.Cui X, Chen Q, Dong Z, Xu L, Lu T, Li D, et al. Inactivation of Sirt1 in mouse livers protects against endotoxemic liver injury by acetylating and activating NF-kappaB. Cell Death Dis (2016) 7(10):e2403. 10.1038/cddis.2016.270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang J, Tian R, Yang Y, Jiang R, Dai J, Tang L, et al. The SIRT1 inhibitor EX-527 suppresses mTOR activation and alleviates acute lung injury in mice with endotoxiemia. Innate Immun (2017) 23(8):678–86. 10.1177/1753425917733531 [DOI] [PubMed] [Google Scholar]
- 34.Carroll B, Dunlop EA. The lysosome: a crucial hub for AMPK and mTORC1 signalling. Biochem J (2017) 474(9):1453–66. 10.1042/BCJ20160780 [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Lou J, Mao YY, Lai TW, Liu LY, Zhu C, et al. Activation of mTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy (2016) 12(12):2286–99. 10.1080/15548627.2016.1230584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lorne E, Zhao X, Zmijewski JW, Liu G, Park YJ, Tsuruta Y, et al. Participation of mammalian target of rapamycin complex 1 in toll-like receptor 2- and 4-induced neutrophil activation and acute lung injury. Am J Respir Cell Mol Biol (2009) 41(2):237–45. 10.1165/rcmb.2008-0290OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen D, Jiao G, Ma T, Liu X, Yang C, Liu Z. The mechanism of rapamycin in the intervention of paraquat-induced acute lung injury in rats. Xenobiotica (2015) 45(6):538–46. 10.3109/00498254.2014.995149 [DOI] [PubMed] [Google Scholar]
- 38.Meng L, Li L, Lu S, Li K, Su Z, Wang Y, et al. The protective effect of dexmedetomidine on LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-kappaB and PI3K/Akt/mTOR pathways. Mol Immunol (2018) 94:7–17. 10.1016/j.molimm.2017.12.008 [DOI] [PubMed] [Google Scholar]
- 39.Liu YC, Gao XX, Chen L, You XQ. Rapamycin suppresses Abeta25-35- or LPS-induced neuronal inflammation via modulation of NF-kappaB signaling. Neuroscience (2017) 355:188–99. 10.1016/j.neuroscience.2017.05.005 [DOI] [PubMed] [Google Scholar]
- 40.Ding Y, Zhang J, Wang R. Inhibition of tissue transglutaminase attenuates lipopolysaccharide-induced inflammation in glial cells through AKT/mTOR signal pathway. Biomed Pharmacother (2017) 89:1310–9. 10.1016/j.biopha.2017.03.027 [DOI] [PubMed] [Google Scholar]
- 41.Guo F, Zou Y, Zheng Y. Moracin M inhibits lipopolysaccharide-induced inflammatory responses in nucleus pulposus cells via regulating PI3K/Akt/mTOR phosphorylation. Int Immunopharmacol (2018) 58:80–6. 10.1016/j.intimp.2018.03.015 [DOI] [PubMed] [Google Scholar]
- 42.Weichhart T, Saemann MD. The multiple facets of mTOR in immunity. Trends Immunol (2009) 30(5):218–26. 10.1016/j.it.2009.02.002 [DOI] [PubMed] [Google Scholar]
- 43.Saemann MD, Haidinger M, Hecking M, Horl WH, Weichhart T. The multifunctional role of mTOR in innate immunity: implications for transplant immunity. Am J Transplant (2009) 9(12):2655–61. 10.1111/j.1600-6143.2009.02832.x [DOI] [PubMed] [Google Scholar]
- 44.Ren C, Zhang H, Wu TT, Yao YM. Autophagy: a potential therapeutic target for reversing sepsis-induced immunosuppression. Front Immunol (2017) 8:1832. 10.3389/fimmu.2017.01832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crisan TO, Plantinga TS, van de Veerdonk FL, Farcas MF, Stoffels M, Kullberg BJ, et al. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One (2011) 6(4):e18666. 10.1371/journal.pone.0018666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456(7219):264–8. 10.1038/nature07383 [DOI] [PubMed] [Google Scholar]
- 47.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 168(6):960–76. 10.1016/j.cell.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabanal-Ruiz Y, Otten EG, Korolchuk VI. mTORC1 as the main gateway to autophagy. Essays Biochem (2017) 61(6):565–84. 10.1042/EBC20170027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem (2011) 286(11):9587–97. 10.1074/jbc.M110.202911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giegerich AK, Kuchler L, Sha LK, Knape T, Heide H, Wittig I, et al. Autophagy-dependent PELI3 degradation inhibits proinflammatory IL1B expression. Autophagy (2014) 10(11):1937–52. 10.4161/auto.32178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chuang SY, Yang CH, Chou CC, Chiang YP, Chuang TH, Hsu LC. TLR-induced PAI-2 expression suppresses IL-1beta processing via increasing autophagy and NLRP3 degradation. Proc Natl Acad Sci U S A (2013) 110(40):16079–84. 10.1073/pnas.1306556110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiao M, Ren F, Zhou L, Zhang X, Zhang L, Wen T, et al. Peroxisome proliferator-activated receptor alpha activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death Dis (2014) 5:e1397. 10.1038/cddis.2014.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meng N, Wu L, Gao J, Zhao J, Su L, Su H, et al. Lipopolysaccharide induces autophagy through BIRC2 in human umbilical vein endothelial cells. J Cell Physiol (2010) 225(1):174–9. 10.1002/jcp.22210 [DOI] [PubMed] [Google Scholar]
- 54.Papinski D, Kraft C. Regulation of autophagy by signaling through the Atg1/ULK1 complex. J Mol Biol (2016) 428(9 Pt A):1725–41. 10.1016/j.jmb.2016.03.030 [DOI] [PubMed] [Google Scholar]
- 55.Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem (2017) 86:225–44. 10.1146/annurev-biochem-061516-044820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol (2012) 32(1):2–11. 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu Y, Liu J, Wu YF, Lou J, Mao YY, Shen HH, et al. mTOR and autophagy in regulation of acute lung injury: a review and perspective. Microbes Infect (2014) 16(9):727–34. 10.1016/j.micinf.2014.07.005 [DOI] [PubMed] [Google Scholar]
- 58.Wan J, Ma J, Anand V, Ramakrishnan S, Roy S. Morphine potentiates LPS-induced autophagy initiation but inhibits autophagosomal maturation through distinct TLR4-dependent and independent pathways. Acta Physiol (Oxf) (2015) 214(2):189–99. 10.1111/apha.12506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung KW, Kim KM, Choi YJ, An HJ, Lee B, Kim DH, et al. The critical role played by endotoxin-induced liver autophagy in the maintenance of lipid metabolism during sepsis. Autophagy (2017) 13(7):1113–29. 10.1080/15548627.2017.1319040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yen YT, Yang HR, Lo HC, Hsieh YC, Tsai SC, Hong CW, et al. Enhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injury. Surgery (2013) 153(5):689–98. 10.1016/j.surg.2012.11.021 [DOI] [PubMed] [Google Scholar]
- 61.Marshall JC. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med (2001) 29(7 Suppl):S99–106. 10.1097/00003246-200107001-00032 [DOI] [PubMed] [Google Scholar]